HAL Id: cea-03134426
https://hal-cea.archives-ouvertes.fr/cea-03134426
Submitted on 8 Feb 2021
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Distributed under a Creative Commons Attribution| 4.0 International License
Complexes-Solid-State Interactions
Young Lee, Jee Kim, Sotaro Kusumoto, Hitomi Ohmagari, Miki Hasegawa,
Pierre Thuéry, Jack Harrowfield, Shinya Hayami, Yang Kim
To cite this version:
Young Lee, Jee Kim, Sotaro Kusumoto, Hitomi Ohmagari, Miki Hasegawa, et al.. Functionalised
Terpyridines and Their Metal Complexes-Solid-State Interactions. Chemistry , Wiley, 2021, 3 (1),
pp.199-227. �10.3390/chemistry3010016�. �cea-03134426�
Article
Functionalised Terpyridines and Their Metal Complexes—
Solid‐State Interactions
†Young Hoon Lee 1, Jee Young Kim 2, Sotaro Kusumoto 3, Hitomi Ohmagari 4, Miki Hasegawa 4, Pierre Thuéry 5,
Jack Harrowfield 6,*, Shinya Hayami 3 and Yang Kim 3
1 Department of Chemistry, University of Ulsan, Ulsan 44610, Korea; [email protected] 2 Department of Chemistry & Advanced Materials, Kosin University, 194 Wachiro, Yongdo‐Gu, Busan 49104, Korea; [email protected] 3 Department of Chemistry, Graduate School of Science and Technology, Kumamoto University, 2‐39‐1 Kurokami, Chuo‐ku, Kumamoto 860‐8555, Japan; [email protected]‐u.ac.jp (S.K.); hayami@kumamoto‐u.ac.jp (S.H.); ykim@kumamoto‐u.ac.jp (Y.K.) 4 Department of Chemistry & Biological Science, College of Science & Engineering, Aoyama Gakuin University, Sagamihara, Kanagawa 252‐5258, Japan; [email protected] (H.O.); [email protected] (M.H.) 5 Université Paris–Saclay, CEA, CNRS, NIMBE, 91191 Gif‐sur‐Yvette, France; [email protected] 6 ISIS, Université de Strasbourg, 8 allée Gaspard Monge, 67083 Strasbourg, France * Correspondence: [email protected]; Tel.: +33‐03‐6885‐5165 † Dedicated to Professor Christoph Janiak in recognition of his seminal contributions to the study of weak interactions in crystalline coordination complexes. Abstract: Analysis of the weak interactions within the crystal structures of 33 complexes of various 4′‐aromatic derivatives of 2,2′:6′,2″‐terpyridine (tpy) shows that interactions that exceed dispersion are dominated, as expected, by cation...anion contacts but are associated with both ligand–ligand and ligand–solvent contacts, sometimes multicentred, in generally complicated arrays, probably largely determined by dispersion interactions between stacked aromatic units. With V(V) as the coordinating cation, there is evidence that the polarisation of the ligand results in an interaction exceeding dispersion at a carbon bound to nitrogen with oxygen or fluorine, an interaction unseen in the structures of M(II) (M = Fe, Co, Ni, Cu, Zn, Ru and Cd) complexes, except when 1,2,3‐trimethoxyphenyl substituents are present in the 4′‐tpy. Keywords: terpyridines; metal complexes; crystal structures; Hirshfeld surfaces; weak interactions 1. Introduction Ligands based on the 2,2′:6′,2″‐terpyridine (tpy) substructure have been employed in an extraordinary variety of studies concerning their metal‐ion coordination chemistry [1– 7]. Much of this chemistry has concerned condensed phases—crystals, liquid crystals, monolayers on surfaces, intercalation complexes, and films—where interactions between complex ion units and between them and their environment can be particularly important, as is well‐known for 2,2′:6′,2″‐terpyridine itself [8]. The postulate of the “terpyridine embrace” [9–12] as a means of rationalising the forms of the lattices of many crystalline terpyridine complexes provides a long‐known example of the recognition of this fact. This form of supposed aromatic–aromatic interaction, however, is known not to be universally operative for such complexes [11,13,14], and more importantly, it is well recognised that terms, such as “π‐stacking” and “π–π interactions” (as well as “OFF” = offset face‐to‐face and “EF” = edge‐to‐face aromatic interactions), widely applied to the description of putative interactions between aromatic entities [15–17] can be rather eclectically interpreted and that there may be infrequent reason to attribute special significance to the association of small aromatic systems beyond the recognition of dispersion interactions
Citation: Lee, Y.H.; Kim, J.Y.; Kusumoto, S.; Ohmagari, H.; Hasegawa, M.; Thuéry, P.; Harrowfield, J.; Hayami, S.; Kim, Y. Functionalised Terpyridines and Their Metal Complexes—Solid‐State Interactions. Chemistry 2021, 3, 199– 227. https://doi.org/10.3390/ chemistry3010016 Received: 12 January 2021 Accepted: 27 January 2021 Published: 5 February 2021 Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. Copyright: © 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses /by/4.0/).
[18–24]. An even broader issue is that of whether intermolecular interactions should be considered parallel to molecular bonding in being the result of the combination of proximal two‐centre interactions [25–27]. Thus, the dissection of a crystal lattice in terms of atom– atom “contacts”, which may or may not be indicative of attractive, labile interactions, is not a straightforward task [25–32], so in the work now reported, we crystallographically characterised a series of complexes of related 4′ derivatives of 2,2′:6′,2″‐terpyridine (Figure 1) in the hope that systematic structural variations might facilitate the identification of major interactions. To assess the interactions within the lattices, we employed the calculation of Hirshfeld surfaces [29] using the program CrystalExplorer [30]. This work is a complement to other structural studies of functionalised terpyridines and their complexes that we have reported [14,33–37] and provides results to be integrated with the present and with the extant literature in general. The ultimate objective, in general, is, of course, to use such information in the design and synthesis of functional materials [38]. Figure 1. 4′‐substituted derivatives of 2,2′:6′,2″‐terpyridine studied in the present work. 2. Experimental Section 2.1. General
Dichloromethane and acetonitrile (spectrophotometric grade) for the measurement of electronic absorption and emission spectra were purchased from Merck (Seoul, S. Korea). Other organic solvents were purchased from Aldrich (Basel, Switzerland). Reagents were
purchased from Wako (Osaka, Japan) (pyridin‐4‐yl‐4‐boronic acid and
4‐(ethoxycarbonyl)phenylboronic acid), Tokyo Chemical Industry (Tokyo, Japan) (benzylbromide, 2,2′:6′,2″‐terpyridine (tpy), 1‐bromo‐4‐iodobenzene, Pd(PPh3)4 and
1,2,3,4,5‐pentafluoro‐6‐iodobenzene), Alfa Aesar (Sulzbach, Germany)
((trimethylsilyl)acetylene and 1‐ethynylbenzene) and Aldrich (Basel, Switzerland) (diisopropylamine, CuI, ZnCl2, CdCl2, RuCl3∙xH2O, Co(BF4)2∙6H2O, Fe(ClO4)2∙H2O,
Ni(ClO4)2∙6H2O, Cu(ClO4)2∙6H2O, Zn(ClO4)2∙6H2O, Cd(ClO4)2∙H2O and K2CO3) and used without further purification. Elemental analyses (C, H and N) were carried out at the
Instrumental Analysis Centre of Kumamoto University, Japan. 1H‐NMR spectra were
recorded with JEOL 500‐ECX (500 MHz) and Bruker 300 AM spectrometers (300.13 MHz) in deuterated solvents using TMS as an internal reference. Electronic absorption spectra were recorded on SCINCO S‐2100 and Varian Cary 100 spectrophotometers. Fluorescence spectra were recorded on Perkin Elmer LS55 and HORIBA FluoroMax‐4P instruments.
