ECOLE NATIONALE DE MEDECINE VETERINAIRE
SIDI THABET Année 2016-2017NOTIONS
DE PHARMACOCINETIQUE
VETERINAIRE
PHARMACIE & TOXICOLOGIE
Pr Agrégé Samir BEN YOUSSEF Dr Jamel BELGUITH Dr Rim HADIJICINETIQUE DES XENOBIOTIQUES
DANS L'ORGANISME
Introduction – Importance 4
PREMIERE PARTIE :
BIODISPONIBILITE, BIOEQUIVALENCE,
MECANISMES DE PASSAGE TRANSMEMBRANAIRE
DES XENOBIOTIQUES
I. BIODISPONIBILITE, BIOEQUIVALENCE 7 1. Définitions 7 1.1 Biodisponibilité absolue 7 1.2 Biodisponibilité relative 8 2. Evaluation de la biodisponibilité 8 3. Facteurs de variation de la biodisponibilité 103.1. Facteurs pharmaceutiques 10
3.1.1. Facteurs liés au principe actif 10 3.1.2. Facteurs liés à la forme pharmaceutique 11
3.1.3. Interactions médicamenteuses 12
3.2. Facteurs physiopathologiques 13
II. MECANISMES DE PASSAGE TRANSMEMBRANAIRE 13
1. La membrane cellulaire 13
2. Les processus de passage transmembranaire 14
2.1. Transports passifs 15 2.1.1. Diffusion simple 15 2.1.2. La filtration 18 2.1.3. La diffusion facilitée 19 2.2. Transport actif 19 2.3. La pinocytose 20
3. Facteurs influençant le passage transmembranaire 20
DEUXIEME PARTIE : DEVENIR DES XENOBIOTIQUES
DANS L'ORGANISME
I. LA RESORPTION 25 1. La résorption digestive 26 1.1. La résorption gastrique 26 1.1.1. Monogastriques 26 1.1.2. Polygastriques 26 1.1.3. Oiseaux 27 1.2. La résorption intestinale 27 Avantages et inconvénients de la voie digestive 28
2. Résorption transcutanée et muqueuse 29
2.1. Voie transcutanée 29
2.2. Résorption par les muqueuses 29
3. Résorption pulmonaire 31
4. Résorption parentérale 32
4.1. Voie intraveineuse 32
4.2. Voie intra-péritonéale 33
4.3. Voies intramusculaire et sous cutanée 33 II. LA DISTRIBUTION 36
1. Transport sanguin 36 1.1. Fixation sur les protéines plasmatiques 36 1.2. Fixation sur les éléments figurés du sang 39
2. Diffusion tissulaire et cellulaire 39
3. Particularités d'organes 42 3.1. Pénétration dans le système nerveux central 42
3.2. Passage transplacentaire 43
III. BIOTRANSFORMATIONS 46
1. Réactions de biotransformations 46
1.1. Lieu des réactions 46
1.2. Principales réactions 47
1.2.1. Réactions de dégradation 47
1.2.2. Réactions de conjugaison 53
2. Facteurs de variation des biotransformations 57
2.2. Facteurs intrinsèques 57 2.3. Facteurs extrinsèques 59 2.3.1. L’induction enzymatique 59 2.3.2. L’inhibition enzymatique 61 IV. ELIMINATION 62 1. Elimination rénale 62 1.1. Filtration glomérulaire 62 1.2. Sécrétion tubulaire 63 1.3. Réabsorption tubulaire 64 2. Elimination digestive 66 2.1. Excrétion biliaire 66 2.2. Excrétion salivaire 68
2.3. Excrétion par la muqueuse digestive 69
3. Autres voies d'élimination 69
3.1. Elimination mammaire 69
3.2. Elimination par les œufs 72
3.3. Elimination pulmonaire 73
3.4. Elimination par la peau et les larmes 73
CINETIQUE DES XENOBIOTIQUES
DANS L'ORGANISME
INTRODUCTION
On entend par cinétique des médicaments et des toxiques ou pharmacocinétique (toxicocinétique pour les toxiques), l'étude quantitative et qualitative du devenir d'un xénobiotique dans l'organisme en fonction du temps après son introduction par l'une des voies d’administration.
Lorsqu'un xénobiotique est introduit dans l’organisme, après une phase de contact dite phase galénique, il subit un cheminement qui se résume en quatre étapes fondamentales (Figure 1).
La résorption (Absorption*) : c’est à dire le passage du site d'application dans la circulation générale
La distribution à partir de la circulation générale dans les différents tissus et organes.
Les biotransformations qui correspondent à des modifications biochimiques que subit le xénobiotique dans l'organisme
L'élimination du xénobiotique par les différentes voies d'excrétion
Le terme de xénobiotique (du grec xénos = étranger), désigne toute substance chimique etrangère à l’organisme, qu'elle soit médicamenteuse ou toxique.
Figure 1 : Place de la pharmacocinétique
(Pr. Jean-Dominique PUYT, ONIRIS)
Le terme d'absorption, synonyme d'ingestion en français est consacré par l’usage employé ; c’est le terme anglais ; il, s’agit en fait de résorption.
Le terme de métabolisme était autrefois synonyme de pharmacocinétique, à une époque où les biotransformations étaient pratiquement la seule étape pharmacocinétique que l'on connaissait.
IMPORTANCE
L'étude de la pharmacocinétique et de la toxicocinétique des xénobiotiques est importante à plus d'un titre.
Elle conditionne le choix des formes pharmaceutiques, de la voie d'administration et de la posologie chez les différentes espèces.
A ce titre, les études pharmacocinétiques constituent une partie importante du dossier pharmaco-toxicologique nécessaire l'obtention de l’autorisation de mise sur le marché (AMM) d'un médicament vétérinaire.
Par ailleurs, elles permettent la connaissance du niveau en résidus dans les denrées alimentaires d’origine animale, lequel est nécessaire pour la détermination du temps d’attente afin de garantir l’innocuité de ces denrées pour le consommateur.
Pour qu’un médicament agisse, il faut qu’il soit libéré de sa forme pharmaceutique et qu’il puisse traverser les barrières cellulaires pour atteindre son site d’action (Fig. 1bis), aussi le présent polycopié est divisé en deux parties :
La première partie est consacrée à l’étude de la biodisponibilité des xénobiotiques ainsi qu’à l’étude du passage transmembranaire des substances chimiques.
La deuxième partie, traite des quatre étapes du devenir d’un
xénobiotique dans l’organisme à savoir : la résorption, la distribution, les biotransformation et l’élimination.
Figure 1bis : Phase galénique et phase pharmacocinétique
PREMIERE PARTIE
BIODISPONIBILITE
ET BIOEQUIVALENCE
MECANISMES
DE PASSAGE
TRANSMEMBRANAIRE
DES XENOBIOTIQUES
Afin de mieux comprendre les quatre grandes étapes du devenir d’un médicament ou d’un toxique dans l’organisme, il est intéressant d’étudier dans un premier temps la notion de biodisponibilité et les mécanismes de passage transmembranaire.
I.
BIODISPONIBILITE, BIOEQUIVALENCE
1. Définitions
La biodisponibilité d’un xénobiotique correspond à la quantité de principe actif libérée de la forme pharmaceutique administrée qui parvient dans la circulation générale et à la vitesse avec laquelle elle y parvient.
Avant le développement de la pharmacocinétique, on admettait le concept ancien « dose administrée = dose résorbée ». Les études pharmacocinétiques ont prouvé que seulement une fraction de la quantité administrée est résorbée et atteint les sites d’action biologiques ou toxiques.
Par ailleurs, on a démontré que cette fraction résorbée dépend de la présentation du médicament, c’est à dire de la forme pharmaceutique.
