Single-domain antibodies and their utility
Texte intégral
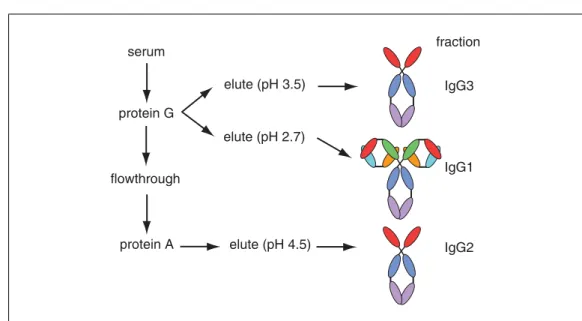
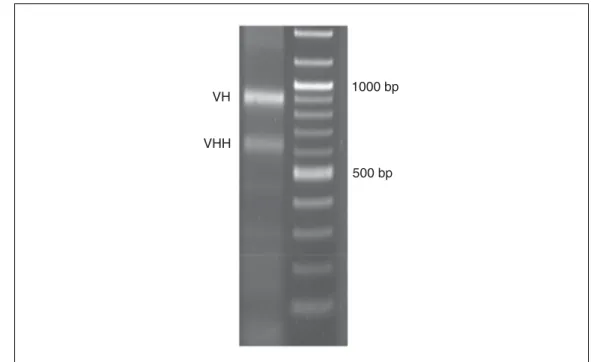
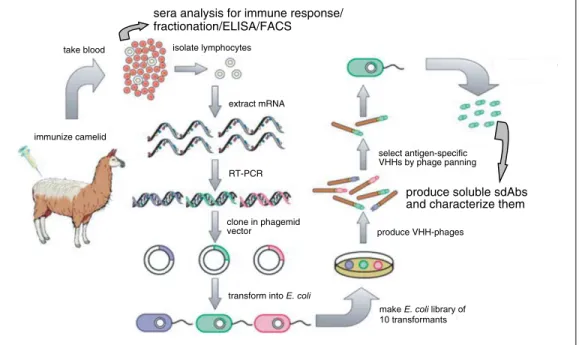
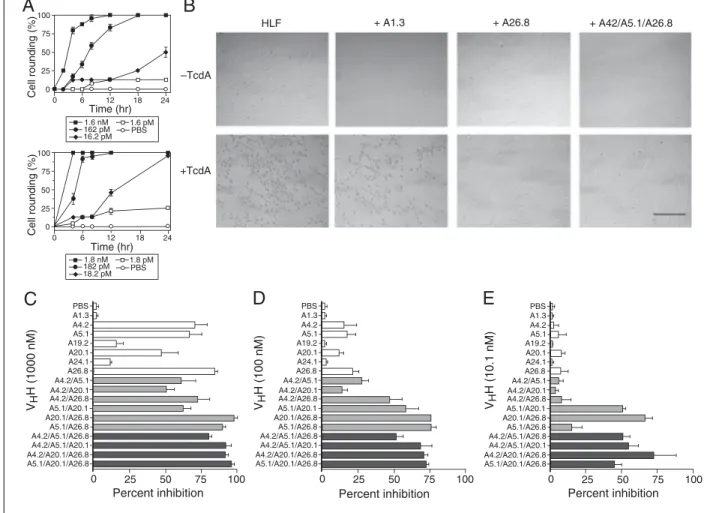
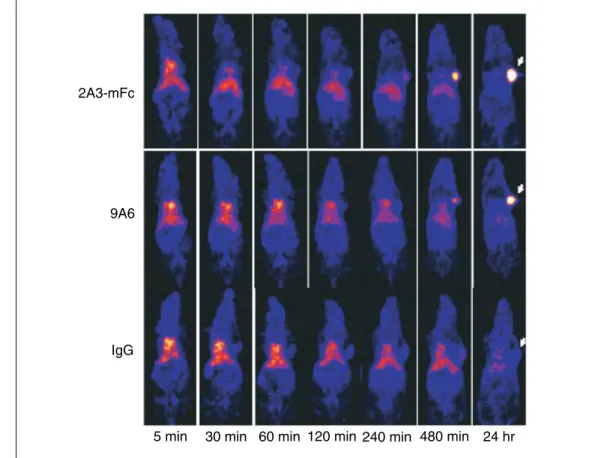
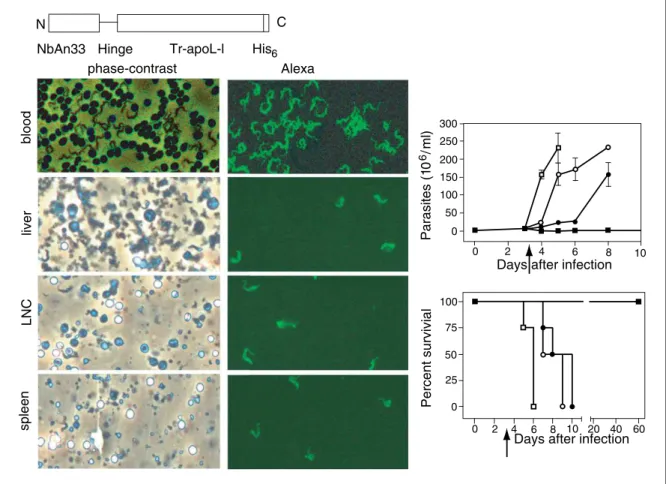
Figure


Documents relatifs
If anonymous users have read and write access to the same area, the server can be used as a communication mechanism to anonymously exchange information.. Servers which
As with UBR connections, entities using VBR-nrt connections for IP over ATM should take advantage of parameter negotiation by specifying PCR equal to the link rate in the
Ethernet technology, as defined by the 802.3 Working Group of the IEEE, continues to evolve, with scalable increases in speed, new types of cabling and interfaces, and
When a node receives a Multicast-Address-Specific Query, if it is listening to the queried Multicast Address on the interface from which the Query was received, it sets a
Even if in this memo we focus on e-mail addresses, a number of elements defined in this specification can also be used for other specifications dealing with embedding
4 - If the server supports this sorting control but for some reason cannot sort the search results using the specified sort keys and the client specified FALSE for
If the CEK from MSG1 is to be used to derive the KEK for MSG2 then MSG1 MUST contain an unprotectedAttrs Attribute of type id-aa-CEKReference with a single value using
Several objectives for this methods have been identified, like to enable an e-mail user to send and receive faxes from his/her e-mail interface, to allow some kind of