1‐bromo‐4‐(2‐phenylethynyl)benzene [39], 2,2′:6′,2″‐terpyridine‐4′‐ol [40], 2,6‐bis(pyridin‐2‐yl)pyridin‐4‐yl trifluoromethanesulfonate [41], 2‐(4‐ethynyl‐6‐(pyridin‐2‐yl)pyridin‐2‐yl)pyridine (4′‐ethynyl‐2,2′:6′,2′′‐terpyridine) [42], 4′‐(biphenyl‐4‐yl)‐2,2′:6′,2′′‐terpyridine (bptpy) [43], 4′‐(tolyl)‐2,2′:6′,2′′‐terpyridine (ttpy) [44], 4′‐(3,4,5‐trimethoxy‐phenyl)‐2,2′:6′,2′′‐terpyridine (tmptpy) [45], 2‐(4‐(2‐phenylethynyl)‐6‐(pyridin‐2‐yl)pyridin‐2‐yl)pyridine (patpy) [46–48], 4′‐((ethoxycarbonyl)biphenyl‐4‐yl)‐2,2′:6′,2″‐terpyridine (ebptpy) [49], 4′‐((ethoxycarbonyl)terphenyl‐4‐yl)‐2,2′:6′,2″‐terpyridine (etptpyb) [33], 4′‐(benzyloxy)‐2,2′:6′,2″‐terpyridine, (bzOtpy) [33],
4′‐(pentafluorophenyl)‐2,2′:6′,2′′‐terpyridine (pfpatpy) [33], 4′‐terphenyl‐2,2′:6′,2′′‐terpyridine (tptpy) [34], 4′‐(4′‐pyridin‐4‐yl‐biphenyl‐4‐yl)‐2,2′:6′,2′′‐terpyridine (pybptpy) [35],
4′‐(4‐bromobiphenyl‐4‐yl)‐2,2′:6′,2′′‐terpyridine (Brbptpy) [36],
2‐(4‐(2‐(3,4,5‐trimethoxyphenyl)ethynyl)‐6‐(pyridin‐2‐yl)pyridin‐2‐yl)pyridine (tmpatpy)
[37] and [Ru(tpy)Cl3] [50] were prepared by literature methods. The synthesis of the ligand 4′‐(4″′‐(ethynylphenyl)phenylethynyl)‐2,2′:6′,2′′‐terpyridine (papatpy) is described below along with that of its complexes. 2.2. Synthesis CAUTION! Most of the complexes described were isolated as perchlorate salts and are thus potential explosives. Although no difficulties were encountered, such materials should always be prepared in the minimum possible quantities. 2.2.1. Vanadium(V) Complex of 4′‐(Biphenyl)‐2,2′:6′,2′′‐terpyridine, Bptpy
(1) [VO2(bptpy)]ClO4: Bptpy (120 mg, 0.3 mmol) was dissolved in CHCl3/CH3OH (20 mL, 1:1, v/v) and VOSO4∙3H2O (65 mg, 0.3 mmol) in methanol (10 mL), added with stirring. Under exposure to a normal atmosphere, the initially blue solution slowly changed colour to yellow–green over several hours, after which NaClO4 (0.4 g) was dissolved in it, and the solution was allowed to slowly evaporate under ambient temperature to produce yellow crystals suitable for an X‐ray diffraction study (yield: 90 mg, 53%). Anal. calc. for C27H19ClN3O6V: C, 57.11; H, 3.37; N, 7.40. Found: C, 56.88; H, 3.50; N, 7.26%. CCDC number, 2055133.
2.2.2. Complexes of 4′‐Terphenyl‐2,2′:6′,2′′‐terpyridine, Tptpy (and Mixed Complexes with 2,2′:6′,2′′‐Terpyridine)
(2) [Fe(tptpy)2](ClO4)2∙CH3OH: Fe(ClO4)2∙6H2O (9.0 mg, 0.025 mmol) in DMF (5 mL) was added to a solution of tptpy (30 mg, 0.065 mmol) in hot DMF (5 mL). After stirring the solution for 2 h at 100 °C, the solvent was removed under reduced pressure. The deep purple residue was washed with methanol and chloroform, and dried at ambient temperature (yield: 25 mg, quantitative). Anal. calc. for C67H49Cl2N6FeO9: C, 66.57; H, 4.09; N, 6.95. Found: C, 66.52; H, 4.07; N, 6.98%. 1H‐NMR (300 MHz) in CD3CN: δ 9.26 (s, 4H), 8.66 (d, J = 8.0 Hz, 4H), 8.46 (d, J = 8.0 Hz, 4H), 8.17 (d, J = 8.0 Hz, 4H), 7.99 (d, J = 8.0 Hz, 4H), 7.94 (t, J = 8.5 Hz, 4H), 7.88 (d, J = 8.0 Hz, 4H), 7.78 (d, J = 7.5 Hz, 4H), 7.55 (t, J = 7.5 Hz, 4H), 7.45 (t, J = 7.5 Hz, 2H), 7.22 (d, J = 5.5 Hz, 4H), 7.11 (t, J = 7.0 Hz, 4H). The vapour diffusion of methanol into a solution of the complex in a minimum volume of DMF produced purple crystals suitable for X‐ray diffraction studies. CCDC number, 2055134.
(3) [Ni(tptpy)2](ClO4)2∙CH3OH: Ni(ClO4)2∙6H2O (8.0 mg, 0.022 mmol) in methanol (5 mL) was added to a solution of tptpy (20 mg, 0.043 mmol) in CHCl3/CH3OH (6 mL, 1:1, v/v). After stirring the solution for 1 h at 60 °C, the solvent was removed under reduced pressure. The yellow residue was washed with methanol and chloroform, and then dried at ambient temperature (yield: 20 mg, 91%). Anal. calc. for C67H49Cl2N6NiO9: C, 66.41; H, 4.08; N, 6.94. Found: C, 66.50; H, 4.02; N, 7.01%. The vapour diffusion of methanol into a solution of the complex in a minimum volume of DMF produced yellow crystals suitable for X‐ray diffraction studies. CCDC number, 2055135.
(4) [Ru(tptpy)2](PF6)2∙3CH3CN: A mixture of tptpy (100 mg, 0.22 mmol) and
RuCl3∙xH2O (50 mg, 0.22 mmol) in ethanol (30 mL) was heated at 70 °C for 6 h to produce a turbid, reddish solution. After cooling to ambient temperature, the brown precipitate formed was collected by filtration, washed with cold ethanol and then dried under vacuum. The elemental analyses were consistent with its formulation as [Ru(tptpy)Cl3] (yield: 80 mg). [Ru(tptpy)Cl3] (30 mg, 0.045 mmol), tptpy (30 mg, 0.065 mmol) and
N‐ethylmorpholine (five drops) were added to methanol/chloroform (30 mL, 1:1, v/v), and
the mixture was heated at 70 °C for 2 h. The resulting deep red solution was filtered through Celite to remove a small amount of purple precipitate. The filtrate was loaded on a short silica column, and the column eluted with acetonitrile/saturated aqueous KNO3/water at 7:1:0.5, v/v. The major red component was collected, and methanolic NH4PF6 was added to the eluate to produce a red powder within 1 h. This was collected by filtration, washed with methanol and then dried under vacuum (yield: 30 mg, 50%). Anal. calc. for C72H55F12N9P2Ru: C, 60.17; H, 3.86; N, 8.77. Found: C, 60.22; H, 3.83; N, 8.80%. Electronic absorption spectrum (in CH3CN): λmax (εmax/M−1 cm−1) = 285 nm (58,900), 312 nm (75,500), 330 nm (72,300), 494 nm (32,100). 1H‐NMR (500 MHz) in CD3CN: δ 9.08 (s, 4H), 8.69 (d, J = 7.5 Hz, 4H), 8.35 (d, J = 8.0 Hz, 4H), 8.13 (d, J = 8.0 Hz, 4H), 7.98 (m, 8H), 7.87 (d, J = 7.5 Hz, 4H), 7.54 (t, J = 8.0 Hz, 4H), 7.46 (m, 6H), 7.21 (t, J = 6.5 Hz, 4H). The slow evaporation of an acetonitrile solution of the complex at ambient temperature produced red crystals suitable for X‐ray structure determination. CCDC number, 2055219.
(5) [Ru(tptpy)(tpy)](PF6)2∙2DMF: Tptpy (60 mg, 0.13 mmol), [Ru(tpy)Cl3] (50 mg, 0.11 mmol) and N‐ethylmorpholine (five drops) were added to methanol/chloroform (30 mL, 1:1, v/v), and the mixture was heated at 70 °C for 2 h. The resulting deep red solution was filtered through Celite to remove a small amount of brown precipitate, and the filtrate was subjected to chromatography on silica using acetonitrile/saturated aqueous KNO3/water at 7:1:0.5, v/v, as the eluant. The major red component was collected, and methanolic NH4PF6 was added to the eluate to produce a red precipitate, which was filtered and washed with methanol, and then dried in a vacuum (yield: 23 mg, 20%). Anal. calc. for C54H48F12N8O2P2Ru: C, 52.64; H, 3.93; N, 9.10. Found: C, 52.55; H, 3.85; N, 9.05%. IR spectrum (KBr disc, cm−1): 3106, 3038, 3030 (Ar CH), 863 (PF6). Electronic absorption spectrum (in CH3CN): λmax (εmax, M−1 cm−1) = 275 nm (43,700), 282 nm (43,900), 309 nm (73,900), 485 nm (24,800). 1H‐NMR (500 MHz) in CD3CN: δ 9.06 (s, 2H), 8.75 (d, J = 8.0 Hz, 2H), 8.67 (d, J = 8.0 Hz, 2H), 8.50 (d, J = 8.5 Hz, 2H), 8.42 (t, J = 8.5 Hz, 1H), 8.33 (d, J = 8.0 Hz, 2H), 8.12 (d, J = 8.0 Hz, 2H), 7.96 (m, 7H), 7.86 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 7.5 Hz, 2H), 7.54 (t, J = 7.5 Hz, 2H), 7.44 (d, J = 5.0 Hz, 2H), 7.35 (d, J = 5.5 Hz, 2H), 7.18 (m, 4H). Upon dissolving the complex in hot DMF and then allowing the solution to stand for one week at ambient temperature, red crystals suitable for X‐ray structure determination were obtained. CCDC number, 2055220.