Le concept de biodisponibilité s’est développé initialement pour évaluer l’efficacité thérapeutique de spécialités pharmaceutiques différentes, renfermant les mêmes quantités de principe actif après administration par la même voie. La biodisponibilité s’intéresse essentiellement aux évènements qui précèdent la résorption ; c'est-à-dire à la phase galénique.
Il s’agit pour un comprimé de son délitement et de la dissolution du principe actif dans le contenu du tube digestif, pour une suspension de sa dispersion dans le tissu conjonctif sous cutané ou intermusculaire puis de la dissolution du principe actif.
On définit deux types de biodisponibilité : la biodisponibilité relative et la biodisponibilité absolue.
Biodisponibilité absolue
Biodisponibilité relative
1.1. Biodisponibilité absolue
C’est la biodisponibilité d’un même principe actif administré par des voies différentes, elle permet de définir le rapport de la quantité de principe actif résorbé par une voie quelconque par comparaison à la voie intraveineuse
1.2. Biodisponibilité relative
C’est la biodisponibilité d’un principe actif sous des formes pharmaceutiques différentes administrés par la même voie.
Elle permet de comparer deux spécialités pharmaceutiques et d’évaluer l’intérêt de l’une sur l’autre. Elle permet également d’évaluer le rôle que jouent les excipients et les techniques de fabrication (pulvérisation, micronisation…etc.). La biodisponibilité relative a pour but de comparer les différences de résorption par une même voie d’administration entre des spécialités pharmaceutiques différentes de même composition qualitative et quantitative et présentées sous des formes galéniques identiques.
A une équivalence chimique ou à une équivalence galénique (même forme galénique) ne correspond pas toujours une équivalence thérapeutique (ou bioéquivalence). Ainsi deux spécialités pharmaceutiques seront dites
bioéquivalentes si elles procurent dans l'organisme des courbes d'évolution
des concentrations dans le temps similaires.
La bioéquivalence s'applique à un principe actif dans une forme galénique donnée et ne vaut que pour une voie d'administration précise.
Elle est surtout étudiée pour les spécialités pharmaceutiques destinées à la voie orale. On peut également comparer la bioéquivalence de lots de fabrication successifs d'une même spécialité.
On peut enfin évaluer la bioéquivalence de spécialités pharmaceutiques dans des formes galéniques différentes et apprécier l'intérêt d'une forme galénique sur une autre. On peut à partir de là établir le rôle possible des excipients ou de la technique de fabrication.
2. Evaluation de la biodisponibilité
La biodisponibilité est évaluée essentiellement à partir des courbes pharmacocinétiques d’évolution des concentrations sanguines en fonction du temps (Fig. 2)
La surface sous la courbe est directement proportionnelle à la quantité de principe actif qui atteint la circulation générale. Le temps qui sépare l’administration, du temps de pic de concentration plasmatique (Cmax.) est
appelé : Tmax. Il permet d’apprécier la vitesse de résorption.
La voie intraveineuse présente une biodisponibilité de 100% puisque toute la dose administrée parvient à la circulation générale.
Le coefficient de biodisponibilité : F, se calcule par rapport à la voie intraveineuse selon la formule suivante :
F% = (Surface sous la courbe IM / dose IM)
(Surface sous la courbe IV / dose IV)
On procède de la même manière pour la détermination du coefficient de biodisponibilité par les autres voies (orale, sous cutanée…etc.)
Cependant, le coefficient de biodisponibilité, s’il apporte une bonne indication sur la quantité de principe actif résorbée, n’apporte aucune information sur la vitesse du phénomène. Il est alors important de l’accompagner de Tmax. et Cmax.
Ceci est d’autant plus important que des spécialités ayant des coefficients de biodisponibilité identiques, peuvent avoir des effets biologiques ou toxiques différents.
Le coefficient de biodisponibilité absolue permet de comparer la résorption par différentes voies d’administration.
Le coefficient de biodisponibilité relative déterminé selon le même principe, permet quant à lui de comparer la résorption par la même voie d'un principe actif à partir de formes pharmaceutiques différentes spécialités.
La valeur du coefficient de biodisponibilité est insuffisante à elle seule pour comparer valablement des spécialités pharmaceutiques différentes. Si elle est une bonne indication de la quantité de principe actif résorbée, elle n'apporte aucune information sur la vitesse du phénomène.
On peut très bien obtenir avec deux spécialités pharmaceutiques différentes des courbes délimitant des surfaces identiques ; les coefficients de biodisponibilité sont alors identiques. Ces médicaments peuvent pourtant exercer des effets biologiques très différents. C'est pourquoi il faut prendre en compte simultanément le pic de concentration Cmax et le temps au bout duquel il est
En pratique, on admet que des spécialités pharmaceutiques (formulations galéniques identiques) sont bioéquivalentes et donc interchangeables s'il y a
moins de 20 % de différences entre :
leur AUC (surface sous la courbe) ou leurs cœfficients de
biodisponibilité F,
leur Tmax
leur Cmax
Pour compléter les études de bioéquivalence réalisées in vivo, on a mis au point des tests in vitro simples et rapides, susceptibles de donner une information complémentaire sur des différences éventuelles de biodisponibilité et donc garantir une bioéquivalence.
Les différences de biodisponibilité sont liées essentiellement à des différences de vitesse de dissolution dans l'eau du principe actif à partir de sa forme galénique. C'est en partie dans ce but qu'ont été mis au point des tests de
désagrégation (délitement) pour les comprimés ainsi que des tests de dissolution. En effet les différences de biodisponibilité ont principalement pour
origine des différences de mise en solution aqueuse des principes actifs avant leur résorption dans la circulation générale, condition préalable et nécessaire à tout passage transmembranaire.
3. Facteurs de variation de la biodisponibilité
La libération du principe actif à partir de la forme pharmaceutique, étape préalable à la résorption dépend de plusieurs facteurs à savoir des facteurs pharmaceutiques et des facteurs biologiques.
3.1. Facteurs pharmaceutiques
Ce sont des facteurs liés à la forme pharmaceutique, à ses caractéristiques physiques et chimiques.
3.1.1. Facteurs liés au principe actif a. Taille des particules
La biodisponibilité est inversement proportionnelle à la taille des particules , ce sont les particules les plus petites offrant le plus de surface de contact avec le milieu aqueux de l'organisme qui se dissolvent le mieux dans le tube digestif , dans le conjonctif sous cutané ou intramusculaire (cas des suspensions).
L'acétylsalicylate de lysine (ASPEGIC®) est une forme micronisée qui illustre
bien ce cas. Sa biodisponibilité est supérieure à celle des comprimés d'acide acétylsalicylique.
b. Etat d'hydratation
Les formes hydratées sont souvent moins solubles que les formes anhydres. Pour l'ampicilline par exemple, la biodisponibilité croit avec la croissance du taux d'hydratation de la forme pharmaceutique.
c. Etat cristallin
Les poudres amorphes sont en général plus hydrosolubles que les formes cristallines.
d. Forme chimique
Les esters et les sels d'un même principe actif n'ont pas la même solubilité d'où une biodisponibilité différente et des comportements pharmacocinétiques différents.
3.1.2. Facteurs liés à la forme pharmaceutique
Les excipients et les adjuvants des formes pharmaceutiques influent sur la dissolution du principe actif et par conséquent sur la biodisponibilité, le développement de "nouveaux systèmes d'administration " est à ce titre important à considérer.
a. Excipients
Les excipients aqueux sont ceux qui retiennent le moins le principe actif, leur dissolution est immédiate dans le contenu du tube digestif dans le liquide sous cutané ou intermusculaire.
