HAL Id: tel-03106800
https://hal.archives-ouvertes.fr/tel-03106800
Submitted on 12 Jan 2021
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Importance de la cinétique dans les procédés
Isabelle Ziegler-Devin
To cite this version:
Isabelle Ziegler-Devin. Importance de la cinétique dans les procédés. Génie des procédés. Université
de Lorraine, 2020. �tel-03106800�
HABILITATION À DIRIGER DES RECHERCHES
UNIVERSITÉ DE LORRAINE
École doctorale SIMPPÉ
_____________________________________________________________________________________________________
Importance de la cinétique chimique
dans les procédés
_____________________________________________________________________________________________________
Discipline : Génie des Procédés et des Produits et des Molécules
Présentée et soutenue publiquement le 11 décembre 2020
Par :
Isabelle ZIEGLER-DEVIN
Maître de conférences ENSIC - Université de Lorraine
Laboratoire d'Études et de Recherche sur le MAtériau Bois
Jury
Rapporteurs :
Mr Abdel CHAKIR, Professeur, GSMA UMR 7331, Reims
Mr Stéphane GRELIER, Professeur, LCPO UMR 5629, Bordeaux
Mme Gwénaëlle TROUVÉ, Professeur, LGRE EA UHA 233, Mulhouse
Examinateurs :
Mr Nicolas BROSSE, Professeur, LERMAB EA INRAE 4370, Nancy
Mr René FOURNET, Professeur, LRGP UMR7274, Nancy
Invité :
À mes parents,
À Charline, Yorick et Vincent
i
R E M E R C I E M E N T S
Les travaux présentés dans ce manuscrit ont été menés au Laboratoire Réactions et Génie des Procédés (LRGP) ainsi qu’au Laboratoire d’Études et de Recherche sur le Matériau Bois (LERMAB).
****
Avant d’entrer dans le vif du sujet, je tiens à remercier celles et ceux qui m’ont accompagnée depuis maintenant bien longtemps…
****
En premier lieu je tiens à remercier René Fournet et Paul-Marie Marquaire pour m’avoir accueillie en tant que doctorante au Département de Chimie-Physique des Réactions (DCPR) dès 2001, puis au LRGP.
Merci de m’avoir donné ma chance, de m’avoir tant appris.
Je vous suis reconnaissante de m’avoir aidée à devenir Maître de Conférences. C’est avant tout grâce à vous si je présente ce travail !
Merci à Paul-Marie, même si nos points de vue ont parfois divergé et même si un contexte pesant a séparé nos routes, je suis profondément reconnaissante et respectueuse de tout ce que vous m’avez apporté et appris.
Je vous dois tout mon savoir-faire expérimental ; vous m’avez appris la patience et surtout la rigueur nécessaire en recherche.
Également un grand merci à René, avec qui j’ai la chance de travailler depuis 2001. Grâce à toi, j’ai tant appris en cinétique-chimique et surtout en modélisation.
Merci de ta disponibilité, de ta motivation, de ta créativité et de ta persévérance pour que les modèles que nous avons développés aboutissent.
Je réalise également la chance que j’ai de t’avoir comme collègue en enseignement à l’ENSIC !
Je tiens également à remercier Nicolas Brosse de m’avoir ensuite accueillie au LERMAB et surtout de m’avoir permis de commencer une nouvelle et belle aventure en recherche. Merci Nicolas pour ta gentillesse, ton ingéniosité, ton éternelle bonne humeur et ta disponibilité ! Je te remercie également pour la confiance que tu m’accordes et pour tous tes encouragements qui ont mené
(enfin !) à l’aboutissement de ce travail. *****
Ce travail ne pourrait exister sans les valeureux thésards que j’ai eu la chance de co-encadrer. Ce travail est également et avant tout le vôtre !
ii *****
Ma profonde gratitude va également aux membres du jury
qui me font l’honneur de lire, de rapporter ou d’examiner l’ensemble de ce (long !) manuscrit. Je vous remercie d’autant plus que j’ai bien conscience que cette évaluation vous mobilise à une période
où le temps est précieux….
Je remercie également le professeur Guy Furdin d’avoir la gentillesse d’accepter l’invitation à participer à ce jury.
*****
Enfin je remercie les directeurs de laboratoires, les industriels, les instances et organismes ayant permis matériellement la réalisation de ces travaux de recherche.
*****
La réussite, la motivation et l’épanouissement dans l’enseignement et la recherche passent avant tout par un quotidien et « une vie de laboratoire » plaisants.
Je remercie donc tous ceux, enseignants-chercheurs, chercheurs, personnels BIATSS, thésards, invités et stagiaires, qui ont su (et savent) égayer par leur présence et leur gentillesse mon quotidien au LRGP puis au
LERMAB mais également à l’ENSIC, l’ENSGSI, au CPP de Nancy et au CPI de Strasbourg. La liste de personnes que je souhaiterais remercier est tellement longue qu’il faudrait
un autre tome à ce travail !
A l’ENSIC, je pense plus particulièrement à mes collègues enseignants Yves Simon et Laurent Marchal-Heussler.
Au LERMAB, je pense en tout premier lieu à Christelle Perrin (dommage que nos routes professionnelles se soient séparées…), Laurent Chrusciel (merci pour ta gentillesse, ta bonne humeur et ton café), Hubert Chapuis
Nicolas Houssement, Emmanuel Martin et Corinne Courtehoux. *****
Merci à mes parents, vous êtes formidables et je vous dois tout ! Merci à mon mari, Vincent.