(6) [Ru(tptpy)(tpy)](ClO4)2∙2DMF: [Ru(tptpy)(tpy)](PF6)2 (20 mg, 0.018 mmol) was dissolved in DMF (3 mL), and LiClO4 (0.01 g) was added. The slow deposition of red crystals suitable for X‐ray structure determination occurred upon leaving the solution to stand at ambient temperature. Anal. calc. for C54H48Cl2N8O10Ru: C, 56.84; H, 4.24; N, 9.82. Found: C, 56.78; H, 4.27; N, 9.78%. Electronic absorption spectrum in CH3CN: λmax (εmax, M−1 cm−1) = 274 nm (44,000), 281 nm (43,300), 308 nm (71,400), 484 nm (23,600). 1H‐NMR (500 MHz) in CD3CN: δ 9.06 (s, 2H), 8.75 (d, J = 8.5 Hz, 2H), 8.67 (d, J = 8.0 Hz, 2H), 8.50 (d,
(m, 7H), 7.86 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 8.5 Hz, 2H), 7.54 (t, J = 7.5 Hz, 2H), 7.44 (d, J = 6.0 Hz, 2H), 7.35 (d, J = 5.0 Hz, 2H), 7.18 (m, 4H). CCDC number, 2055221.
2.2.3. Zinc(II) Complex of 2‐(4‐(2‐Phenylethynyl)‐6‐(pyridin‐2‐yl)yyridin‐2‐yl)pyridine, Patpy
(7) [Zn(patpy)2](ClO4)2 (YK758): Zn(ClO4)2∙6H2O (30 mg, 0.08 mmol) and patpy (50 mg, 0.15 mmol) were dissolved in acetonitrile (10 mL) and heated at 70 °C for 2 h. After cooling to ambient temperature, the solvent was removed by evaporation under reduced pressure. The colourless residue was washed with methanol and dichloromethane and dried at ambient temperature (yield: 39 mg, 53%). Anal. calc. for C46H30Cl2N6ZnO8: C, 59.34; H, 3.25; N, 9.03. Found: C, 59.41; H, 3.32; N, 9.20%. 1H‐NMR (300 MHz) in CD3CN:
δ 8.90 (s, 4H), 8.61 (d, J = 8.0 Hz, 4H), 8.22 (td, J = 7.9, 1.6 Hz, 4H), 7.86 (m, 8H), 7.62 (m,
6H), 7.46 (dd, J = 5.1, 0.9 Hz, 2H), 7.43 (dd, J = 5.1, 0.9 Hz, 2H). The slow evaporation of an acetonitrile solution of the complex at ambient temperature provided colourless crystals suitable for X‐ray diffraction studies. CCDC number, 2055222.
2.2.4. Complexes of 4′‐(4″′‐(Ethynylphenyl)phenylethynyl)‐2,2′:6′,2′′‐terpyridine, Papatpy
(8) papatpy: 2‐(4‐ethynyl‐6‐(pyridin‐2‐yl)pyridin‐2‐yl)pyridine (0.25 g, 0.97 mmol),
1‐bromo‐4‐(2‐phenylethynyl)benzene (0.30 g, 1.12 mmol), Pd(PPh3)4 (0.10 g, 0.086 mmol) and CuI (0.020 g, 0.11 mmol) were placed into a three‐necked round‐bottom flask under a nitrogen atmosphere. Degassed i‐Pr2NH/THF (30 mL, 1:1, v/v) was added to the mixture and heated under reflux for 12 h under nitrogen. The resultant dark brown, turbid solution was filtered, and the filter was washed with THF. The filtrate and wash solution were evaporated to dryness under reduced pressure, and then dissolved in a minimum volume of dichloromethane. Adsorption on a short silica column and elution with ethylacetate/CH2Cl2 at 1:4, v/v, produced a colourless eluate, which was evaporated under reduced pressure to produce a white powder (yield: 0.20 g). 1H‐NMR (300 MHz) in CDCl3: δ 8.78 (dq, J = 4.1, 0.8 Hz, 2H), 8.69 (d, J = 8.0 Hz, 2H), 8.12 (s, 2H), 7.96 (td, J = 7.8, 1.8 Hz, 2H), 7.59 (m, 6H), 7.43 (m, 5H). Anal. calc. for C31H19N3∙0.5H2O: C, 84.14; H, 4.56; N, 9.50. Found: C, 83.70; H, 4.48; N, 9.33%.
(9) [Co(papatpy)2](BF4)3∙2CH3CN: Co(BF4)2∙6H2O (0.0080 g, 0.023 mmol) in DMF (5 mL) was mixed with papatpy (20 mg, 0.046 mmol) in DMF (10 mL). After heating this solution for 2 h at 100 °C, the solvent was removed under reduced pressure to produce a brown residue, which was washed with methanol and chloroform, and then dried at ambient temperature (yield: 20 mg, 69%). Anal. calc. for C66H44B3CoF12N8: C, 62.49; H, 3.50; N, 8.83. Found: C, 62.55; H, 3.42; N, 8.90%. The vapour diffusion of diethyl ether into an acetonitrile solution of the complex provided yellow, rod‐like crystals suitable for X‐ray diffraction studies. The structure determination and colour of the complex showed that the oxidation of Co(II) to Co(III) had occurred during the synthesis. CCDC number, 2055223.
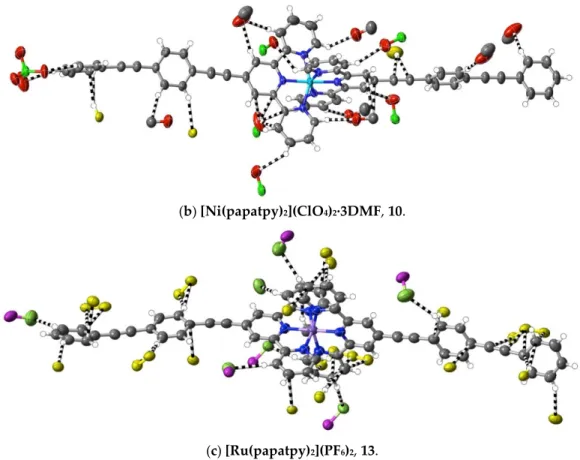
(10) [Ni(papatpy)2](ClO4)2∙3DMF: Ni(ClO4)2∙6H2O (9.0 mg, 0.025 mmol) in DMF (5 mL) was mixed with papatpy (20 mg, 0.046 mmol) in DMF (10 mL). After heating this solution for 2 h at 100 °C, the solvent was removed under reduced pressure to produce a yellow residue, which was washed with methanol and chloroform, and then dried at ambient temperature (yield: 20 mg, 63%). Anal. calc. for C71H59Cl2N9NiO11: C, 63.45; H, 4.43; N, 9.38. Found: C, 63.56; H, 4.42; N, 9.42%. The slow evaporation of a DMF solution of the complex at ambient temperature led to the deposition of yellow, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055137.
(11) [Cu(papatpy)2](ClO4)2∙2CH3CN∙0.5H2O: Cu(ClO4)2∙6H2O (9.0 mg, 0.024 mmol) in DMF (5 mL) was mixed with papatpy (20 mg, 0.046 mmol) in DMF (10 mL). After heating this solution for 2 h at 100 °C, the solvent was removed under reduced pressure. The green residue was washed with methanol and chloroform, and then dried at ambient temperature (yield: 19 mg, 66%). Anal. calc. for C132H90Cl4Cu2N16O17: C, 64.95; H, 3.72; N, 9.18. Found: C, 65.06; H, 3.52; N, 9.22%. The slow evaporation of an acetonitrile solution
of the complex at ambient temperature led to the deposition of green, block‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055138.
(12) [Zn(papatpy)2](ClO4)2∙3DMF: Zn(ClO4)2∙6H2O (9.0 mg, 0.023 mmol) in DMF (5 mL) was mixed with papatpy (20 mg, 0.046 mmol) in DMF (10 mL). After heating this solution for 2 h at 100 °C, the solvent was removed under reduced pressure to produce a colourless residue, which was washed with methanol and chloroform, and then dried at ambient temperature (yield: 21 mg, 68%). Anal. calc. for C71H59Cl2N9ZnO11: C, 63.14; H, 4.40; N, 9.33. Found: C, 63.10; H, 4.45; N, 9.30%. 1H‐NMR (300 MHz; CD3CN): δ = 8.90 (s, 4H), 8.60 (d, J = 9.0 Hz, 4H), 8.23 (td, J = 7.8, 1.5 Hz, 4H), 7.86 (m, 8H), 7.76 (d, J = 8.5 Hz, 4H), 7.65 (m, 4H), 7.50 (m, 6H), 7.47 (dd, J = 5.1, 0.9 Hz, 2H), 7.43 (dd, J = 5.1, 1.0 Hz, 2H). The slow evaporation of a DMF solution of the complex at ambient temperature led to the deposition of colourless, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055139.