Les solutions aqueuses sont mieux résorbées que les solutions huileuses qui sont en général des formes retards.
L'effet retard maximal est obtenu avec les formes solides ou implants sous cutanés (cas des anabolisants) permettant une libération continue et durable du principe actif.
b. Liants et désintégrants
Les liants des comprimés ralentissent la vitesse de leur désintégration diminuent la biodisponibilité. Les désintégrants ont un effet inverse
c. Nouveaux systèmes d'administration (NSA)
Voie orale
Les innovations les plus élaborées, concernent la voie digestive et la voie percutanée. Les formes pharmaceutiques développées visent la présence du
principe actif pendant la période de risque durant laquelle on veut protéger l'animal et sont dites "longue durée d'action".
Pour la prévention de la météorisation spumeuse chez les bovins au moment de la mise en herbe, on utilise des bolus de 165mm/38mm refermant un gel tensioactif.
La taille du bolus empêche son passage à travers l'orifice réticulo-omasal ou le rejet à travers le cardia d'où une localisation réticulo-ruminale stricte.
La libération du gel tensioactif s'étale sur 3 mois à partir cette forme.
D'autres formes pharmaceutiques sont développées pour les antiparasitaires, les anti-infectieux systémiques, les antibiotiques ionophores et les oligo-éléments.
Le système à l'origine de toute une famille est le PARATECT® (bolus qui libère
du morantel) à action préventive stricte.
Pour les anthelminthiques chez les bovins, des systèmes de "Pulse Release Bolus" sont développés. Ils assurent une libération de principes actifs systémiques à doses thérapeutiques sans intervention humaine.
Ces bolus sont administrés par la voie orale lors de la mise en pâturage des veaux. Ils se localisent alors dans le réseau. La première dose thérapeutique est libérée en 21-23 jours pour le SYNTHEX® puis les suivantes tous les 21-31
jours, la cinquième et dernière dose étant libérée au 115è - 120è jour après l'administration.
Pour l'électronic bolus " : VALBAZEN E BOLUS® constitué de " tubes collés sur
un socle commun et lourd, son arrivée dans l'humidité rumen-réseau déclenche un système électronique. Exactement 31jours plus tard, il y'a libération d'une dose d'anthelminthique, aussitôt s'enclenche la libération de la deuxième charge qui se produit 31 jours après, puis la 3ème et dernière dose thérapeutique est libérée 93 jours après l'ingestion avec une erreur de plus ou moins 15 secondes.
Voie percutanée
Pour la voie percutanée, les techniques dites "pour on" ou "spot on" (lorsque la quantité appliquée est faible) permettent la résorption de principes actifs appliqués le long de la région dorsolombaire avec une excellente biodisponibilité. Ces techniques seront développées dans l'étude de la résorption des médicaments et des toxiques.
3.1.3. Interactions médicamenteuses
L'association de plusieurs médicaments peut être à l'origine de modifications de la biodisponibilité par suite d'incompatibilités physico-chimiques ou de phénomènes de compétition lors de la résorption.
Incompatibilités physico-chimiques
Les cas des tétracyclines qui forment des chélates insolubles avec le calcium illustre bien les incompatibilités physico-chimiques à l'origine
d'une mauvaise résorption. C’est le cas également du charbon actif absorbant n'importe quel médicament, couramment utilisé dans le traitement des intoxications pour réduire la résorption des toxiques par la voie digestive.
3.2. Facteurs physio-pathologiques
Ce sont des facteurs liés à l'individu qui reçoit la forme pharmaceutique.
La vacuité, le pH du tube digestif, une accélération du transit lors de diarrhée sont autant des facteurs biologiques qui modifient la biodisponibilité.
II. Mécanismes de passage transmembranaire des xénobiotiques
Le cheminement d’un xénobiotique dans l’organisme repose sur la traversé de nombreuses membranes cellulaires : épithélium gastro-intestinal, paroi vasculaire, cellules de différents organes, épithélium des tubules rénaux…etc. Cette diversité des barrières membranaires recouvre cependant une unité structurale commune : la membrane cellulaire.
1. La membrane cellulaire
La membrane cellulaire est de nature lipoprotéique. Sa conception la plus récente est celle dite en « mosaïque fluide » de SINGER et NICHOLSON (1972). Elle apparait constituée par une double couche de phospholipides discontinue dans laquelle sont intercalés des protéines globulaires occupant la totalité ou partie de l’épaisseur de la membrane (Fig. 3).
Les molécules lipidiques sont faites d’un pôle hydrophobe dirigé vers l’intérieur et d’un pôle hydrophile dirigé vers l’extérieur (milieu extracellulaire ou intracellulaire).
La membrane cellulaire comporte des pores situés au niveau des axes centraux des protéines globulaires.
Ce modèle, met en évidence le caractère lipidique prédominant de la membrane cellulaire et l’existence de pores protéiques. Ces deux caractéristiques sont fondamentales pour comprendre les mécanismes de passage transmembranaire.
2. Les processus de passage transmembranaire
La traversée des membranes biologiques (diffusion transcellulaire) par les xénobiotiques s'opère par les mêmes mécanismes biochimiques mis en jeu par les cellules pour les transports des constituants endogènes. On distingue ainsi (Fig. 4 & 4bis) :
des transports passifs : - la diffusion simple
- la diffusion paracellulaire (entre deux cellules) = filtration, - la diffusion facilitée,
des transports actifs,
la pinocytose (endocytose et exocytose).
Figure 4 : Processus de passage transmembranaire des xénobiotiques.
Figure 4bis : La pinocytose
Transports passifs
2.1. Transports passifs 2.1.1. Diffusion simple
C’est le processus le plus fréquent, les substances en solution traversent la membrane par simple dissolution dans les phospholipides membranaires.
C’est un transport passif ne nécessitant aucune dépense d’énergie de la part de la cellule.
Les substances dissoutes migrent du milieu le plus concentré vers le milieu le moins concentré jusqu'à équilibre de part et d’autre de la membrane (notion de gradient de concentration). La vitesse de diffusion est donnée par la loi de FICK (Figure 5) :
Figure 5 : La loi de FICK
Du fait du caractère lipidique marqué de la membrane cellulaire, ce sont les
molécules liposolubles, peu ionisées qui diffusent le plus facilement.
La liposolubilité est le principal facteur limitant de la résorption des xénobiotiques par diffusion passive.
Ceci nous amène à considérer deux paramètres importants, jouant un rôle essentiel dans la diffusion à savoir le pH du milieu qui influe sur le degré d’ionisation d’une molécule et le coefficient de partage octanol/eau de cette molécule renseignent sur sa liposolubilité.
a. pH du milieu
On distingue d’une manière générale deux catégories de xénobiotiques :
- Des molécules neutres non ionisables,
- Des molécules ionisables qui sont soit des acides soit des bases définies par leur pKa ou logarithme négatif de leur constante de dissociation Ka.
L’équation d’HENDERSON-HASSELBACH permet de déterminer la concentration relative de la forme ionisée (fi) et de la forme non ionisée (fni) d’une substance en fonction du pH du milieu de dissolution :
log fni/fi = pH – pKa
Lorsque le pH est égal au pKa ; il ya équilibre entre les deux formes (log 1 = 0).
En pratique, dans le tube digestif, dans le milieu extracellulaire ou dans le milieu intracellulaire, ces substances se trouvent sous forme non ionisée et sous forme ionisée. C’est sous la forme non ionisée (liposoluble) que les
xénobiotiques diffusent bien à travers les membranes cellulaires, sous
forme ionisée ils pénètrent très peu.
Les molécules neutres, ne subissent aucune influence du pH.