iii
T A B L E D E S M A T I E R E S
R E M E R C I E M E N T S ... I T A B L E D E S M A T I E R E S ... III T A B L E D E S I L L U S T R A T I O N S ... IX L I S T E D E S T A B L E A U X ... XIII A V A N T P R O P O S ... XV: CV ET ACTIVITES D’ENSEIGNEMENT ET DE RECHERCHE ... 1
1 CURRICULUM VITAE ... 1
1.1 SITUATION ADMINISTRATIVE ... 1
1.2 EXPERIENCES PROFESSIONNELLES ... 2
1.3 CURSUS UNIVERSITAIRE ... 2
2 ACTIVITES D’ENSEIGNEMENT ... 3
2.1 ENSEIGNEMENT A L’ÉCOLE NATIONALE SUPERIEURE DES INDUSTRIES CHIMIQUE ... 3
2.2 ENSEIGNEMENT AU CYCLE PREPARATOIRE POLYTECHNIQUE DE NANCY ... 3
2.3 ENSEIGNEMENT AU CYCLE PREPARATOIRE INTEGRE DE STRASBOURG ... 3
2.4 ENSEIGNEMENT A L’ÉCOLE NATIONALE SUPERIEURE EN GENIE DES SYSTEMES ET DE L’INNOVATION ... 3
2.5 ENSEIGNEMENT EN FORMATION CONTINUE SPECIALISATION ÉCONOMIE CIRCULAIRE ... 4
2.6 RECAPITULATIF HORAIRE DES ENSEIGNEMENTS SUR LES CINQ DERNIERES ANNEES ... 4
3 ACTIVITES DE RECHERCHE ... 4
: ACTIVITES DE RECHERCHE DEVELOPPEES AU LRGP ... 5
CHAPITRE 1 : OBJECTIFS ET METHODOLOGIE ... 5
CHAPITRE 2 : QUELQUES RAPPELS DE CINETIQUE ... 9
1 LES REACTEURS IDEAUX DE LABORATOIRE ... 9
2 LA MESURE DE LA VITESSE ... 10
3 LA LOI DE VITESSE ... 11
4 LA LOI D’ARRHENIUS ... 13
5 LES PROCESSUS ELEMENTAIRES ... 13
6 LES REACTIONS COMPLEXES ... 14
CHAPITRE 3 : LES MONTAGES EXPERIMENTAUX DEVELOPPES ... 15
1 LA ZONE REACTIONNELLE : LES REACTEURS UTILISES ET LEUR MODE DE CHAUFFAGE ... 15
1.1 LES REACTEURS ... 15
1.1.1 Le réacteur ouvert auto-agité par jets gazeux ... 16
1.1.2 Le réacteur auto-agité pour les études hétéro-homogènes. ... 18
1.1.3 Le réacteur ouvert tubulaire à écoulement piston ... 20
1.2 CHAUFFAGE DU REACTEUR ... 21
1.2.1 Utilisation d’éléments chauffants ... 21
1.2.2 Utilisation d’un four tubulaire ... 22
1.3 LE CONTROLE DE LA PRESSION DU REACTEUR ... 23
2 ZONE PRE REACTIONNELLE ... 23
2.1 CAS DES REACTIFS GAZEUX ... 23
2.2 CAS DES REACTIFS LIQUIDES ... 24
2.3 CAS DES REACTIFS SOLIDES ... 25
3 ZONE POST-REACTIONNELLE ... 27
4 SYNTHESE ... 27
CHAPITRE 4 : DEVELOPPEMENT DE MODELES CINETIQUES ... 29
1 LES ESPECES IMPLIQUEES DANS LES MECANISMES ... 29
2 LES REACTIONS IMPLIQUEES DANS LE MECANISME ... 29
3 ÉCRITURE DES MECANISMES ... 30
4 LES DONNEES CINETIQUES DU MECANISME... 31
5 LES DONNEES THERMODYNAMIQUES ... 32
6 RELATIONS ENTRE GRANDEURS THERMODYNAMIQUES ET CINETIQUES ... 33
7 SIMULATIONS AVEC CHEMKIN II ... 34
iv
CHAPITRE 5 : DU PROCEDE A SA MODELISATION CINETIQUE ... 37
1 ÉLABORATION DES COMPOSITES C/C : MODELISATION CINETIQUE HETERO-HOMOGENE DU DEPOT DE PYROCARBONE OBTENU PAR PYROLYSE DE PROPANE. ... 37
1.1 LES COMPOSITES C/C ... 37
1.1.1 Les fibres de carbone ex-PAN ... 38
1.1.2 Le pyrocarbone ... 41
1.1.3 Elaboration des composites C/C ... 43
1.1.4 Principales propriétés et applications des composites C/C ... 44
1.2 OBJECTIFS DE L’ETUDE ... 45
1.3 LE PILOTE EXPERIMENTAL ... 46
1.3.1 La zone préréactionnelle ... 47
1.3.2 La zone réactionnelle ... 48
1.3.3 La zone post-réactionnelle : ... 48
1.3.4 Les études paramétriques réalisées ... 52
1.4 LE MODELE CINETIQUE... 52
1.4.1 Le modèle homogène ... 52
1.4.1.1 Les espèces impliquées dans le mécanisme ... 53
1.4.1.2 Les réactions impliquées dans le mécanisme ... 53
1.4.1.3 Les données cinétiques du mécanisme ... 53
1.4.1.4 Les données thermodynamiques du mécanisme ... 54
1.4.1.5 Écriture et classement du mécanisme ... 54
1.4.2 Le modèle hétérogène ... 54
1.4.2.1 Hypothèses envisagées pour décrire les réactions de dépôt de pyrocarbone ... 54
1.4.2.2 Les processus élémentaires hétérogènes pris en compte dans le mécanisme ... 56
1.4.2.3 Nomenclature des espèces et écriture des réactions de surface au format Chemkin ... 59
Réactions unimoléculaires de surface... 60
Réactions bimoléculaires gaz/surface. ... 60
Réactions bimoléculaires surface/surface ... 60
1.4.2.4 Méthode et stratégie utilisée pour l’écriture du mécanisme hétérogène ... 61
1.4.2.5 Les données thermodynamiques des espèces du mécanisme hétérogène ... 61
1.4.2.6 Détermination des paramètres cinétiques des réactions du mécanisme hétérogène ... 62
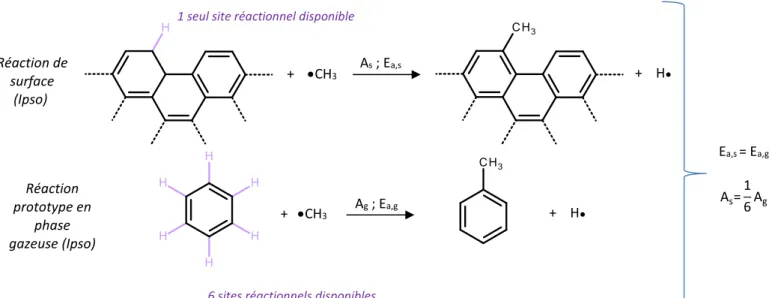
Utilisation de réactions prototypes en phase gazeuse ... 62
Estimation des paramètres cinétiques des réactions de surface ... 63
1.4.2.7 Détermination de la densité de site ... 64
1.4.2.8 Bilan sur l’écriture du mécanisme hétérogène ... 64
1.4.3 Simulations ... 64
1.4.4 Modélisation de la vitesse de dépôt de pyrocarbone ... 65
1.4.5 Modélisation de la composition de la phase gazeuse ... 67
1.4.5.1 Étude paramétrique en fonction de la température ... 68