(13) [Ru(papatpy)2](PF6)2: A mixture of papatpy (40 mg, 0.092 mmol) and RuCl3∙xH2O (0.020 g) in ethanol (30 mL) was heated at 70 °C for 5 h to produce a turbid, reddish‐purple solution. The precipitate formed after cooling to ambient temperature was collected and washed on a filter with cold ethanol to produce a brown powder, taken to be [Ru(papatpy)Cl3]. [Ru(papatpy)Cl3] (25 mg, 0.039 mmol), papatpy (20 mg, 0.046 mmol) and
N‐ethylmorpholine (five drops) in CH3OH/CHCl3 (40 mL, 3:1, v/v) were heated at 70 °C for 4 h. The resultant deep red solution was filtered through Celite to remove some insoluble purple material, and the solvent was evaporated off under reduced pressure. The dark red residue was dissolved in a minimum volume of acetonitrile and subjected to
chromatography on a short silica column using acetonitrile/saturated aqueous KNO3/water
at 7:1:0.5, v/v, as the eluent. The major red component was collected, and methanolic NH4PF6 was added to this eluate. After 1 h, the red precipitate was filtered off, washed with methanol and then dried under vacuum (yield: 23 mg, 47%). Anal. calc. for C62H38F12N6P2Ru: C, 59.19; H, 3.04; N, 6.68. Found: C, 59.10; H, 3.12; N, 6.72%. 1H‐NMR (300 MHz) in CD3CN: δ 8.92 (s, 4H), 8.55 (dq, J = 8.1, 0.4 Hz, 4H), 8.01 (td, J = 7.8, 1.5 Hz, 4H), 7.85 (d, J = 8.6 Hz, 4H), 7.76 (d, J = 8.6 Hz, 4H), 7.66 (m, 4H), 7.50 (m, 6H), 7.45 (dq, J = 5.5, 0.6 Hz, 4H), 7.25 (dd, J = 5.6, 1.3 Hz, 2H), 7.22 (dd, J = 5.6, 1.3 Hz, 2H). The slow evaporation of a solution of the complex in CH3CN at ambient temperature produced red crystals suitable for X‐ray structure determination. CCDC number, 2055140.
(14) [Ru(papatpy)(tpy)](PF6)2∙3CH3CN: [Ru(tpy)Cl3] (50 mg, 0.11 mmol) and
papatpy (60 mg, 0.14 mmol) were dissolved in methanol/chloroform (30 mL, 1:1, v/v), and
N‐ethylmorpholine (five drops) was added. The mixture was heated under reflux for 2 h
and filtered through Celite to remove a small amount of brown precipitate. The deep red filtrate was chromatographed with a short silica‐gel column using a mixed eluent (acetonitrile/saturated aqueous KNO3/water at 7:1:0.5, v/v). The major red band was collected, and excess methanolic NH4PF6 was added. The red precipitate obtained was filtered, washed with methanol and dried under vacuum (yield: 28 mg, 22%). The
product was dissolved in CH3CN and left for one week at room temperature to afford red crystals suitable for X‐ray diffraction studies. Anal. calc. for C52H39F12N9P2Ru: C, 52.89; H, 3.33; N, 10.67. Found: C, 52.95; H, 3.20; N, 10.81%. 1H‐NMR (300 MHz, CD3CN): δ = 8.90 (s, 2H), 8.78 (d, J = 8.2 Hz, 2H), 8.54–8.50 (m, 4H), 8.45 (t, J = 8.2 Hz, 1H), 7.94 (qd, J = 7.9, 1.5 Hz, 4H), 7.83 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.5 Hz, 2H), 7.65–7.62 (m, 2H), 7.50–7.47 (m, 3H), 7.42–7.37 (m, 4H), 7.24–7.16 (m, 4H). CCDC number, 2055141.
(15) [Cd(papatpy)2](ClO4)2∙2CH3CN∙H2O: Cd(ClO4)2∙H2O (8.0 mg, 0.026 mmol) in DMF (5 mL) was mixed with papatpy (20 mg, 0.046 mmol) in DMF (10 mL). After heating this solution for 2 h at 100 °C, the solvent was removed under reduced pressure. The colourless residue was washed with methanol and chloroform, and then dried at ambient temperature (yield: 18 mg, 55%). Anal. calc. for C66H46Cl2N8CdO9: C, 62.01; H, 3.63; N, 8.76. Found: C, 61.97; H, 3.55; N, 8.82%. 1H‐NMR (300 MHz) in CD3CN: δ 8.85 (s, 4H), 8.64 (d, J = 8.1 Hz, 4H), 8.26 (td, J = 7.8, 1.6 Hz, 4H), 8.13 (s, 4H), 7.82 (d, J = 8.4 Hz, 4H), 7.75 (d, J = 8.4 Hz, 4H), 7.65 (m, 4H), 7.56 (d, J = 5.1 Hz, 2H), 7.53 (d, J = 5.1 Hz, 2H), 7.49 (m, 6H). The vapour diffusion of
diethyl ether into an acetonitrile solution of the complex provided colourless, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055142.
(16) [Cd(papatpy)Cl2]∙CH2Cl2: CdCl2 (13 mg, 0.071 mmol) in CH3CN/CHCl3 (10 mL, 1:1, v/v) was mixed with papatpy (30 mg, 0.069 mmol) in chloroform (5 mL). After heating this solution at 70 °C for 2 h, the solvent was removed under reduced pressure. The colourless residue was washed with methanol and chloroform, and then dried at ambient temperature (yield: 32 mg, 64%). Anal. calc. for C32H21Cl4N3Cd: C, 54.77; H, 3.02; N, 5.99. Found: C, 54.67; H, 3.11; N, 5.88%. 1H‐NMR (300 MHz) in DMSO‐d6: δ 8.79 (d, J = 4.3 Hz, 4H), 7.78 (d, J = 8.1 Hz, 4H), 7.73 (d, J = 8.4 Hz, 4H), 7.62 (m, 3H), 7.48 (m, 4H). The slow evaporation of a solution of the complex in CH3CN/CH2Cl2 (1:1, v/v) produced colourless, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055224.
2.2.5. Iron(II) Complex of 4′‐(Pentafluorophenylethynyl)‐2,2′:6′,2′′‐terpyridine, pfpatpy
(17) [Fe(pfpatpy)2](ClO4)2∙CH3CN∙H2O: Fe(ClO4)2∙6H2O (9.0 mg, 0.025 mmol) in DMF (5 mL) was added to a solution of pfpatpy (30 mg, 0.065 mmol) in hot DMF (5 mL). After stirring for 2 h at 100 °C, the solvent was removed under reduced pressure, and the deep purple residue was washed with CH3OH and CHCl3, and then dried at room temperature (yield: 25 mg, 89%). It was dissolved in DMF, and CH3CN vapour was diffused in to produce purple crystals suitable for X‐ray diffraction studies. Anal. calc. for C46H20Cl2F10FeN6O8∙0.5CH3CN∙H2O: C, 49.56; H, 2.08; N, 7.99. Found: C, 49.98; H, 1.91; N, 7.70%. (There was a partial loss of CH3CN by comparison with the crystal.) CCDC number, 2055225.
2.2.6. Complexes of 4′‐(4′‐Pyridin‐4‐yl‐biphenyl‐4‐yl)‐2,2′:6′,2′′‐terpyridine, pybptpy
(18) [Ni(pybptpy)2](ClO4)2∙2DMF∙H2O: Ni(ClO4)2∙6H2O (20 mg, 0.055 mmol) in DMF (5 mL) was added to pybptpy (50 mg, 0.11 mmol) in DMF (10 mL). After stirring the solution for 2 h at 100 °C, the solvent was removed under reduced pressure, and the yellow residue was washed with methanol and chloroform, and then dried at ambient temperature (yield: 54 mg, 82%). Anal. calc. for C70H60Cl2N10NiO11: C, 62.42; H, 4.49; N, 10.40. Found: C, 62.4; H, 4.45; N, 10.21%. The vapour diffusion of methanol into a DMF solution of the complex produced yellow crystals suitable for X‐ray diffraction studies. CCDC number, 2055226.
(19) [Cu(pybptpy)2](ClO4)2∙3DMF∙H2O: Cu(ClO4)2∙6H2O (20 mg, 0.055 mmol) in DMF (5 mL) was added to pybptpy (50 mg, 0.11 mmol) in DMF (10 mL). After stirring the solution for 2 h at 100 °C, the solvent was removed under reduced pressure, and the green residue was washed with methanol and chloroform, and then dried at ambient temperature (yield: 58 mg, 74%). Anal. calc. for C73H67Cl2CuN11O12: C, 61.54; H, 4.74; N, 10.81. Found: C, 61.33; H, 4.82; N, 10.88%. The vapour diffusion of methanol into a DMF solution of the complex produced green crystals suitable for X‐ray diffraction studies. CCDC number, 2055227.