Cas d’un acide faible
L’équation de dissolution d’un acide faible est la suivante :
AH A- + H+
L’équation d’HENDERSON-HASSELBACH donne les concentrations en fni et fi selon la formule suivante :
pKa - pH = Log [AH]/A- = log fni/fi
En conséquence,
Un acide faible sera d’autant moins ionisé que le milieu est acide
Ceci explique la très bonne résorption gastrique des acides faibles (Pénicilline G, salicylés, anticoagulants coumariniques et dérivés de l’indane-dione … etc.)
Cas d’une base faible
BH+ B + H+
L’équation d’HENDERSON-HASSELBACH donne les concentrations en fni et fi selon la formule suivante :
pH - pKa = Log [B]/BH+ = log fni/fi
En conséquence,
Une base faible sera d’autant moins ionisée que le milieu est basique
Ceci explique la résorption duodénale des bases faibles (tétracyclines, alcaloïdes...).
Le pH étant variable d’un compartiment à l’autre dans l’organisme, c'est-à-dire de part et d’autre d’une barrière membranaire, ceci entraine des déplacements des molécules chaque fois qu’elles se trouvent dans un milieu favorisant la fni (Fig 5).
Une différence d'une unité de pH entre les deux compartiments entraîne à l'équilibre environ 10 fois plus de molécules dans un compartiment que
dans l'autre.
On comprend alors le déplacement des molécules ionisables par suite de différences de pH entre des secteurs différents de l'organisme.
Figure 5’ : Diffusion passive d’un xénobiotique acide faible du sang vers un tissu
b. Coefficient de portage octanol-eau (huile-eau)
Le coefficient de partage octanol-eau permet de déterminer le degré de liposolubilité d’une substance. Celle-ci jouant un rôle fondamental dans le passage transmembranaire.
Ce coefficient est mesuré par évaluation de la distribution d’une substance entre l’octanol et l’eau (ou le chloroforme et l’eau).
Plus sa valeur est grande, plus la substance est liposoluble et plus elle traversera facilement la membrane cellulaire.
L’exemple des barbituriques, montre la relation parallèle entre le coefficient octanol-eau du composé et l’intensité de l’activité pharmacologique traduisant une meilleure traversée transmembranaire (Tableau I).
Cependant, ce coefficient de partage octanol-eau ne doit pas avoir des valeurs très élevées. En effet, lors de la diffusion passive, la substance passe
obligatoirement dans le milieu aqueux pour atteindre la membrane, de ce fait elle doit posséder un certain degré d’hydrosolubilité pour pouvoir se déplacer. Les substances exclusivement liposolubles sont incapables de franchir les barrières cellulaires (Vaseline, paraffine, …etc.) et sont utilisés pour leurs effets locaux (laxatifs).
Tableau I
Liposolubilité et activité pharmacologique de quelques barbituriques
BARBITURIQUE COEFFICIENT DE PARTAGE
OCTANOL-EAU ACTIVITE Phénobarbital Pentobarbital Penthiobarbital 3 39 580 Sédatif-hypnotique action lente Anesthésique général action rapide Anesthésique général action très rapide
Au bilan, ce sont les molécules liposolubles et légèrement hydrophiles qui sont les mieux diffusibles. Les composés exclusivement liposolubles ou insolubles dans l’eau comme de nombreux métaux ne traversent pas les membranes par diffusion passive.
2.1.2. Filtration
La filtration constitue le passage des substances chimiques à travers les pores membranaires ou les espaces intercellulaires.
Par conséquent, seules les substances hydrosolubles ou à un moindre degré hydrosolubles, de faible poids moléculaire sont intéressés par ce processus. Etant donné la nature protéique des pores et leur dimension, les deux facteurs qui conditionnent la filtration sont l’hydrosolubilité du composé et son
poids moléculaire (intéresse les médicaments de petite taille).
Le processus de filtration est un processus passif qui dépend du diamètre des pores, de leur nombre ainsi que du gradient de concentration de part et d’autre de la membrane.
En pratique, dans la plupart des cas le diamètre de ces pores est compris entre et A (1 A = 10-10 mètre), ce qui laisse filtrer uniquement les petites molécules de PM ‹ D environ. Il ne s’agit que d’ions minéraux (Na+, k+, Ca++,…etc.), les molécules organiques sont toutes de taille plus importante.
La majorité des xénobiotiques a une masse relative comprise entre 200 et 500 D, ce qui interdit habituellement leur passage par transport par les canaux ioniques.
On note cependant deux exceptions anatomiques : les capillaires sanguins des territoires hépatique, rénaux et ceux musculaires, au niveau desquels le diamètre des pores est plus important. Au niveau des reins, le diamètre est de 8 A permettant la filtration de molécules jusqu’à un PM voisin de 69000 Daltons.
2.1.3. Diffusion facilitée
Elle diffère du transfert actif dans le fait qu’elle s’effectue dans le sens du gradient de concentration, sans dépense d'énergie, mais à une vitesse supérieure à celle d’un simple phénomène de diffusion passive. Le glucose, la vitamine B12 pénètrent par ce mécanisme. La diffusion de molécules de glucose
à travers la membrane cytoplasmique nécessite une protéine « transporteur spécifique au glucose ».
2.2. Transport actif : Transfert actif par l’intermédiaire de systèmes de transport
Contrairement aux deux mécanismes précédents, le transfert par l’intermédiaire de systèmes de transport constitue un mécanisme actif nécessitant de l’énergie. Il fait intervenir des molécules transporteuses spécifiques et peut se faire contre un gradient de concentration.
Les transports actifs nécessitent une participation active de la part des cellules sous forme d'énergie. Ils impliquent également des transporteurs
spécifiques et agissent contre le gradient de concentration.
On distingue des transporteurs d’efflux et d’influx ; parmi les plus importants figurent :
des glycoprotéines P qui sont surtout des transporteurs d’efflux, encore appelées pompes à efflux,
des transporteurs d’anions organiques (OAT), spécialisés dans le transport de xénobiotiques acides,
des transporteurs de cations organiques (OCT), spécialisés dans le transport de xénobiotiques basiques.
Les molécules transporteuses se caractérisent par :
Leur spécificité pour certaines structures.
Leur saturation, leur nombre étant fixe, Au-delà de certaines teneurs, leurs capacités de transport, en particulier pour les substances acides, peuvent être dépassées
La possibilité de compétition de deux composés pour le même transporteur (Pénicilline G et probénicide).
La possibilité d’une inhibition par combinaison irréversible avec une substance inhibitrice.
Le transfert actif est un mécanisme se faisant contre un gradient de concentration c'est-à-dire du milieu le moins concentré vers le milieu le plus concentré. Il nécessite de l’énergie fournie par hydrolyse de l’ATP.
Ce transfert intervient principalement au niveau du rein (sécrétion tubulaire active), des hépatocytes (excrétion biliaire) et des plexus choroïdes.
Les pompes à efflux jouent un rôle majeur dans la barrière hémato-méningée au niveau des plexus choroïdes du cerveau pour l'élimination des xénobiotiques présents dans le système nerveux central.
Les teneurs en xénobiotiques dans l’urine ou la bile peuvent atteindre fois et plus les teneurs sanguines.
il existe des glycoprotéines membranaires P (P-gp) rejettent dans la lumière intestinale les molécules qui ont diffusé passivement dans les entérocytes. Ainsi ces pompes vont à l'encontre de la résorption intestinale de certaines molécules.
2.3. La pinocytose
La pinocytose est un processus analogue à la phagocytose qui se produit pour des éléments solides.