1.4.5.2 Étude paramétrique en fonction du temps de passage ... 70
1.4.5.3 Étude paramétrique en fonction du rapport surface des fibres sur le volume de gaz (A/V) ... 72
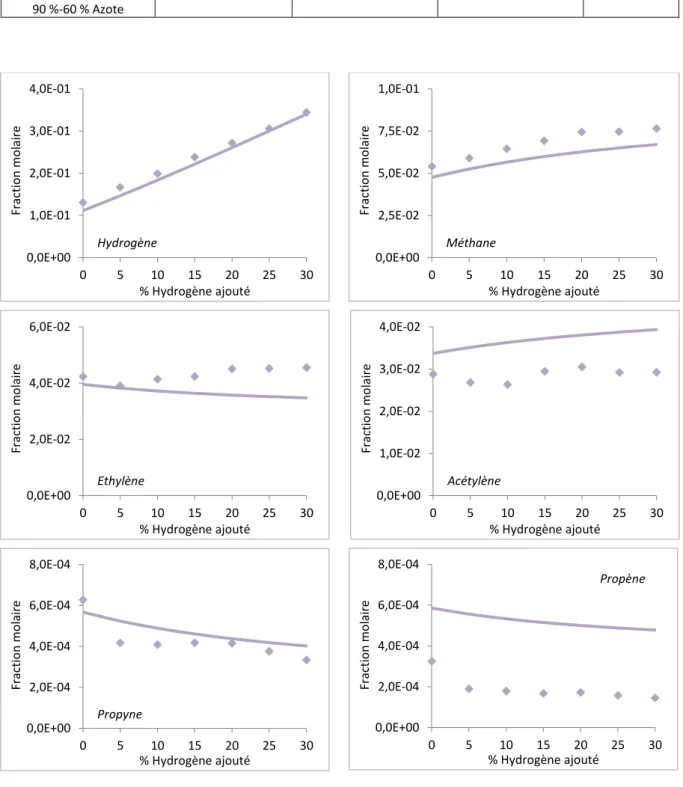
1.4.5.4 Étude paramétrique en fonction du pourcentage d’hydrogène ajouté ... 74
1.4.5.5 Étude paramétrique en fonction du pourcentage d’acétylène ajouté ... 75
1.4.5.6 Étude paramétrique en fonction de la température en présence de benzène ... 77
1.4.6 Validation globale du modèle ... 79
1.5 EXPLOITATION DU MECANISME : ANALYSE DES PRINCIPAUX PRECURSEURS DU PYROCARBONE ET VOIES DE DEPOTS ... 80
1.6 CONCLUSION ET VALORISATION DE CE PROJET ... 84
2 MODELISATION CINETIQUE DE L’ABATTEMENT D’UNE MOLECULE MODELE DE DIOXINES ... 85
2.1 PROBLEMATIQUE DES DIOXINES. ... 85
2.1.1 Les dioxines ... 85
2.1.2 Fonctionnement d’une Usine d’Incinération d’Ordures Ménagères ... 88
2.1.3 Formation des dioxines dans les procédés d’incinération ... 89
2.1.4 La règle des 3T ... 90 2.2 OBJECTIFS DE L’ETUDE ... 90 2.3 LE PILOTE EXPERIMENTAL ... 92 2.3.1 La zone préréactionnelle ... 93 2.3.2 La zone réactionnelle ... 94 2.3.3 La zone post-réactionnelle ... 94
v
2.4 LE MODELE CINETIQUE... 97
2.4.1 Le modèle ... 97
2.4.1.1 Les espèces impliquées dans le mécanisme ... 98
2.4.1.2 Les réactions impliquées dans le mécanisme ... 98
2.4.1.3 Les données cinétiques du mécanisme ... 98
2.4.1.4 Les données thermodynamiques du mécanisme ... 99
2.4.1.5 Écriture et classement du mécanisme ... 99
2.4.1.6 Simulations ... 100
2.4.2 Validation sur les travaux d’A. Tritz - résultats de pyrolyse du DBF à forte dilution ... 100
2.4.2.1 Conversion du DBF ... 100
2.4.2.2 Composition de la phase gazeuse - espèces « lourdes » ... 101
2.4.3 Validation sur les travaux d’A. Tritz - résultats d’oxydation du DBF à forte dilution ... 103
2.4.3.1 Conversion du DBF ... 103
2.4.3.2 Composition de la phase gazeuse - espèces « lourdes » ... 104
2.4.4 Validation sur les travaux de R. Wörner - résultats d’oxydation du DBF à faible dilution ... 105
2.4.4.1 Conversion du DBF ... 105
2.4.4.2 Composition de la phase gazeuse - espèces « légères » et « lourdes » ... 106
2.4.5 Validation globale du modèle ... 107
2.5 EXPLOITATION DU MECANISME ... 108
2.5.1 Voies de consommation du DBF ... 108
2.5.2 Extrapolation du mécanisme... 109
2.5.2.1 Influence de traces d’oxygène sur la pyrolyse du DBF... 110
2.5.2.2 Influence de traces d’oxygène sur l’oxydation du DBF ... 110
2.5.2.3 Influence d’un ajout de méthane sur l’oxydation du DBF ... 111
2.6 CONCLUSION ET VALORISATION DE CE PROJET ... 112
3 MODELISATION CINETIQUE DU REFORMAGE THERMIQUE DES GAZ ISSUS DE LA BIOMASSE ... 113
3.1 CONVERSION THERMOCHIMIQUE DE LA BIOMASSE LIGNOCELLULOSIQUE ... 114
3.1.1 Composition élémentaire de la biomasse ... 114
3.1.2 Valorisation énergétique de la biomasse ... 114
3.1.3 La valorisation thermochimique de la biomasse ... 116
3.1.3.1 La pyrolyse de la biomasse ... 116
3.1.3.2 La gazéification de la biomasse ... 119
Le syngas ... 119
Le procédé de gazéification de la biomasse ... 120
3.1.4 L’épuration des gaz issus de la gazéification de la biomasse ... 124
3.2 OBJECTIFS DE L’ETUDE ... 126 3.3 LE PILOTE EXPERIMENTAL ... 126 3.3.1 La zone pré-réactionnelle ... 128 3.3.2 La zone réactionnelle ... 128 3.3.3 La zone post-réactionnelle ... 129 3.3.3.1 Produits « légers » ... 129 3.3.3.2 Produits « lourds »... 129
3.3.4 Les études paramétriques réalisées ... 129
3.4 LE MODELE CINETIQUE... 130
3.5 SIMULATIONS ... 131
3.6 VALIDATION DU MECANISME CINETIQUE ... 133
3.6.1 Validation en condition vaporeformage du méthane ... 133
3.6.2 Validation en condition de reformage de l’ethylène en mélange complexe ... 135
3.6.3 Validation en condition de vaporeformage du benzène et du méthane en mélange complexe 137 3.6.4 Conclusion et validation globale du modèle. ... 138
3.7 EXPLOITATION DU MECANISME ... 139
3.7.1 Analyse de flux et schéma réactionnel ... 139
3.7.2 Etude paramétrique autour d’une composition type de synags ... 140
3.7.3 Pilote PEGASE (CEA) ... 141
3.8 CONCLUSION ET VALORISATION DE CE PROJET ... 143
4 CONCLUSION GENERALE ... 145
REFERENCES BIBLIOGRAPHIQUES ... 147
A ... 147
vi C ... 149 D ... 149 E ... 150 F ... 150 G ... 150 H ... 151 K ... 151 L ... 152 M ... 153 N ... 153 P ... 153 Q ... 153 R ... 154 S ... 154 T ... 154 V ... 155 W ... 155 Y ... 156 Z... 156
: ACTIVITES DE RECHERCHE DEVELOPPEES AU LERMAB ... 159
FAISABILITE DE LA VALORISATION D’UN LIGNEUX (SALIX VIMINALIS) ISSU DE PHYTOTECHNOLOGIES EN BIOCARBURANT ... 159
1 CONTEXTE : LA PROBLEMATIQUE DES BIOMASSES CONTAMINEES AUX ETM ... 159