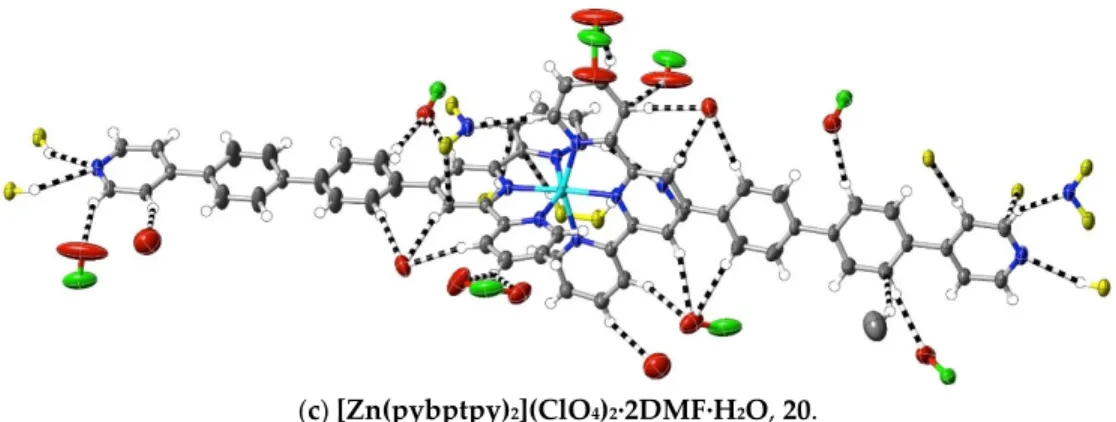
(20) [Zn(pybptpy)2](ClO4)2∙2DMF∙H2O: Zn(ClO4)2∙6H2O (20 mg, 0.055 mmol) in DMF (5 mL) was added to pybptpy (50 mg, 0.11 mmol) in DMF (10 mL), and the solution was stirred for 2 h at 100 °C before the solvent was removed under reduced pressure. The white residue was washed with methanol and chloroform, and then dried at ambient temperature (yield: 49 mg, 63%). Anal. calc. for C70H60Cl2N10O11Zn: C, 62.11; H, 4.47; N, 10.35. Found: C, 62.07; H, 4.55; N, 10.51%. 1H‐NMR (300 MHz) in DMSO‐d6: δ 9.50 (s, 4H), 9.23 (d, J = 8.2 Hz, 4H), 8.73 (d, J = 5.6 Hz, 4H), 8.63 (d, J = 7.8 Hz, 4H), 8.35 (t, J = 7.6 Hz, 4H), 8.22 (d, J = 8.2 Hz, 4H), 8.12 (d, J = 8.3 Hz, 4H), 8.05 (m, 8H), 7.86 (d, J = 5.8 Hz, 4H), 7.56 (t, J = 6.5 Hz, 4H). The vapour diffusion of methanol into a DMF solution of the complex produced colourless crystals suitable for X‐ray diffraction studies. CCDC number, 2055228.
2.2.7. Complexes of 4′‐(4‐Bromobiphenyl‐4‐yl)‐2,2′:6′,2′′‐terpyridine, Brbptpy
(21) [Co(Brbptpy)2](BF4)2∙2CH3CN: Co(BF4)2∙6H2O (20 mg, 0.060 mmol) in dimethylformamide (DMF; 5 mL) was added to a solution of Brbptpy (50 mg, 0.11 mmol) in hot DMF (10 mL). After stirring for 2 h at 100 °C, the solvent was removed under reduced pressure. The brown residue was washed with methanol and dried at ambient temperature (yield: 0.048 g, 71%). Anal. calc. for C58H42B2Br2CoF8N8: C, 56.03; H, 3.40; N, 9.0. Found: C, 56.18; H, 3.36; N, 8.95%. The vapour diffusion of diethyl ether into a solution of the complex in a minimum volume of acetonitrile produced brown crystals suitable for X‐ray diffraction studies. CCDC number, 2055148.
(22) [Ni(Brbptpy)2](ClO4)2∙DMF∙H2O: Ni(ClO4)2∙6H2O (40 mg, 0.12 mmol) in methanol
(10 mL) was added to a solution of Brbptpy (100 mg, 0.22 mmol) in CHCl3/CH3OH (10 mL,
1:1, v/v). After stirring the solution for 1 h at 60 °C, the solvent was removed under reduced pressure, and the yellow residue was washed with cold methanol and chloroform, and then dried at ambient temperature (yield: 100 mg, 68%). Anal. calc. for C111H79Br4 Cl4N13Ni2O18: C, 54.16; H, 3.2; N, 7.40. Found: C, 54.32; H, 3.30; N, 7.45%. The vapour diffusion of methanol into a solution of the complex in a minimum volume of DMF produced yellow crystals suitable for X‐ray diffraction studies. CCDC number, 2055149.
(23) [Cu(Brbptpy)2](ClO4)2∙2CH3OH: Cu(ClO4)2∙6H2O (45 mg, 0.12 mmol) in methanol (10 mL) was added to a solution of Brbptpy (100 mg, 0.22 mmol) in CHCl3/CH3OH (10 mL, 1:1, v/v). After stirring the solution for 1 h at 60 °C, the solvent was removed under reduced pressure, and the green residue was washed with cold methanol and chloroform, and then dried at ambient temperature (yield: 110 mg, 75%). Anal. calc. for C56H43Br2Cl2CuN6O10: C, 53.63; H, 3.46; N, 6.70. Found: C, 56.28; H, 3.38; N, 6.75%. The complex was dissolved in boiling methanol, and the solution was allowed to evaporate slowly at ambient temperature to provide brown crystals suitable for X‐ray diffraction studies. CCDC number, 2055150. (24) [Ru(Brbptpy)2](PF6)2∙3CH3CN: With the substitution of Brbptpy for tptpy, the same procedure as used with complex 4 provided red crystals (with a 51% yield) of the complex suitable for structure determination. Anal. calc. for C60H45Br2F12N9P2Ru: C, 49.95; H, 3.14; N, 8.74. Found: C, 49.35; H, 3.30; N, 8.82%. Due to the loss of the sample, an NMR spectrum was not obtained. CCDC number, 2055151. 2.2.8. Complexes of 4‐((Ethoxycarbonyl)biphenyl‐4‐yl)‐2,2′:6′,2″‐terpyridine, Ebptpy
(25) [Ni(ebptpy)2](ClO4)2∙3DMF: Ni(ClO4)2∙6H2O (8.0 mg, 0.022 mmol) in DMF (5 mL) was added to ebptpy (20 mg, 0.044 mmol) in DMF (10 mL). The resulting solution was heated for 2 h at 100 °C before the solvent was removed under reduced pressure. The yellow residue was washed with methanol and chloroform and then dried at ambient temperature (yield: 21 mg, 69%). Anal. calc. for C69H67Cl2N9NiO15: C, 59.54; H, 4.85; N, 9.06. Found: C, 59.60; H, 4.53; N, 9.21%. The vapour diffusion of methanol into a DMF solution of the complex produced yellow, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055152.
(26) [Cu(ebptpy)2](ClO4)2∙CH3CN: Cu(ClO4)2∙6H2O (8.1 mg, 0.022 mmol) in DMF (5 mL) was added to ebptpy (20 mg, 0.044 mmol) in DMF (10 mL). The resulting solution was heated for 2 h at 100 °C before the solvent was removed under reduced pressure. The green residue was washed with methanol and chloroform, and dried at ambient temperature (yield: 0.018 g, 67%). Anal. calc. for C62H49Cl2N7CuO12: C, 61.11; H, 4.05; N, 8.05. Found: C, 60.99; H, 4.02; N, 8.10%. The vapour diffusion of diethyl ether into an acetonitrile solution of the complex provided green, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055153.
2.2.9. Zinc(II) Complex of 4‐((Ethoxycarbonyl)terphenyl‐4‐yl)‐2,2′:6′,2″‐terpyridine, etptpy
(27) [Zn(etptpy)2](ClO4)2∙2DMF∙H2O: Zn(ClO4)2∙6H2O (7.0 mg, 0.016 mmol) in DMF (5 mL) was added to etptpy (20 mg, 0.037 mmol) in DMF (10 mL). After stirring the
resulting solution for 2 h at 100 °C, the solvent was removed under reduced pressure. The white residue was washed with methanol and chloroform and then dried at ambient temperature (yield: 19 mg, 84%). Anal. calc. for C75H61Cl2N7O13Zn: C, 64.13; H, 4.38; N, 6.98. Found: C, 64.08; H, 4.52; N, 6.70%. 1H‐NMR (300 MHz) in DMSO‐d6: δ 9.50 (s, 4H), 9.23 (d, J = 7.5 Hz, 4H), 8.62 (d, J = 7.5 Hz, 4H), 8.33 (t, J = 6.8 Hz, 4H), 8.21 (d, J = 7.7 Hz, 4H), 8.13 (t, J = 8.0 Hz, 8H), 7.99 (m, 12H), 7.55 (t, J = 5.5 Hz, 4H), 4.41 (q, J = 6.7 Hz, 4H,– CH2–), 1.40 (t, J = 7.2 Hz, 6H, –CH3). The vapour diffusion of methanol into a DMF solution of the complex produced pale‐yellow, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055154.