Elle correspond à une invagination de la membrane cellulaire pour englober une gouttelette lipidique du milieu extérieur, former une vésicule qui traverse toute la membrane pour libérer son contenu dans le cytoplasme ou le milieu intérieur. Elle intéresse des molécules liposolubles (vitamines A, D, E, et K) et elle est particulièrement développée chez le jeune.
3. Facteurs biologiques influençant le passage transmembranaire
Des facteurs biologiques liés à l'organisme animal interviennent surtout dans les transports passifs mais aussi, pour certains, dans les transports actifs.
Ils sont variés. Les plus importants sont la surface d'échange, le temps de contact, le gradient de concentration et les interférences avec des constituants endogènes.
a. Surface d'échange
L'intensité des échanges de substances au travers des membranes biologiques entre deux territoires (compartiments) est proportionnelle à la surface de
cette membrane.
Plus la surface membranaire qui sépare deux compartiments est importante, plus les échanges sont favorisés. Le contact est en effet ainsi facilité.
Cette différence de surface entre la paroi stomacale et celle de l'intestin grêle avec ses microvillosités contribue à ce que l'intestin soit le principal site de résorption des médicaments administrés par voie orale, tout comme des aliments. La très grande surface d’échange représentée par l’épithélium pulmonaire adapté pour l’oxygénation du sang permet des échanges gazeux très intenses.
b. Temps de contact
L'intensité des échanges de substances au travers des membranes biologiques entre deux territoires (compartiments) est également proportionnelle au
temps de contact des molécules avec la membrane qui sépare les deux
compartiments.
Ainsi l'accélération du transit digestif lors de diarrhée est défavorable à la résorption aussi bien des nutriments que des xénobiotiques.
c. Gradient de concentration
L'intensité des échanges de substances au travers des membranes biologiques est enfin proportionnelle au gradient de concentration, c'est-à dire à la différence de concentrations de xénobiotique de part et d'autre de la membrane. Ce gradient a une importance prépondérante dans les transports passifs car les échanges s'opèrent conformément à la loi de FICK, du milieu le plus concentré vers le moins concentré, et ce jusqu'à équilibre de concentration de part et d'autre de la membrane.
Tout facteur qui tend à diminuer ce gradient contribue à réduire les passages transmembranaires. Ainsi les médicaments pris en cours de repas subissent une dilution dans le contenu alimentaire, ce qui abaisse le gradient de concentration et, de là, réduit la résorption digestive des principes actifs.
Ainsi, le contenu ruminal entraîne une dilution très importante des médicaments par voie orale, d'où le faible recours à cette voie d'administration médicamenteuse chez les ruminants.
Inversement, la présence d'une vascularisation importante au niveau de l'intestin grêle au cours de la digestion favorise l'entraînement immédiat des nutriments et des xénobiotiques qui viennent de franchir la barrière digestive dans le courant sanguin, ce qui tend à maintenir un gradient de concentration élevé.
d. Interférences avec des constituants endogènes
Un certain nombre de constituants endogènes peuvent interférer avec les xénobiotiques et avoir une influence directe sur le taux de passages transmembranaires. C'est le cas notamment des lipides, des protéines et du calcium.
• Interférences avec les lipides
Certains organes sont particulièrement riches en lipides ; c’est le cas notamment du système nerveux et du foie. Les xénobiotiques liposolubles présentent une très forte affinité pour les lipides et s’y concentrent. Ainsi tous les médicaments psychotropes sont liposolubles ; leurs teneurs dans le système nerveux qui conditionne directement leur puissance d’action est proportionnelle à leur liposolubilité.
• Interférences avec les protéines
Les protéines sont des constituants importants du sang (albumine, globulines) et de nombreux tissus et liquides biologiques. Beaucoup de molécules présentent une affinité pour les protéines.
Cette propriété peut exercer une influence sur la distribution tissulaire des xénobiotiques. Cette influence est connue sous le nom d'effet DONNAN.
En fonction de l'affinité pour les protéines et des différences de concentrations en protéines de part et d'autre de la membrane biologique, le passage transmembranaire des xénobiotiques peut être facilité ou retardé.
Ce phénomène est bien connu pour les pénicillines qui, dans les conditions normales, ne sont pas présentes dans le liquide céphalorachidien ; mais lors de méningite le liquide céphalo-rachidien contient des protéines d'origine inflammatoire sur lesquelles elles se fixent, ce qui permet d'atteindre des teneurs assez importantes.
Certaines protéines à groupements thiols tels que la cystine des phanères ou les métallothionéines, localisées notamment dans le foie et le rein, présentent une affinité élevée pour les métaux et métalloïdes thioloprives (arsenic, plomb, mercure, cuivre…etc.). Ceci explique la concentration dans ces organes de ces métaux lors d'intoxication.
• Interférences avec le calcium
Une affinité de certains xénobiotiques pour le calcium endogène des dents ou des os explique leur distribution particulière dans ces tissus, voire leur accumulation préférentielle. Ils forment avec le calcium des chélates insolubles qui précipitent. C'est le cas de certains antibiotiques comme les tétracyclines qui forment des complexes avec les métaux divalents ; c'est le cas encore du fluor qui est à l'origine de lésions osseuses et dentaires lors de fluorose en rapport avec son affinité pour le tissu osseux.
De même, la présence de calcium non endogène, mais alimentaire (lait) peut limiter la résorption de certains xénobiotiques administrés par voie orale ; ils forment également des chélates insolubles qui sont directement éliminés par voie fécale sans pouvoir être résorbés.
L’étude de la biodisponibilité d’une part et des mécanismes de passage transmembranaire d’autre part nous permet de mieux comprendre les différentes étapes du devenir d’un xénobiotique dans l’organisme objet de la deuxième partie de ce polycopié.
DEUXIEME PARTIE
CINETIQUE DES XENOBIOTIQUES
DANS L’ORGANISME
I.
LA RESORPTION DES XENOBIOTIQUES
La résorption correspond à l’étape par laquelle un xénobiotique passe de son site d’application dans la circulation générale.
Selon le mode d’administration, on distingue deux types de résorption :
La résorption médiate : le médicament est administré à l’extérieur de l’organisme ou dans une lumière (administration cutanée ou muqueuse ou par la voie digestive).
La résorption immédiate : le médicament est directement introduit dans le liquide extracellulaire au sein d’un tissu (administration parentérale).
Lors d’administration intraveineuse (IV) où le médicament est introduit directement dans le sang, la phase de résorption est supprimée (shuntée). La vitesse de résorption est variable selon les différentes voies d’administration. Le choix de celle-ci dépend souvent de l’indication d’urgence de l’intervention (Fig. 6).
Figure 6 : Vitesse de résorption d’un xénobiotique
et concentration plasmatique selon différentes voies d’administration. IV IP IM SC Per os Temps Concentration plasmatique
1. LA RESORPTION DIGESTIVE
Après ingestion d'un xénobiotique, la résorption digestive, dite encore orale ou entérale, peut s'effectuer à tous les étages du tractus digestif mais en pratique surtout dans l’intestin grêle et l’estomac. La voie rectale est moins utilisée en médecine vétérinaire.
La résorption digestive des xénobiotiques s'opère surtout par diffusion
simple, ce qui suppose une liposolubilité marquée du principe actif.
Afin de gagner la circulation générale, un xénobiotique administré per os est appelé à franchir la muqueuse digestive. Celle-ci peut être considérée comme une membrane lipidique à pores favorable à la résorption des substances liposolubles.
La résorption gastrique
En regard des particularités anatomo-physiologiques des différentes espèces, la résorption gastrique est différente chez les monogastriques, les polygastriques et les oiseaux.
Monogastriques
Les monogastriques présentent les particularités suivantes :
Un pH très acide du milieu stomacal, voisin de 1 à 2 chez le chien et le chat et de 4 à 5 chez le cheval et le veau.