1.1 SOURCES DE POLLUTION AUX ETM ... 160
1.2 IMPACT SUR LES ORGANISMES ... 160
2 LA PHYTOREMEDIATION ... 161
2.1 DEFINITION DE LA PHYTOREMEDIATION ... 161
2.2 STRATEGIES DE PHYTOREMEDIATION ... 161
2.3 LA PHYTOEXTRACTION ... 162
2.4 LES LIMITES DE LA PHYTOEXTRACTION ... 164
2.5 LE VOIES DE VALORISATION DES PLANTES ISSUES DE LA PHYTOREMEDIATION ... 165
3 LA BIOMASSE LIGNOCELLULOSIQUE ... 166
3.1 LES CELLULES ET LA PAROI PECTOCELLULOSIQUE ... 167
3.2 LES BIOPOLYMERES CONSTITUTIFS DE LA BIOMASSE LIGNOCELLULOSIQUE ... 168
3.2.1 La cellulose ... 169
3.2.2 Les hémicelluloses ... 170
3.2.3 La lignine ... 174
4 LE BIOETHANOL ... 176
4.1 LE BIOETHANOL DE PREMIERE GENERATION ... 176
4.1.1 Le procédé de production du bioéthanol 1G ... 176
4.1.2 Utilisation du bioéthanol 1G en tant que carburant ... 177
4.1.3 Problématique du bioéthanol 1G ... 178
4.2 LE BIOETHANOL DE DEUXIEME GENERATION OU ETHANOL CELLULOSIQUE ... 179
5 LE PRETRAITEMENT PAR EXPLOSION A LA VAPEUR OU STEAM-EXPLOSION ... 180
5.1 LE PRINCIPE ... 181
5.1.1 Le vapocraquage ... 181
5.1.2 La décompression explosive ... 182
5.1.3 Paramètres clé de la steam-explosion ... 183
5.2 LE PILOTE D’EXPLOSION A LA VAPEUR DU LERMAB ... 183
5.3 CINETIQUE D’HYDROLYSE DES POLYSACCHARIDES EN MILIEU ACIDE ... 186
5.3.1 Cas de l’hydrolyse de la cellulose ... 186
5.3.2 Cas de l’hydrolyse des hémicelluloses ... 188
5.4 SEVERITE DU PRETRAITEMENT ... 189
5.4.1 Historique du facteur de sévérité ... 189
5.4.2 Calcul du facteur de sévérité selon Overend et Chornet ... 192
5.4.3 Les limitations du facteur de sévérité proposé par Overend et Chornet ... 192
vii
7 LA FERMENTATION ETHANOLIQUE ... 195
8 FAISABILITE DE LA PRODUCTION DE BIOETHANOL-2G A PARTIR DE SAULE CONTAMINE AUX ETM ... 196
8.1 COMPOSITION CHIMIQUE DU SAULE ... 197
8.2 PRETRAITEMENT DU SAULE CONTAMINE PAR EXPLOSION A LA VAPEUR ... 197
8.2.1 Rendement en pâte cellulosique... 198
8.2.2 Composition chimique de la pâte ... 199
8.2.3 Distribution des ETMs ... 200
8.2.4 Conclusion sur l’effet du prétraitement par explosion à la vapeur... 201
8.3 HYDROLYSE ENZYMATIQUE DES PATES ... 202
8.3.1 Protocole d’hydrolyse enzymatique... 202
8.3.2 Cinétique d’hydrolyse ... 202
8.4 FERMENTATION DES HYDROLYSATS ... 204
8.4.1 Protocole de fermentation des hydrolysats ... 204
8.4.2 Résultats de fermentation ... 205
9 CONCLUSION ET VALORISATION DE CE PROJET ... 206
REFERENCES BIBLIOGRAPHIQUES ... 209 A ... 209 B ... 210 C ... 211 D ... 211 E ... 212 F ... 212 G ... 213 H ... 213 I ... 214 J ... 214 K ... 214 L ... 214 M ... 215 N ... 216 O ... 216 P ... 216 R ... 217 S ... 217 T ... 219 U ... 219 V ... 219 W ... 220 Y ... 220 Z... 220 : BILAN ET PERSPECTIVES ... 161 BILAN ... 221
BILAN GLOBAL DES PUBLICATIONS ET THESES CO-ENCADREES ... 226
PERSPECTIVES ... 231
1 PERSPECTIVES A COURT TERME : VERS UNE VALORISATION DES DECHETS DE BOIS DE CLASSE B EN BIOETHANOL ... 232
2 PERSPECTIVES A MOYEN TERME : VERS UNE OPTIMISATION DE LA VALORISATION DES FRACTIONS ISSUES DU PROCEDE DE STEAM-EXPLOSION EN VUE D’UNE PRODUCTION DE BIOETHANOL - CAS DES EXTRACTIBLES. 233 3 PERSPECTIVES A LONG TERME : SCALING-UP DU PROCEDE D’EXPLOSION A LA VAPEUR ... 237
ix
T A B L E D E S I L L U S T R A T I O N S
Figure 1 : Méthodologie utilisée pour l’étude cinétique des réactions thermiques en phase gazeuse. ... 6
Figure 2 : Schéma d’un réacteur quelconque. ... 10
Figure 3 : Schéma de principe des montages réactionnels développés. ... 15
Figure 4 : Photographie du réacteur auto-agité par jets gazeux. ... 16
Figure 5 : Schéma de principe du réacteur auto-agité par jets gazeux [Tritz 2014a]... 18
Figure 6 : Schéma et photo du réacteur continu agité pour l’étude des réactions de CVD/CVI [Ziegler 2004]. ... 19
Figure 7 : Schéma et photo du réacteur continu agité catalytique [Barbé 1996]. ... 19
Figure 8 : Principe de fonctionnement d’un régulateur de débit massique thermique [Hardy 1999]. ... 24
Figure 9 : Schéma du réservoir d’alimentation d’un RDM liquide. ... 25
Figure 10 : dispositif de mélange et d’évaporation du réactif liquide (CEM). ... 25
Figure 11 : Dynacalibrator VICI 150 (source VICI Metronics - [Susaya 2012]). ... 26
Figure 12 : Principe de fonctionnement du Dynacalibrator VICI 150 (source VICI Metronics). ... 26
Figure 13 : Schéma et photo d’un tube de diffusion. ... 26
Figure 14 : Principe de fonctionnement simplifié de CHEMKIN II. ... 35
Figure 15 : Monomère et polymère d’acrylonitrile. ... 38
Figure 16 : Principe du procédé de fabrication des fibres de carbone ex-PAN selon Luyckx ... 38
Figure 17 : Mécanisme d’oxydation du PAN [Mascia 1991]. ... 39