2.2.10. Complexes of 4′‐(Benzyloxy)‐2,2′:6′,2″‐terpyridine, bzOtpy
(28) [Fe(bzOtpy)2](ClO4)2: A mixture of Fe(ClO4)2∙6H2O (12 mg, 0.033 mmol) and
bzOtpy (30 mg, 0.088 mmol) in CH2Cl2/CH3OH (20 mL, 1:1, v/v) was heated under reflux for 2 h. Upon cooling, the solvent was evaporated off under reduced pressure. The purple residue was washed with methanol and dichloromethane and dried at ambient temperature (yield: 30 mg, 97%). Anal. calc. for C44H34Cl2N6FeO10: C, 56.61; H, 3.67; N, 9.00. Found: C, 56.48; H, 3.89; N, 9.11%. 1H‐NMR (300 MHz) in CD3CN: δ 8.60 (s, 4H), 8.47 (d, J = 7.8 Hz, 4H), 7.92 (t, J = 7.7 Hz, 4H), 7.77 (d, J = 7.5 Hz, 4H), 7.64 (m, 6H), 7.20 (d, J = 5.4 Hz, 4H), 7.13 (t, J = 6.4 Hz, 4H), 5.72 (s, 4H, –CH2–). The slow evaporation of an acetonitrile solution of the complex at ambient temperature produced purple crystals suitable for X‐ray diffraction studies. CCDC number, 2055155.
(29) [Ni(bzOtpy)2](ClO4)2: Ni(ClO4)2∙6H2O (17 mg, 0.047 mmol) and bzOtpy (30 mg, 0.088 mmol) were dissolved in CH2Cl2/CH3OH (20 mL, 1:1, v/v) and heated under reflux for 2 h. Upon cooling, the solvent was evaporated off under reduced pressure. The yellow residue was washed with methanol and dichloromethane and then dried at ambient temperature (yield: 36 mg, 82%). Anal. calc. for C44H34Cl2N6NiO10: C, 56.44; H, 3.66; N, 8.98. Found: C, 56.70; H, 3.74; N, 9.08%. The slow evaporation of an acetonitrile solution of the complex at ambient temperature produced purple crystals suitable for X‐ray diffraction studies. CCDC number, 2055156.
2.2.11. 4′‐(3,4,5‐Trimethoxy‐phenyl)‐2,2′:6′,2′′‐terpyridine, tmptpy, and its Zn(II) Complex
(30) Tmptpy [34] in its diprotonated form as the perchlorate salt, hemi‐tetrahydrofuran solvate, [H2tmptpy](ClO4)2∙0.5THF: tmptpy (100 mg) was
dissolved in THF (30 mL), and 1 mL of 1 M HClO4 was added with stirring over 30 min,
after which the solution was allowed to slowly evaporate under ambient temperature to produce colourless crystals suitable for structure determination. Anal. calc. for C52H54Cl4N6O23: C, 49.07; H, 4.28; N, 6.60; Found: C, 48.54; H, 4.31; N, 6.48%. CCDC number, 2055157.
(31) [Zn(tmptpy)2](ClO4)2∙CH3CN: Zn(ClO4)2∙6H2O (7.0 mg, 0.016 mmol) in DMF (5 mL) was added to tmptpy (20 mg, 0.037 mmol) in DMF (10 mL). The solvent was removed under reduced pressure after stirring the solution for 2 h at 100 °C. The white residue was washed with methanol and chloroform and then dried at ambient temperature (yield: 19 mg, quantitative). Anal. calc. for C50H45Cl2N7O14Zn: C, 54.39; H, 4.11; N, 8.88. Found: C, 54.44; H, 4.08; N, 8.96%. 1H‐NMR (300 MHz) in CD3CN: δ 9.00 (s, 4H), 8.82 (dt, J = 8.1, 0.8 Hz, 4H), 8.23 (td, J = 7.8, 1.7 Hz, 4H), 7.87 (dq, J = 5.1, 0.6 Hz, 4H), 7.46 (dd, J = 5.1, 1.0 Hz, 2H), 7.44 (s, 4H), 7.43 (dd, J = 5.1, 1.0 Hz, 2H), 4.10 (s, 12H, –CH3),
3.92 (s, 6H, –CH3). The vapour diffusion of methanol into a DMF solution of the complex
produced pale‐yellow, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055158.
2.2.12. Complexes of
2‐(4‐(2‐(3,4,5‐Trimethoxyphenyl)Ethynyl)‐6‐(pyridin‐2‐yl)pyridin‐2‐yl)pyridine, tmpatpy
(32) [Ni(tmpatpy)2](ClO4)2∙CH3CN∙H2O: Ni(ClO4)2∙6H2O (9.0 mg, 0.025 mmol) in
acetonitrile (5 mL) was added to tmpatpy (20 mg, 0.047 mmol) in CH3CN/CHCl3 (10 mL,
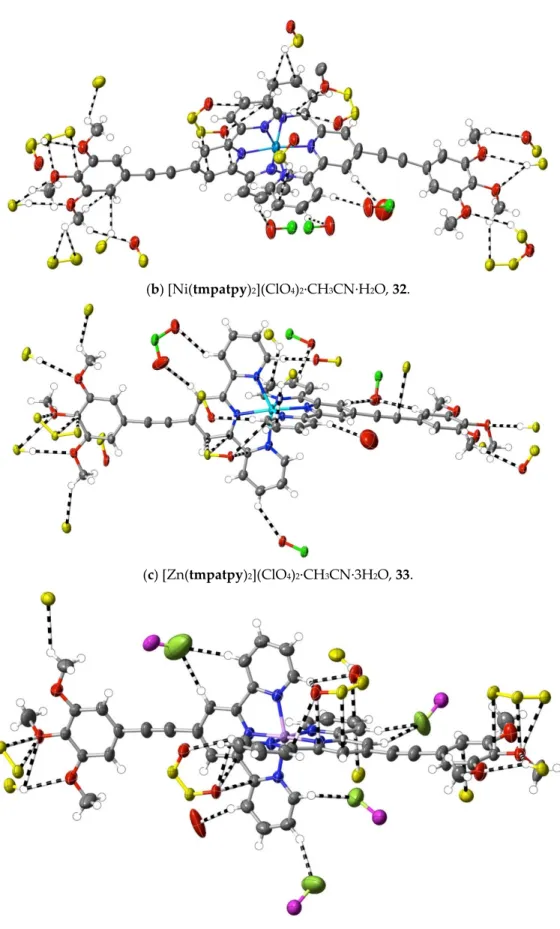
1:1, v/v). After stirring the resulting solution for 1 h at 60 °C, the solvent was removed under reduced pressure. The yellow residue was washed with methanol and chloroform and then dried at ambient temperature (yield: 21 mg, 57%). Anal. calc. for C106H89Cl4N13O29Ni2: C, 56.13; H, 3.96; N, 8.03. Found: C, 56.22; H, 3.98; N, 8.11%. The slow evaporation of an acetonitrile solution of the complex at ambient temperature provided yellow, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055159.
(33) [Zn(tmpatpy)2](ClO4)2∙CH3CN∙3H2O: Zn(ClO4)2∙6H2O (9.0 mg, 0.024 mmol) in
acetonitrile (5 mL) was added to tmpatpy (20 mg, 0.047 mmol) in CH3CN/CHCl3 (10 mL,
1:1, v/v). After stirring the resulting solution for 1 h at 60 °C, the solvent was removed under reduced pressure, and the bright yellow residue was washed with methanol and chloroform, and then dried at ambient temperature (yield: 22 mg, 76%). Anal. calc. for C54H51Cl2N7O17Zn: C, 53.77; H, 4.26; N, 8.13. Found: C, 53.57; H, 4.20; N, 8.18%. 1H‐NMR (300 MHz) in CD3CN: δ 8.86 (s, 4H), 8.60 (dt, J = 8.1, 0.8 Hz, 4H), 8.22 (td, J = 7.7, 1.6 Hz, 4H), 7.86 (dq, J = 5.1, 0.8 Hz, 4H), 7.46 (dd, J = 5.1, 1.0 Hz, 2H), 7.43 (dd, J = 5.1, 0.9 Hz, 2H), 7.10 (s, 4H), 3.95 (s, 12H, –CH3), 3.85 (s, 6H, –CH3). The slow evaporation of an acetonitrile solution of the complex at ambient temperature provided pale yellow, rod‐like crystals suitable for X‐ray diffraction studies. CCDC number, 2055160.
(34) [Ru(tmpatpy)2](PF6)2∙1.5DMF: A mixture of tmpatpy (50 mg, 0.12 mmol) and
RuCl3∙xH2O (25 mg) in ethanol (30 mL) was heated at 70 °C for 4 h to produce a turbid, reddish‐purple solution. This was cooled to ambience to produce a precipitate of a brown
powder, washed on the filter with ethanol, which was assumed to be [Ru(tmpatpy)Cl3].