Un faible volume : 0,1 à 1 litre chez les carnivores, 8 à 15 litres chez le cheval.
Une vidange complète de l’estomac entre les repas chez les carnivores et le porc. Chez le cheval, l’estomac n’est jamais vide. Il en résulte qu’au niveau stomacal chez les monogastriques, ce sont les acides
faibles qui vont se trouver sous la forme non ionisée qui seront les résorbés.
Leur résorption est moins bonne chez le cheval et veau dont le pH du contenu stomacal est relativement plus élevé.
Le faible volume de l’estomac, limite par ailleurs le phénomène de dilution stomacale des principes actifs phénomène défavorable à la résorption.
La connaissance de la vidange stomacale permet de connaitre le moment propice d’administration du médicament per os sachant que la résorption est meilleure dans un estomac vide.
Polygastriques
Les particularités anatomo-physiologiques des polygastriques sont :
Un réservoir gastrique de 150 litres environ chez le bovin et de 50 litres chez les ovins et les caprins ce qui représente un facteur important de dilution.
Un pH de 5,5 à 6,5
La présence d’une µ-flore ruminale importante exerçant une activité enzymatique de dégradation sur de nombreux médicaments.
Ces particularités expliquent la résorption souvent médiocre des médicaments par la voie orale chez les poly gastriques qui est peu utilisée pour obtenir une action générale.
Oiseaux
Les particularités anatomo-physiologiques des oiseaux sont : Un jabot à activité motrice irrégulière, à pH acide. Un système lymphatique intestinal peu développé.
Il en résulte que la résorption des acides faibles se fait au niveau du jabot par diffusion passive avec toutefois une possibilité de stockage de certains médicaments auquel fait suite une libération prolongée dans le temps.
Le système lymphatique intestinal peu développé, ne permet pas une bonne résorption des composés liposolubles et les concentrations plasmatiques efficaces ne sont parfois pas atteintes (chlorotétracycline … etc.)
Il existe des différences de résorption importantes entre les mammifères et les oiseaux. Pour l’ampicilline par exemple, des doses 2 à 8 fois plus importantes sont nécessaires pour atteindre chez les oiseaux des concentrations sanguines équivalentes à celles des mammifères.
La résorption intestinale
La résorption intestinale se fait surtout dans l’intestin grêle et particulièrement au niveau du duodénum (pH 5-6).
La muqueuse intestinale représente une surface d’échange considérable, intensément vascularisée, grâce aux villosités et microvillosités. Ces villosités augmentent cette surface de 10 à 20 fois.
Les bases faibles, présentent une résorption intestinale excellente, la forme non ionisée prédominant dans l’intestin.
Par ailleurs, si théoriquement les acides faibles doivent être moins bien résorbés qu’au niveau de l’estomac, en pratique leur résorption intestinale est meilleure du fait de l’importance de la surface d’échange.
La vascularisation importante en drainant rapidement tout ce qui est résorbé, concourt au maintien du gradient de concentration favorable à la résorption. L'intestin grêle possède une vascularisation porte très importante. Cette irrigation sanguine facilite l'entraînement des xénobiotiques qui viennent de franchir la barrière digestive pour arriver dans la circulation générale. Ceci concourt au maintien d'un gradient de concentration élevé, propice à leur résorption intestinale. La fraction qui franchit la barrière digestive est immédiatement entraînée dans le courant sanguin.
Au bilan, les xénobiotiques qui arrivent dans le tube digestif sont résorbés par diffusion passive au niveau de l’estomac et surtout de l’intestin grêle.
Les autres mécanismes sont moins fréquents. Pour arriver au niveau du sang, la plupart des xénobiotiques filtrent à travers la paroi capillaire sanguine, les grosses molécules lipidiques et protéiques empruntent la voie lymphatique.
Avantages et inconvénients de la voie digestive :
Le principal avantage est la facilité d’administration des médicaments chez les petits animaux (comprimés, gélules, sirops…) et les grands animaux (aliments médicamenteux, additifs, …etc.).
Les inconvénients en revanche sont nombreux :
- Certains composés sont dégradés au niveau de l’estomac par l’acidité importante (ce qui est gênant quand il s’agit de médicaments : pénicilline G, hormones naturelles,…etc.).
- Certains médicaments forment des complexes insolubles non
résorbables avec les cations divalents (tétracyclines – Ca++ ou Mg++) ce qui diminue leur biodisponibilité.
- Tous les composés résorbés par la muqueuse digestive empruntent la veine porte, passent obligatoirement par le foie ou ils subissent l’action d’enzymes de biotransformation, ce processus en général diminue notablement la quantité de médicament actif et donc l’activité
biologique. Ce processus est connu sous le nom de ‘‘first past effect’’
= ‘‘effet de premier passage hépatique’’.
Ceci n’est pas le cas pour la voie rectale, qui présente l’avantage
d’acheminer directement dans la circulation générale le médicament en évitant l’effet de premier passage hépatique par la traversée initiale du foie. La voie rectale peut être intéressante chez les carnivores
domestiques en cas d'urgence du fait de sa facilité d'accès, notamment pour l’administration d’anticonvulsivants tels que le diazépam lors d'intoxication par des convulsivants.
- En toxicologie la voie digestive constitue la voie la plus fréquemment empruntée dans laquelle on observe des fois une activation des toxiques ingérés (réduction des nitrates en nitrites, plus toxiques).
Facteurs de variation de la résorption digestive :
- La résorption digestive est meilleure dans un tractus digestif vide qu’en présence d’aliments qui diluent le composé.
- Les aliments gras facilitent la résorption des médicaments mais aussi des toxiques (ne jamais administrer de lait ou d’huile lors d’intoxication par la voie orale).
- L’accélération du transit intestinal lors de diarrhée est défavorable à la résorption.
2. LA RESORPTION TRANSCUTANEE ET MUQUEUSE
Voie Transcutanée
La peau constitue une barrière naturelle protectrice constituée du derme et de l’épiderme lesquels sont formés par un ensemble de couches cellulaires, l’épiderme comprend une couche cornée de cellules mortes à l’extérieur très peu perméable.
D’une manière générale, l’application cutanée a pour but un effet purement local. En effet, la structure de la peau est telle que seules les substances très liposolubles peuvent franchir la barrière cutanée.
Chez l’animal, la résorption peut se faire au niveau de l’appareil pilo-sébacé très développé (2000 follicules pileux /cm²).
En dehors de la liposolubilité du principe actif, la résorption transcutanée dépend d’autres facteurs :
o Nature des excipients : le rôle des excipients est très important .Ce sont les excipients huileux qui favorisent la pénétration. Le diméthyl-sulfoxyde (DMSO), entraine une augmentation considérable de la résorption, il constitue l’excipient des formes « pour on » et « spot on » qui permettent le traitement de parasitoses internes en versant le produit le long de la région dorsolombaire de l’animal.
Ainsi, le traitement des strongyloses digestives et pulmonaires et de l’hypodermose bovine s’effectuent par cette méthode.
La biodisponibilité du lévamisole « pour on » est comparable voire supérieure à la biodisponibilité des formes orales.
o Facteurs pathologiques : l’inflammation de la peau, la présence de lésions cutanées sont autant de facteurs favorisant la résorption. des précautions d’application (traitement d’une partie du corps seulement à la fois) doivent être prises lors de lésions très étendues sous risque d’accidents médicamenteux avec certains insecticides (organo-phosphorés).
La voie cutanée joue un rôle important en toxicologie, c’est une zone de résorption importante pour les pesticides en solution organique, le solvant favorisant la pénétration.