Figure 18 : Étape de « carbonisation » du PAN « oxydé ». ... 40
Figure 19 : Classement des fibres de carbone en fonction de leurs propriétés mécaniques [Camus 2016]. ... 40
Figure 20 : Disposition des nappes de fibres ex-PAN (a) [Fradet 2015]. ... 41
Figure 21 : Représentation de carbone turbostratique [Bokros 1969]. ... 42
Figure 22 : Cristallites d’un carbone turbostratique [Naslain 1979]. ... 42
Figure 23 : Exemples d’applications de composites C/C dans le domaine spatial. ... 45
Figure 24 : Exemples d’applications des composites C/C dans le domaine aéronautique et automobile. ... 45
Figure 25 : Schéma de principe du pilote expérimental d’élaboration des composites C/C. ... 47
Figure 26 : Exemple de croissance « verticale » par collage de phénanthrène ... 55
Figure 27 : Exemple de croissance « latérale » de pyrocarbone [Lacroix 2009a]. ... 55
Figure 28 : Sites de surface « bateau » et « zig-zag ». ... 56
Figure 29 : Hydrogène de surface en sites de surfaces « bateau » et « zig-zag ». ... 56
Figure 30 : Réaction d’amorçage unimoléculaire à partir d’un site H(S). ... 57
Figure 31 : Réaction d’addition d’une molécule d’éthylène sur un site C.(S). ... 57
Figure 32 : Réaction de métathèse sur un site H(S) par un radical méthyle. ... 57
Figure 33 : Réaction de β-scission de l’espèce de surface CH.CH2(S). ... 58
Figure 34 : Réaction d’isomérisation surfacique. ... 58
Figure 35 : Réaction de substitution d’un atome d’hydrogène H(S) par un groupement méthyle. ... 58
Figure 36 : Réaction de terminaison entre un site CH2.z(S) et un radical méthyle. ... 59
Figure 37 : Réaction de déshydrogénation moléculaire des sites CH2CH2(S). ... 59
Figure 38 : Réaction unimoléculaire de surface. ... 60
Figure 39 : Réaction bimoléculaire gaz/surface. ... 60
Figure 40 : Réaction bimoléculaire surface/surface... 61
Figure 41 : Exemple d’une réaction prototype - cas d’une métathèse. ... 62
Figure 42 : Exemple d’une réaction prototype - cas d’une β-scission. ... 63
Figure 43 : Calcul des paramètres cinétiques d’une réaction de surface ... 63
Figure 44 : Confrontation des valeurs simulées et expérimentales de la vitesse de dépôt du pyrocarbone. ... 66
Figure 45 : Confrontation des valeurs simulées et expérimentales en fonction de la température. ... 70
Figure 46 : Confrontation des valeurs simulées et expérimentales en fonction du temps de passage. ... 71
Figure 47: Confrontation des valeurs simulées et expérimentales en fonction du rapport A/V. ... 73
Figure 48 : Confrontation des valeurs simulées et expérimentales en présence d’un ajout d’hydrogène. ... 75
Figure 49 : Confrontation des valeurs expérimentales et simulées en présence d’un ajout d’acétylène. ... 77
Figure 50 : Confrontation des valeurs simulées et expérimentales en présence d’un ajout de benzène. ... 79
Figure 51 : Schéma de flux de dépôt de pyrocarbone sur des sites bateaux. ... 82
Figure 52 : Schéma de flux de dépôt de pyrocarbone sur des sites zig-zag. ... 83
x
Figure 54 : 2,3,7,8-tétrachlorodibenzodioxine - (dioxine dite de « Seveso »). ... 86
Figure 55 : Évolution de la répartition des secteurs émetteurs de dioxines ... 88
Figure 56 : Exemple de principe de fonctionnement d’une UIOM (Incinérateur du Grand-Dijon). ... 88
Figure 57 : Molécule de dibenzofurane (DBF). ... 91
Figure 58 : Schéma de principe du pilote expérimental d’abattement du DBF. ... 92
Figure 59 : Contrôle du régime stationnaire du flux gazeux de DBF. ... 93
Figure 60 : Cartouche contenant l’adsorbant Tenax®.. ... 96
Figure 61 : Confrontation des valeurs simulées et expérimentales de la fraction molaire du DBF non converti101 Figure 62 : Confrontation des valeurs simulées et expérimentales des espèces gazeuses issues de la pyrolyse102 Figure 63 : Confrontation des valeurs simulées et expérimentales de la fraction molaire du DBF non converti103 Figure 64 : Confrontation des valeurs simulées et expérimentales des espèces gazeuses ... 105
Figure 65 : Confrontation des valeurs simulées et expérimentales de la fraction molaire du DBF non converti106 Figure 66 : Confrontation des valeurs simulées et expérimentales des espèces gazeuses ... 107
Figure 67 : Description des voies de consommation du DBF en condition d’oxydation [Tritz 2014b]. ... 109
Figure 68 : Évolution de la fraction molaire du DBF en condition de pyrolyse et en présence ... 110
Figure 69 : Évolution de la fraction molaire du DBF en condition d’oxydation. ... 111
Figure 70 : Évolution de la fraction molaire du DBF en condition d’oxydation ... 111
Figure 71 : Voies de conversion de la biomasse. ... 115