[Ru(tmpatpy)Cl3] (40 mg, 0.063 mmol), tmpatpy (30 mg, 0.071 mmol) and
N‐ethylmorpholine (five drops) were dissolved in CH3OH/CHCl3 (40 mL, 3:1, v/v) and heated at 70 °C for 2 h. The resultant deep red solution was filtered through Celite to remove some insoluble purple material, and the solvent was evaporated under reduced pressure. The dark red residue was dissolved in a minimum volume of acetonitrile and loaded on a short silica column, and the column was eluted with acetonitrile/saturated
aqueous KNO3/water (7:1:0.5, v/v). The major red component was collected, methanolic
NH4PF6 was added, and 1 h was allowed for the precipitation of a red powder, which was then collected, washed on the filter with methanol and dried under vacuum (yield: 35 mg, 41%). Anal. calc. for C113H105F24N15O15P4Ru2: C, 50.36; H, 3.93; N, 7.80. Found: C, 50.41; H, 3.80; N, 7.78%. 1H‐NMR (300 MHz) in CD3CN: δ 8.88 (s, 4H), 8.54 (dq, J = 8.2, 0.8 Hz, 4H), 8.01 (td, J = 7.7, 1.5 Hz, 4H), 7.45 (dq, J = 5.5, 0.6 Hz, 4H), 7.24 (dd, J = 5.6, 1.3 Hz, 2H), 7.22 (dd, J = 5.6, 1.3 Hz, 2H), 7.09 (s, 4H), 3.96 (s, 12H, –CH3), 3.85 (s, 6H, –CH3). The slow evaporation of an acetonitrile solution of the complex provided red crystals suitable for X‐ray structure determination. CCDC number, 2055161. 3. Crystallography
Diffraction data for crystals of complexes 1–3, 7–18, 20, 21, 23–28 and 30–33 were obtained at 100(2) K using an ADSC Quantum 210 detector at 2D SMC with a silicon (111) double crystal monochromator (DCM) for various synchrotron wavelengths between 0.62988 and 0.75000 Å at the Pohang Accelerator Laboratory, Pohang, South Korea. The ADSC Q210 ADX program [51] was used for data collection (detector distance, 63 mm; omega scan; Δω = 1°; exposure time, 1 s per frame), and HKL3000sm (Ver. 703r) [52] was used for cell refinement, reduction and absorption correction. The structures were solved by direct methods and refined by full‐matrix least‐squares fitting on F2 using SHELXL‐2014 [53]. All the non‐hydrogen atoms were refined with anisotropic displacement parameters. The carbon‐bound hydrogen atoms were introduced at
calculated positions, and all the hydrogen atoms were treated as riding atoms with an isotropic displacement parameter equal to 1.2 times that of the parent atom. Diffraction data for complexes 4–6, 19, 22, 29 and 34 were obtained on a RIGAKU Saturn CCD diffractometer with a confocal mirror, using graphite‐monochromated Mo Kα radiation (λ = 0.71073 Å) or, for 19 only, Cu Kα radiation (λ = 1.54187 Å). The structures were solved by direct methods and refined on F2 by full‐matrix least‐squares treatment [53]. For complexes 7, 9, 13, 14, 18, 20, 28, 29, 32 and 34, the SQUEEZE option of PLATON [54] was used to take into account the contribution of disordered solvents during refinement. The results (the size of the voids, number of electrons present in the voids and possible solvent molecules present) are summarized in Table S1.
Full details of all the structure determinations have been deposited as cifs in the Cambridge Crystallographic Data Collection under CCDC deposition numbers 2055133‐2055135, 2055137‐2055142, 2055148‐2055161 and 2055219‐2055228. Note that for structures 7 and 9, the solutions were unsatisfactory in that the residual (R1 factor) exceeded 0.10, but they have been included for comparative purposes.
The nature of the atoms involved in the interactions exceeding dispersion was established using CrystalExplorer [30], and their specific identities (atom numbers and locations) were established using CrystalMaker [55], which was then used to prepare figures with the interactions shown as dashed lines. In the case of the complexes for which SQUEEZE was applied during the structure refinement (see above), the Hirshfeld surfaces calculated with CrystalExplorer may lack indications of some interactions since not all the solvent molecules present were included in the calculation. 4. Results and Discussion Most of the ligands used in the present work are well‐known species for which the coordination chemistry is well‐established. We have described in detail, elsewhere [33], the syntheses of three new ligands (bzOtpy, etptpy and pfatpy) and, here, that of
papatpy (Figure 1), closely based on methods established for related compounds, as were
the syntheses of their metal‐ion complexes, so a detailed discussion of these aspects of the present work is not warranted. With the exception of the kinetically inert Ru(III) ions, the metal ions used herein are all labile species, so the synthesis of their complexes is trivial. The crystallisation of the complexes in a form suitable for X‐ray diffraction measurements was not necessarily trivial and generally involved the exploration of several methods, although only in the case of complexes of pfpatpy were real difficulties encountered, these being such that only a single complex of this ligand was structurally characterised. In regard to the coordination spheres of the metal ions and the stacking tendency of the cations in the 33 compounds structurally characterised, nothing setting them apart from their extensive context is apparent, and our focus was on just the solid‐state interactions discernible from analysis of the Hirshfeld surfaces.
As a background to the analysis of the crystal structures of complexes of 4′‐substituted 2,2′:6′,2″‐terpyridine, it is useful to consider the structures of free ligands that are available, although these are relatively limited in number, and this we have done in a separate work [37]. All free terpyridines show non‐chelating, bis‐transoid arrays of the three N‐donor atoms, except in the case of the 4′‐hydroxy derivative, which exists as a 1:1 mixture of the hydroxypyridine and pyridone tautomers in the solid state [56–58], where the pyridone tautomer adopts a bis‐cisoid array, while the hydroxypyridine form has a bis‐transoid form. Significantly, although the stacking of the terpyridine units is a common feature of free ligand structures, this appears in most instances other than those where large, polar substituents are present, to be associated simply by dispersion interactions between terpyridine units, and only the pyridone tautomer of 4′‐hydroxy‐2,2′:6′,2″‐terpyridine is clearly involved in additional, specific C...C
interactions between facing aromatic planes, with interactions of the peripheral H‐atoms being the dominant form of interactions, exceeding dispersion in all cases.
While much of the interest in 2,2′:6′,2″‐terpyridine chemistry has been focused on metal‐ion complexes in low oxidation states [1–7], complexes of high‐oxidation‐state metal ions are also known, with vanadium(V) [59] and uranium(VI) (as the uranyl ion, UO22+) [60–63] providing examples of extremes. In that the formation of a coordinate bond is formally a process where the metal ion gains electrons and the ligand loses them, such extreme cases might be expected to be associated with charge redistribution favouring the appreciable interaction of the bound ligand with nucleophiles, akin to the addition to an imine bond. In the case of crystalline [VO2(bptpy)]ClO4, 1, the Hirshfeld surfaces of the two inequivalent cations provide evidence for exactly this, with the most marked region of interaction exceeding dispersion, that being due to the contact of vanadyl‐O (O1) with one of the carbon atoms (C10) adjacent to the central N‐atom of the ligand (Figure 2a). Reciprocal, remarkably short contacts (O1...C10i = O1i...C10, 2.81(1) Å; i = 1 − x, 1 − y, 2 − z) of this type result in the presence of pairs of the two inequivalent cations, where the slightly bowed terpyridine units lie close to parallel, with a separation of ~3.5 Å, and where there are only barely perceptible indications of other interactions beyond the dispersion of the C and H atoms of ligand units lying both close to parallel and nearly perpendicular to the ligand of the cation considered (Figure S1). Multiple interactions involving peripheral aromatic H‐atoms and vanadyl‐ or perchlorate‐O atoms serve to link the whole structure together, but none involve unusually short contacts, and as is seen in the examples discussed later, they appear to be a common feature of structures regardless of the metal ion present. However, as some interactions involve the 4′‐biphenyl substituent on the terpyridine unit, it is unsurprising that there are significant differences in the structure of the “parent” species [VO2(tpy)]ClO4 [59] when compared to that of 1 (Figure 2b). Here, the most obvious interaction beyond the dispersion of a given cation again involves the contact of a O‐atom with a C adjacent to a N, but the O‐atom is from perchlorate, and two adjacent C atoms, one from the central and one from an outer pyridine unit (O4...C6, 3.09(1) Å; O4...C5, 3.162(8) Å), are involved.
Vanadyl‐O...C contacts are still apparent and are associated with the maintenance of a stacked arrangement of (equivalent‐) cation pairs but involve C‐atoms separated by another from N (O1...C4′, 3.162(8) Å; ′ = 1 − x, 1 − y, 2 − z). Note that while in 1, the nearer
phenyl ring of the biphenyl substituent is close to coplanar with the terpyridine unit, indicating, possibly, a degree of delocalisation as an influence upon the electronic properties of the whole, this is also seen in some M(II) complexes of the ligand [43] and appears to be a consequence of CH interactions with counteranions.
(a)
(b)
Figure 2. Interactions beyond dispersion, shown as dashed lines, of the cations present in (a) [VO2(bptpy)]ClO4, 1 and (b)
[VO2(tpy)]ClO4 [44]. O atoms are identified by showing the atoms to which they are attached. Colour coding: C = grey, N
= dark blue, O = red, Cl = green, and V = light blue, except for carbon atoms from adjacent cations, which are shown in yellow. H‐atoms are shown as small, colourless spheres of arbitrary 0.2 Å radius.