Résorption par les muqueuses :
Malgré une résorption plus importante par les muqueuses que par la peau en raison de l’absence de couche cornée et d’une vascularisation plus importante, cette voie est en général utilisée en thérapeutique pour des effets purement locaux.
Voie vaginale et utérine :
Lors de traitements locaux, vaginaux ou utérins soit par dépôt d’oblets gynécologiques d’antibiotiques soit par injection intra-utérine on assiste à une résorption trans-muqueuse.
La nature des composés conditionne la résorption. C’est ainsi que le benzyl-pénicillinate de sodium est facilement résorbé alors que les pénicillines de semi-synthèse le sont faiblement.
La voie vaginale est également utilisée pour réaliser certains traitements hormonaux généraux de synchronisation de l’œstrus chez les ruminants par mise en place :
- Chez les bovins d’une spirale vaginale d’élastomère siliconé qui libère lentement la progestérone.
- Chez les ovins, d’éponges vaginales en mousse de polyuréthane imprégnées d’un progestagène de synthèse (Médroxyprogestérone, Fluorogestone) qui libère le progestagéne, qui est alors résorbé pour produire les effets hormonaux d’inhibition de l’ovulation.
Dès le retrait de la spirale chez les bovins ou de l’éponge chez la brebis, il se produit une élimination du progestagène, libération d’hormones gonadotropes, apparition de chaleurs et ovulation.
Autres muqueuses :
La résorption par les muqueuses oculaire ou auriculaire est très faible. Ce sont des voies utilisées exclusivement pour des traitements locaux.
3. RESORPTION PULMONAIRE
La résorption par la voie respiratoire est favorisée par :
La grande surface d’échange (5 à m² chez l’homme)
L’épaisseur très faible de l’épithélium respiratoire ( ,2 à 2 microns)
Cette résorption concerne les composés gazeux et volatils liposolubles. Elle trouve des applications en anesthésie gazeuse et constitue une voie importante de pénétration des toxiques gazeux et volatils comme le CO2, CO, SO2, SH2, HCN…etc.
La profondeur de la pénétration des xénobiotiques par la voie pulmonaire dépend de la taille des particules, seules les particules dont la taille est de l’ordre de l’ordre de à 3 µm parviennent jusqu’aux alvéoles pulmonaires et sont bien résorbées.
Certains médicaments sont administrés par la voie pulmonaire sous forme d’aérosols.
Les aérosols sont des dispersions dans l’air de très fines gouttelettes dont le diamètre est inférieur à 5 microns, capables de rester en suspension dans l’air. Inspirés elles atteignent les alvéoles pulmonaires.
La pénétration des aérosols chez les volailles se répartit comme suit en fonction de la taille des gouttelettes :
Taille (microns) 10 3 3-1 1
Zone atteinte Narines Trachée Alvéoles Sacs aériens
Les aérosols sont utilisés chez l’homme (asthme, bronchites), le cheval et pour la vaccination chez les volailles.
4. RESORPTION PARENTERALE
La résorption parentérale (étymologiquement, "autre que la voie orale") concerne principalement les voies intraveineuse, intramusculaire, sous-cutanée et intrapéritonéale. Toutes ces voies sont aussi dénommées par abus de langage "injectables".
L’administration des médicaments par la voie parentérale est très utilisée en médecine vétérinaire. Ce mode d’administration trouve des avantages dans le fait qu’il permet : d’éviter « l’effet de premier passage hépatique », d’obtenir des effets rapides avec une posologie précise.
Voie intraveineuse : IV
Lors de l’administration d’un médicament par la voie intraveineuse (directement dans le sang), la phase de résorption est supprimée (shuntée).
Des concentrations sanguines importantes d’anti-infectieux peuvent être atteintes dans les infections graves.
Avantages et inconvénients de la voie intraveineuse
- Des substances irritantes par la voie intramusculaire peuvent être administrées par la voie intraveineuse.
- Cependant, il est nécessaire d’injecter des solutions aqueuses, les suspensions et solutions huileuses sont à proscrire (risque d’embolies).
- Les solutions doivent être isotoniques.
- Une injection trop rapide peut entrainer des troubles cardio-respiratoires graves, en pratique, ces injections doivent être faites lentement.
- Enfin lors d’injection IV, l’élimination est rapide, la filtration glomérulaire étant très sollicitée.
La perfusion intraveineuse
C’est une injection intra veineuse lente, prolongée et à débit constant.
L’augmentation des concentrations sanguines est progressive jusqu’à un état d’équilibre avec la quantité éliminée dans le même intervalle.
La perfusion est utilisée pour administrer des volumes liquidiens importants ou des médicaments à demi-vie brève.
Voie intra-péritonéale
Le péritoine constitue une surface d’échange importante, bien irriguée par des capillaires sanguins et lymphatiques. La résorption par cette voie est presque aussi rapide que par la voie intra veineuse.
La voie intra-péritonéale est parfois employée en médecine vétérinaire, surtout chez les bovins ainsi qu'en toxicologie expérimentale chez les petits rongeurs. C'est une voie très facile, peu dangereuse, indolore et extrêmement rapide dans les effets biologiques produits.
Elle présente cependant l’inconvénient de gagner la circulation générale par la veine porte et donc de faire subir aux médicaments « l’effet de premier passage hépatique ».
• Voies intramusculaire et sous-cutanée
Ce sont les voies les plus utilisées en médecine vétérinaire. Ces deux voies présentent des caractères communs.
Après injection, on distingue deux temps dans la résorption :
o Dans un premier temps, les formes liquides injectées se répartissent dans la trame conjonctive qui entoure les faisceaux musculaires ou dans le tissu conjonctif sous-cutané. Ceci suppose une hydrosolubilité des molécules. Tout excipient hydrophobe gêne cette mise en solution. C'est pourquoi les médicaments en solution aqueuse sont le plus vite résorbés puisque leurs principes actifs sont déjà au départ dans une forme dissoute dans une phase hydromiscible.
Pour faciliter cette mise en solution, des adjuvants comme la hyaluronidase sont d'ailleurs parfois ajoutés dans certaines préparations destinées à la voie sous-cutanée. Elle est destinée à faciliter la diffusion des xénobiotiques dans le tissu conjonctif sous-cutané (T.C.S.C.) en dépolymérisant l'acide hyaluronique, ce qui facilite le relâchement du tissu conjonctif et augmente la mobilité de l'eau interstitielle.
o Dans un second temps, la résorption se fait par diffusion passive ou par filtration à travers les pores des capillaires sanguins.
Les xénobiotiques, une fois dissous dans l'eau extracellulaire, sont tout de suite résorbés aussi bien par filtration au niveau des lames basales entre les cellules endothéliales des vaisseaux capillaires que par diffusion
simple après dissolution dans la couche lipidique membranaire. Les
principes actifs mis au préalable en solution aqueuse sont ainsi immédiatement résorbés. De ce fait, les principes actifs présentés en
solution aqueuse ou dans un excipient hydromiscible ont une résorption
très rapide et exercent un effet immédiat et intense.
En revanche les suspensions aqueuses ou les solutions huileuses forment un dépôt au site de leur injection à partir duquel les principes actifs liposolubles ou insolubles sont progressivement libérés et mis en solution dans la phase aqueuse du T.C.S.C. avant d'être résorbés. C'est pourquoi les médicaments présentés en suspension ou en solution huileuse ont un effet moins rapide mais plus durable que les mêmes présentés en solution aqueuse.
Selon la nature des sels ou des esters ainsi que des excipients employés cette mise en solution aqueuse sera plus ou moins rapide. Un "effet
retard" peut ainsi être recherché pour permettre au médicament d'agir
plus longtemps, ce qui évite le renouvellement trop fréquent des injections.