Figure 72 : Schéma global de pyrolyse de la biomasse [Antal 1983]. ... 116
Figure 73 : Influence des paramètres temps de séjour, température et puissance de chauffe ... 117
Figure 74 : Mécanisme globalisé de la pyrolyse de la cellulose [Bradbury 1979]. ... 118
Figure 75 : Mécanisme globalisé de la pyrolyse de la lignine et des hémicelluloses [Miller 1997]. ... 118
Figure 76 : « Maturation » des goudrons en fonction des températures de pyro-gazéification ... 119
Figure 77 : Gazéifieurs à lit fixe à contre-courant et à co-courant [Sikarwar 2017]. ... 122
Figure 78 : Gazéifieurs à lit fluidisé dense (à gauche) et circulant (à droite) [Sikarwar 2017]. ... 123
Figure 79 : Gazéifieur à lit entraîné [Williams 2007]. ... 123
Figure 80 : Schéma de principe du pilote expérimental de reformage à haute température. ... 127
Figure 81 : Profils de température du four VECSTAR ... 128
Figure 82 : Évolution des fractions molaires du réactif et des produits en fonction de la température ... 134
Figure 83 : Évolution des fractions molaires du réactif et des produits en fonction de la température ... 136
Figure 84 : Évolution des fractions molaires des réactifs et produits en fonction de la température. ... 138
Figure 85 : Schéma réactionnel simplifié de reformage du méthane à 1300°C - [Hiblot 2010] ... 140
Figure 86 : Influence de la composition du syngas sur la fraction molaire de méthane ... 141
Figure 87 : Evolution des fractions molaires des réactifs et produits en fonction de la température. ... 142
Figure 88 : Stratégies de phytoremédiation [Favas 2014]. ... 162
Figure 89 : Exemples de plantes hyperaccumulatrices. ... 163
Figure 90 : Exemples de plantes accumulatrices à faiblement accumulatrices. ... 164
Figure 91 : Voies de valorisations envisageables pour les biomasses issues de phytoremédiation. ... 165
Figure 92 : Arbre phylogénétique des plantes (Plantae). ... 167
Figure 93 : (a) Image MET cellule de peuplier [Joseleau 2004]. ... 168
Figure 94 : Biopolymères constitutifs de la biomasse lignocellulosique. ... 169
Figure 95 : Molécule de de β-D-glucopyranose. ... 169
Figure 96 : Motif de cellobiose constitutif de la cellulose... 169
Figure 97 : Représentation simplifiée de la cellulose. ... 169
Figure 98 : Principaux monosaccharides constitutifs des hémicelluloses. ... 171
Figure 99 : Liaisons engagées dans le squelette des hémicelluloses. ... 171
Figure 100 : Exemple de xyloglucane. ... 172
Figure 101 : Exemple de glucuronoxylane. ... 172
Figure 102 : Exemple d’arabinoxylane. ... 172
Figure 103 : Exemple de glucuronoarabinoxylane. ... 172
Figure 104 : Homoxylane non substitué. ... 173
Figure 105 : Mannane et glucomannane. ... 173
Figure 106 : Exemple de galactomannane. ... 173
Figure 107 : Exemple de galactoglucomannane. ... 173
Figure 108 : Exemple de glucane mixte (⚫ : β-D-glucose). ... 173
Figure 109 : Répartition des hémicelluloses dans les gymnospermes et angiospermes [Terret 2019]. ... 174
xi
Figure 111 : Liaisons majeures dans la lignine [Dorrestijn 2000]. ... 175
Figure 112 : Exemple de liaisons engagées dans la lignine. ... 175
Figure 113 : Principe de production du bioéthanol-1G. (Opérations unitaires en bleu). ... 177
Figure 114 : Principe de production du bioéthanol-2G (schéma FUTUROL). ... 180
Figure 115 : Effet du prétraitement par SE sur la biomasse lignocellulosique - adapté de [Hsu 1996]. ... 181
Figure 116 : Impact de la sévérité du procédé de SE sur la dépolymérisation des hémicelluloses. ... 182
Figure 117 : Réactions de déshydratation des monosaccharides. ... 182
Figure 118 : Photo et schéma synoptique du pilote d’explosion à la vapeur du LERMAB. ... 185
Figure 119 : Mécanisme d’hydrolyse de la cellulose [Saeman 1945]. ... 186
Figure 120 : Modèle cinétique d’hydrolyse de la cellulose proposé par Mok et al. [Mok 1992]. ... 187
Figure 121 : Modèle cinétique d’hydrolyse de la cellulose proposé par Bouchard et al. [Bouchard 1990]. ... 188
Figure 122 : Modèle cinétique d’hydrolyse des hémicelluloses calqué sur le modèle de Saeman. ... 188
Figure 123 : Modèle cinétique d’hydrolyse des hémicelluloses selon Kobayashi and Sakai [Kobayashi 1956]. . 188
Figure 124 : Modèle cinétique d’hydrolyse des hémicelluloses selon Connor and Lorenz [Connor 1986]. ... 189
Figure 125 : Exemple de calcul du facteur H - tracé selon données de Brasch et Free. ... 191
Figure 126 : Corrélation entre le facteur H et le rendement en pâte et en lignine ... 191
Figure 127 : Représentation schématique de l'action des cellulases sur la structure de la cellulose ... 194