In known structures of uranyl‐ion complexes of 2,2′:6′,2″‐terpyridine [60–63] (and, in some cases [60,61], of 4′‐chloro‐2,2′:6′,2″‐terpyridine), the close contact of at least one O‐atom with one or two C‐atoms bound to the N of another complex forming a stacked
array is common but not universal. In [UO2(tpy)(tdc)]2 (tdc =
thiophene‐1,4‐dicarboxylate) and its chloroterpyridine analogue [45], there are such contacts with C6...O2′(uranyl) 3.039(5) Å and C6...O4′(carboxylate) 2.947(8) Å, respectively,
although much more obvious in the former are reciprocal uranyl‐O2...N2′ contacts of 2.783(4) Å (Figure S2a). In [(UO2)2(OH)(tpy)(dcb)3] (dcb = 3,5‐dichlorobenzoate) and its chloroterpyridine analogue [61], there are contacts with C15...O2′(uranyl) 3.014(7) Å and
C15...O2′(uranyl) 2.915(6) Å, respectively, while in [(UO2)2(OH)2(tpy)2](ClO4)2 [62], where there are two inequivalent dimers within the structure, the C...O contacts C5...O8′(uranyl)
2.949(6) Å, C6...O8′(uranyl) 2.959(7) Å and C41...O2′(uranyl) 2.953(6) Å are apparent
(Figure S2b). In [UO2(tpy)(bpdc)] (bpdc = 2,2′‐bipyridine‐3,3′‐dicarboxylate) [63], there is a similarly short contact of C5...O1′(uranyl) 2.937(3) Å, but in the four complexes
[UO2(tpy)(tph)] and [UO2(tpy)(npdc)] (tph = terephthalate {benzene‐1,4‐dicarboxylate}; npdc = naphthalene‐1,4‐dicarboxylate) and their chloroterpyridine analogues, such interactions are not evident. Here, extended stacking arrays of the aromatic units appear to block the access of other species to C‐atoms bound to N in the ligand. This is a timely reminder that any weak interactions beyond dispersion evident in a Hirshfeld surface representation are exactly that, weak, and that the balance between any number of such interactions may be extremely sensitive to compositional differences.
If a positive charge on a bound metal does produce charge depletion on the ligand in the region of the donor atoms, then any effect should diminish with a decreasing charge, and the smallest positive charge is that carried by the proton, H+. The diprotonation of a terpyridine is required to produce the di‐cisoid configuration found in metal‐ion complexes such as 1, and the structure of compound 30, [H2tmptpy](ClO4)2∙0.5THF, adds to several known for such species. (A summary of relevant literature is given in [33].) Here, as is broadly true [33], there is no evidence on the Hirshfeld surface, except for an indication that the interaction of one perchlorate‐O (O17) may be better considered as involving the C25–H25 bond rather than H25 alone, for interactions of nucleophiles with C‐atoms attached to any of the N‐donor atoms, and this is consistent with what is seen in complex 1 and several other species discussed above, being evidence for the significant polarising effects of highly charged cations. Peripheral CH...O interactions of aromatic
CH with perchlorate‐O, however, are numerous, and the cations lie in infinite stacks largely involving dispersion interactions only, except for barely discernible C...C
interactions between alternate pairs of the two inequivalent cations of the structure, with C11...C44i 3.31(4) Å and C18...C29i 3.34(4) Å (i = x + 0.5, y + 0.5, z) (Figure 3). While the atoms C11 and C29 are adjacent to N in the terpyridine unit, C18 and C44 are C‐atoms of the substituent to which methoxy groups are attached, showing that purely dispersive stacking of the terpyridine units as seen in unfunctionalised ligand salts such as [H2tpy]Cl2.H2O [64] can be disrupted by substituents.
Figure 3. Views perpendicular (left) and at 30° (right) to the mean plane of one of the stacked pairs of inequivalent cations
found in the structure of [H2tmptpy](ClO4)2∙0.5THF, 30, showing (dashed lines) the interactions exceeding dispersion of the
cations. Carbon atoms of one cation are shown in black; of the other, in grey. C...C interactions are visible in the 30° view.
The vast majority of the structural studies of complexes of 2,2′:6′,2″‐terpyridine and its derivatives concern metal(II) and metal(III) species, and the present work largely complements such investigations, which range, for Co(II), for example, from early determinations on a single compound for which H‐atom coordinates are not available, e.g., [65], to those of several complexes of various derivatives with full coordinates, e.g., [66], such
variations reflecting, in part, major advances in the ease of the synthesis of the ligands [1–7] and in crystallographic equipment. The complexes 2–6 of the tptpy ligand add Fe(II), Ni(II) and Ru(II) to the series, with Co(II), Cu(II), Zn(II) and Cd(II) studied earlier [34,37], and form part of a rather large family of structurally characterised complexes of 4′ derivatives of 2,2′:6′,2″‐terpyridine, where the substituent is an apolar aromatic unit. An obvious question to ask in relation to the nine structures of 4′‐terphenyl‐2,2′:6′,2″‐terpyridine we studied is whether or not the changes in the electron configurations of the exclusively M(II) cations involved have an effect detectable in the form of the Hirshfeld surfaces of the complexes in the solid state. The answer is that none is clearly evident. A feature of all the structures is that the terphenyl tail is significantly twisted, both within its chain and relative to the terpyridine unit, indicating that there is no extended delocalisation within the whole ligand and, thus, that the addition of the terphenyl tail does not produce a larger, planar aromatic unit that might engage in stronger stacking interactions than in simple 2,2′:6′,2‐terpyridine complexes. It is certainly possible to discern aromatic and aza‐aromatic entities that form arrays of “face‐to‐face” and “edge‐to‐face” types, and these are associated with various C...C and CH...C
(CH...π) interactions that exceed dispersion and are thus evident on the Hirshfeld surfaces. In
the totality of the interactions exceeding dispersion (Figure 4), there is no evidence for nucleophile addition to C‐atoms adjacent to N, so it can be said at this point that M(II) cations seem to be distinguishable from M(V) and M(VI) cations in this respect. Interactions of the cations with the counteranions and lattice solvents occur along with aromatic–aromatic interactions, and even in only the eight cases considered, there are variations in both the counteranions and lattice solvents as well as in the space groups, so it is unsurprising that there are differences in the exact patterns of interactions across the eight, but in every one, there are C...C and CH...C interactions beyond dispersion with both the tpy head groups and
at least one of the phenyl groups of the tails. A comparison of the mixed‐ligand complexes 5 and 6 shows that aromatic...aromatic interactions beyond dispersion are little affected by the
difference in anion, indicating that aromatic...aromatic interactions in general, including
dispersion, may be the dominant factor influencing the array of cations in the crystal. Analysis (Figure S3) of the structures reported [43] for a family of complexes of the ligand 4′‐biphenyl‐2,2′:6′,2″‐terpyridine (bptpy) confirms the observation that for a similar range of M(II) species (M = Co, Ni, Cu, Zn, Cd and Ru), none provides evidence of significant weak interaction with C‐atoms bound to N, though perhaps because of the differences in temperature for the present and these earlier determinations, the C...C and CH...C interactions
beyond dispersion between aromatic/aza‐aromatic units are generally less numerous for the bptpy derivatives. The known structures for the Co(II) [37] and Zn(II) [35] complexes of the very long‐tailed 4′‐quaterphenyl‐2,2′:6′,2″‐terpyridine (qptpy) ligand provide no exceptions to the observation of a lack of any enhanced electrophilicity of C‐atoms bound to coordinated N, just like the very numerous structures of complexes of 2,2′:6′,2″‐terpyridine itself and its 4′‐phenyl and ‐4‐methylphenyl (tolyl) derivatives, where, however, there are examples of M(III) complexes where the generalisation appears to be valid (M = Co, Rh) [67,68] and at least one (M = Cr) [69] where it is not. It is, of course, well‐known [70] that the synthesis of functionalised aza‐aromatic ligands can be based on nucleophilic attack on precisely the C‐atoms adjacent to N.
(b) [Co(tptpy)2](BF4)2∙CH3OH (Co = pink; B = brown) [37].
(c) [Ni(tptpy)2](ClO4)2∙CH3OH (Ni = turquoise), 3.
(d) [Cu(tptpy)2](ClO4)2∙CH3OH (Cu = violet) [34].
(e) [Zn(tptpy)2](ClO4)2∙DMF (Zn = sky blue) [34].
(f) [Ru(tptpy)2](PF6)2 (Ru = mauve), 4. (g) [Ru(tptpy)(tpy)](ClO4)2∙2DMF (Ru = mauve), 5. (h) [Ru(tptpy)(tpy)](PF6)2∙2DMF (Ru = mauve), 6. (Non‐disordered cation.) (i) [Cd(tptpy)2](ClO4)2∙DMF∙0.5H2O (Cd = dark green) [34].
Figure 4. Interactions exceeding dispersion (dashed lines) of cations complexed by 4′‐terphenyl‐2,2′:6′,2″‐terpyridine. As
![Figure 2. Interactions beyond dispersion, shown as dashed lines, of the cations present in (a) [VO 2 (bptpy)]ClO 4 , 1 and (b) [VO 2 (tpy)]ClO 4 [44]. O atoms are identified by showing the atoms to which they are attached. Colour coding: C = grey, N](https://thumb-eu.123doks.com/thumbv2/123doknet/13165658.390216/14.918.154.763.127.806/figure-interactions-dispersion-cations-present-identified-showing-attached.webp)
 2 ∙0.5THF, 30, showing (dashed lines) the interactions exceeding dispersion of the](https://thumb-eu.123doks.com/thumbv2/123doknet/13165658.390216/15.918.159.766.689.937/figure-perpendicular-stacked-inequivalent-structure-interactions-exceeding-dispersion.webp)