C'est le cas d'un antibiotique comme la pénicilline G qui existe sous forme de sel de sodium hydrosoluble d'action immédiate (3 heures), de sels organiques peu dissociables et donc peu solubles dans l'eau ; ainsi le sel de procaïne en suspension aqueuse possède une action retard qui se prolonge une dizaine d’heures et le sel de benzathine en suspension aqueuse retard exerce un effet pendant 15 jours à 3 semaines. De telles
présentations sont dites"semi-retard", "retard" ou "à longue action
(LA)" selon les cas.
Notez qu’il arrive que l’on recherche à retarder la résorption sous-cutanée de certains médicaments en réduisant le débit sanguin local par addition de vasoconstricteurs. C'est le cas de certains anesthésiques locaux comme la procaïne qui est parfois associée à de l'adrénaline (procaïne adrénalinée) afin de prolonger l’anesthésie locale. La rapidité de la résorption est voisine pour les deux voies sous cutanée et intra musculaire, la voie intra musculaire étant un peu plus rapide par suite d’une meilleure vascularisation des muscles.
Les macromolécules (Toxines, venins) sont résorbées par voie lymphatique. Facteurs de variation de la résorption
La vitesse de résorption varie essentiellement en fonction de la forme pharmaceutique et de l’association des principes actifs avec d’autres composés.
o Forme pharmaceutique
Ce sont les solutions aqueuses qui sont les plus rapidement résorbées par filtration à travers les espaces intracellulaires (de diamètre important 30 à 40 A°) qui séparent les cellules endothéliales des capillaires sanguins. Les molécules par la suite franchissent la paroi capillaire par diffusion passive. Les solutions huileuses permettent-elles de ralentir la résorption des principes actifs.
Les suspensions aqueuses ou huileuses dans lesquelles le principe actif est insoluble procurent un effet retard important. Le principe actif étant libéré lentement par hydrolyse. Cette hydrolyse est d’autant plus lente que le radical salifiant ou estérifiant est plus long.
L’effet retard maximum est obtenu par la pose d’implants ou de pellets solides en sous-cutanée. C’est un mode d’administration des hormones permettent une imprégnation faible et continue de l’organisme, l’implant se dissolvant lentement.
o Associations avec d’autres composés
L’association d’un principe actif avec la hyaluronidase qui hydrolyse la substance fondamentale (acide hyaloronique) augmente la résorption suite à une diminution de la viscosité au niveau du tissu conjonctif.
A l’inverse, l’association d’anesthésiques locaux avec la noradrénaline diminue la résorption et augmente l’action locale de l’anesthésique. La noradrénaline étant vasoconstrictrice.
Certains composés ne sont pas résorbés par la voie intramusculaire comme les hétérosides cardiotoniques (digitaline, ouabaïne) en raison de leur forte affinité pour les fibres musculaires.
La résorption des xénobiotiques est une étape très importante à connaitre pour le vétérinaire. Celui-ci selon la rapidité de l’action souhaitée, les propriétés chimiques du principe actif, les caractéristiques de la forme pharmaceutiques opte pour telle ou telle voie d’administration.
En toxicologie sa connaissance permet de mettre en route la première composante du traitement des intoxications visant une limitation de la pénétration du toxique.
La résorption par ailleurs prépare la deuxième étape de la cinétique des médicaments et des toxiques dans l’organisme, à savoir la distribution.
II. LA DISTRIBUTION DES XENOBIOTIQUES
La distribution constitue la deuxième étape du devenir d’un xénobiotique dans l’organisme après sa résorption. Le médicament ou le toxique pour rejoindre leur site d’action sont véhiculés principalement par le sang qui les diffuse dans les différents tissus.
Cette distribution comprend deux aspects que nous envisagerons successivement :
o Le transport sanguin o La diffusion tissulaire
1. Le transport sanguin
Les xénobiotiques sont véhiculés sous deux formes dans le sang :
• Une forme libre, seule capable de diffuser dans les tissus et d’atteindre les sites d’action.
•
• Une forme liée, essentiellement par fixation sur les protéines plasmatiques, secondairement sur les éléments figurés du sang.
La proportion de ces deux formes et ses variations jouent un rôle essentiel. • La forme libre concerne surtout des formes chimiques hydrosolubles,
par conséquent soit les substances neutres ou ionisables (ionisées ou non) hydrosolubles, soit les formes ionisées et dissociées des molécules acides ou basiques liposolubles. Une partie des formes liposolubles (en proportion de leur coefficient de partage lipides/eau) est également solubilisée dans la phase aqueuse sanguine.
• La forme liée concerne surtout les fractions liposolubles ou insolubles dans le sang qui ne peuvent être transportées que fixées sur certains constituants sanguins, protéines et/ou cellules du sang.
1.1. Fixation sur les protéines plasmatiques
Les composés qui se fixent aux protéines plasmatiques sont les composés liposolubles ou insolubles dans le sang qui ne peuvent être transportés sous la forme libre.
La fixation protéique se fait essentiellement sur l’albumine, protéine plasmatique la plus importante (≈40g/L ; 50 % des protéines plasmatiques).
Certains composés se fixent sur des globulines particulières. C’est le cas de l’hydrocortisone qui se fixe sur la transcortine, c’est aussi le cas des androgènes et des œstrogènes qui se fixent sur la Testostérone Estradiol Binding Globulin (T.E.B.G.).
Cette fixation se fait habituellement par des liaisons de faible énergie (liaison hydrogène, hydrophobe, électrostatique ou de VAN DER WAALS). Elle est immédiatement réversible.
Les deux formes libre et liée se trouvent toujours en équilibre dans le sang. Lorsque la liaison est covalente, stable, des processus toxiques peuvent apparaitre.
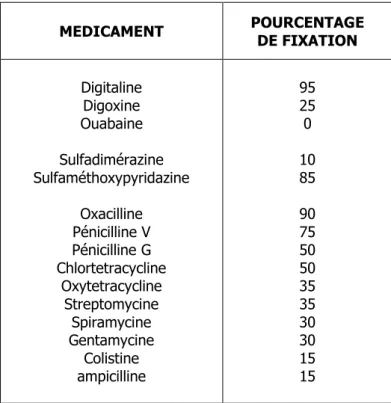
Le tableau II indique le pourcentage de fixation protéique de quelques principes actifs usuels.
Conséquences de la fixation protéique
- Des substances liposolubles ou insolubles dans le sang sont ainsi véhiculées, leurs taux de fixation sont élevés.
- La forme libre est la seule forme diffusible aux travers des membranes biologiques ; en effet sous sa forme liée, hydrosoluble et de poids moléculaire élevée, le xénobiotique ne peut franchir les membranes biologiques ni par diffusion simple, ni par filtration. Il s'ensuit que la forme libre est la seule forme biologiquement active voire toxique.
- La forme liée des xénobiotiques constitue une forme de "stockage" dans l'organisme ; le sang exerce ainsi un certain rôle de réservoir. Le sang par sa richesse en protéines par rapport aux liquides extracellulaires retient ainsi de nombreux xénobiotiques. Ce phénomène rend compte en partie, de l'action retard des sulfamides liposolubles méthoxylés.
- L’administration simultanée de deux composés (surtout les acides en raison du nombre limité des sites de fixation sur l'albumine) à fort taux de fixation protéique peut conduire à des phénomènes de compétition. Il en résulte une augmentation de la forme libre active qui peut engendrer des effets toxiques ou une diminution de la demi-vie plasmatique par suite d’une élimination accrue. Ainsi, les anticoagulants sont facilement déplacés par l’acide acétylsalicylique ou la phénylbutazone. La pénicilline G est de la même façon déplacée par les sulfamides.