Figure 128 : Rendement en pâte cellulosique (% MS initiale) en fonction de la sévérité du traitement. ... 199
Figure 129 : Teneur en lignine (a) et en sucres résiduels (b) dans la pâte (% MS de pâte)... 199
Figure 130 : Teneur en glucose (a) et en xylose (b) résiduels dans la pâte (% MS de pâte) ... 200
Figure 131 : Taux d’ETM résiduel dans les pâtes en fonction du facteur de sévérité. ... 201
Figure 132 : Cinétique d'hydrolyse enzymatique des six pâtes obtenues après SE du Saule ... 202
Figure 133 : Modélisations des interactions Cellulase (Trichoderma Reseei) - Cellulose - Lignine... 203
Figure 134 : Suivi cinétique de fermentation des trois hydrolysats récupérés après hydrolyse ... 205
Figure 135 : Schéma du dispositif « oxygen uptake » développé par Vaglmimigli et al. [Amorati 2017] ... 236
xiii
L I S T E D E S T A B L E A U X
Tableau 1 : Principales espèces identifiées par GC/MS ... 50
Tableau 2 : Estimation de paramètres cinétiques par RPD. ... 53
Tableau 3 : Conditions opératoires et de modélisation. ... 65
Tableau 4 : Rappel des conditions de l’étude paramétrique en fonction de la température. ... 68
Tableau 5 : Rappel des conditions de l’étude paramétrique fonction du temps de passage. ... 70
Tableau 6 : Rappel des conditions de l’étude paramétrique fonction du temps de passage. ... 72
Tableau 7 : Rappel des conditions de l’étude paramétrique en présence d’un ajout d’hydrogène. ... 74
Tableau 8 : Rappel des conditions de l’étude paramétrique en présence d’un ajout d’acétylène. ... 75
Tableau 9 : Rappel des conditions de l’étude paramétrique en présence de benzène. ... 77
Tableau 10 : Principaux flux de dépôt de pyrocarbone. ... 81
Tableau 11 : Répartition des différents congénères des PCDD et PCDF. ... 86
Tableau 12 : Évolution des émissions dans l’air en PCDD et PCDF en France métropolitaine. ... 87
Tableau 13 : Classement des sous-secteurs les plus émetteurs de PCDD et PCDF en 2018 en % en France... 87
Tableau 14 : Principaux produits identifiés par GC/MS lors de l’oxydation ou de la pyrolyse du DBF. ... 95
Tableau 15 : Estimation de paramètres cinétiques par RPD. ... 99
Tableau 16 : Conditions opératoires utilisées pour la validation du mécanisme. ... 100
Tableau 17 : Composition élémentaire de quelques biomasses (% massiques). ... 114
Tableau 18 : Valeurs des paramètres cinétiques issus des mécanismes globaux. ... 119
Tableau 19 : Composition moyenne du syngas [Demirbas 2007]. * donnée sur gaz sec et sans diazote. ... 119
Tableau 20 : Composition chimique des goudrons en fonction de la température ... 120
Tableau 21 : Exemples de qualité requise pour la valorisation du syngas (inspiré de [ADEME 2001]). ... 120
Tableau 22 : Composition typique du syngas en sortie de procédé (n.d. : non déterminé). ... 124
Tableau 23 : Critère de Péclet du réacteur en mullite pour un temps de passage de 0,68 s [Hiblot 2010]. ... 128
Tableau 24 : Produits issus du vaporeformage du benzène. ... 130
Tableau 25 : Réactions apportées au mécanisme initial. ... 131
Tableau 26 : Réaction hétérogène apportées au mécanisme initial. ... 131
Tableau 27 : Conditions opératoires utilisées pour la validation du mécanisme. ... 132
Tableau 28 : Teneurs en ETM dans certains plantes hyperaccumulatrices [Chaney 2010.] ... 163
Tableau 29 : Hémicelluloses dans les parois primaires et secondaires des végétaux [Vibe Scheller 2010]. ... 174
Tableau 30 : Exemple de répartition des liaisons dans la lignine de conifères et feuillus [Dorrestijn 2010]. ... 176
Tableau 31 : Énergies d’activation de l’hydrolyse cellulose obtenue à partir du modèle de Saeman. ... 187
Tableau 32 : Exemples de valeurs d’énergies d’activation d’hydrolyse d’hémicelluloses. ... 189
Tableau 33 : Vitesse relative d’autohydrolyse ... 190
Tableau 34 : Comparaison des paramètres T et ts pour une même sévérité. ... 193
Tableau 35 : Effet de certains ETM sur les différentes familles d'enzymes cellulolytiques. ... 195
Tableau 36 : Composition chimique du Saule et teneurs en ETM (en mg.kg-1). ... 197
Tableau 37 : Composition de l’holocellulose du Saule (%). ... 197
Tableau 38 : Conditions de prétraitement par explosion à la vapeur. ... 198
Tableau 39 : Teneur en métaux et hydrolysabilité des pâtes cellulosiques en fonction des conditions ... 204
xv
A V A N T P R O P O S
Ce travail construit autour du « rôle et de l’importance de la cinétique chimique dans les procédés » retrace des travaux réalisés entre 2004 et 2018 dans deux laboratoires de l’université de Lorraine, à savoir le Laboratoire Réactions et Génie des Procédés (LRGP) et le Laboratoire d'Étude et de Recherche sur le Matériau Bois (LERMAB).
Le manuscrit s’articule autour de trois parties :
La première partie retrace le parcours universitaire m’ayant conduite aux fonctions de Maître de Conférences à l’École Nationale Supérieure des Industries Chimiques.
La seconde partie présente une partie de mes travaux de recherche effectués au LRGP et qui portent sur la cinétique des réactions thermiques en phase gazeuse mises en jeu dans des procédés industriels ou de potentiels procédés industriels.
La troisième partie présente une partie des travaux de recherche réalisés au LERMAB et qui s’intéressent aux procédés de valorisation chimique de la biomasse par voie humide et plus particulièrement à l’optimisation de procédés de prétraitement de la biomasse.
: CV et activités d’enseignement et de
recherche
1
1
CURRICULUM VITAE
Précisions préalables :
Le Département de Chimie Physique des Réactions (DCPR) a fusionné avec trois autres laboratoires de l’École Nationale Supérieure des Industries Chimiques (ENSIC) pour devenir le Laboratoire Réactions et Génie des Procédés (LRGP) en 2010.
L’Institut National Polytechnique de Lorraine (INPL), l’Université Henri Poincaré ont fusionné en 2012 avec deux autres universités lorraines pour devenir l’Université de Lorraine.
1.1 Situation administrative
Isabelle Devin, née Ziegler Née le 5 mai 1975 à Lunéville
Grade : Maître de Conférences depuis le 1er octobre 2005
Section CNU : 31
Université : Université de Lorraine
Composante : École Nationale Supérieure des Industries Chimiques - Nancy Unités de recherche :
2001-2014 : Département de Chimie Physique des Réactions - Nancy Devenu, Laboratoire Réactions et Génie des Procédés
Depuis 2014 : Laboratoire d'Étude et de Recherche sur le Matériau Bois - Vandoeuvre lès Nancy
Adresse : ENSIC LERMAB
1 rue Grandville Faculté des Sciences et Techniques, BP 20451 BP 239
54000 Nancy 54506 Vandoeuvre lès Nancy Mail : [email protected]
2
1.2 Expériences professionnelles
Depuis octobre 2005 : Maître de Conférences - INPL puis Université de Lorraine 01/05 - 09/05 :ATER à temps plein
Enseignement : École Européenne d’Ingénieurs en Génie des Matériaux - INPL Recherche réalisée au DCPR
07/04 - 01/05 : Post-Doctorat
Financé par Snecma Propulsion Solide Réalisé au DCPR
04/01 - 07/04 : Thèse
Financée par Snecma Propulsion Solide Réalisée au DCPR
1.3 Cursus universitaire
2001-2004 : Doctorat en Génie des Procédés de l’Institut National Polytechnique de Lorraine Sujet « Modélisation cinétique des dépôts de pyrocarbone obtenus par pyrolyse d’hydrocarbures » Financement : Snecma Propulsion Solide
Directeurs de thèse : Paul-Marie Marquaire et René Fournet
Laboratoire d’accueil : Département de Chimie-Physique des Réactions - ENSIC - Nancy Durée : 3 ans et 3 mois
Ce travail a obtenu le prix de thèse de l’INPL
Jury : Rapporteurs : Gérard Vignoles - Professeur Bordeaux I Jean Pierre Sawerysyn - Professeur Lille Examinateurs : Guy Furdin - Professeur Université de Lorraine
Cédric Descamps - Ingénieur R&D Snecma Propulsion Solide René Fournet - Professeur Université de Lorraine
Paul-Marie Marquaire - Directeur de Recherche CNRS 1997- 1998 : DEA Sciences et Ingénierie des Matériaux
École des Mines de Nancy
Stage : « Propriétés de surface et désagglomération des poudres de TiO2(II) obtenues par broyage à
haute-énergie »
Laboratoire de Science et Génie des Matériaux et de Métallurgie - École des Mines de Nancy 1996-1997 : Maîtrise de Chimie Physique
2ème année de Magistère de Génie Moléculaire Matériaux et Procédés
Faculté des Sciences - Université Henri Poincaré - Nancy 1995-1996 : Licence de Chimie Physique
1ère année de Magistère de Génie Moléculaire Matériaux et Procédés
Faculté des Sciences - Université Henri Poincaré - Nancy 1993-1995 : DEUG Sciences des Structures et de la Matière Faculté des Sciences - Université Henri Poincaré – Nancy
3
2
Activités d’enseignement
J’enseigne dans différentes écoles d’Ingénieurs, formation et classes préparatoires de l’Université de Lorraine ainsi que de l’Université de Strasbourg. Mes enseignements portent essentiellement sur la chimie physique, et plus particulièrement la cinétique chimique appliquée en génie des procédés.
2.1 Enseignement à l’École Nationale Supérieure des Industries Chimique
L’ENSIC est une école qui forme des ingénieurs ayant des compétences à la fois en chimie et chimie-physique mais également en génie chimique et génie des procédés. Elle délivre deux diplômes d’Ingénieur, le diplôme d’Ingénieur des Industries Chimiques (I²C) et le diplôme d’Ingénieur des Techniques de l'Industrie (FITI). Elle permet également à des étudiants inscrits en filière industrie des facultés de pharmacie d’obtenir le diplôme d’Ingénieur des Industries Chimiques (Pharma+). J’y enseigne en cinétique chimique dans la filière I²C du module « systèmes réactifs et procédés » lors de séances d’apprentissage par problème. J’encadre également des TP de cinétique chimique homogène (en phase liquide et en phase gazeuse). J’enseigne également dans le module de projet « système réactif et informatique ». Ce projet vise à rendre les étudiants aptes à dimensionner un réacteur industriel complexe en utilisant conjointement des compétences de cinétique, de génie de la réaction chimique, de méthodes numériques et informatique. Enfin, je suis en général chaque année, un à trois étudiants (I²C ou FITI) au cours de leur stage ingénieur.
2.2 Enseignement au Cycle Préparatoire Polytechnique de Nancy
Le CPP de Nancy est un des cycles préparatoires de la Prépa des INP. C’est une formation de deux ans sur contrôle continu à l’issue de laquelle les étudiants peuvent intégrer une des écoles d’ingénieurs du réseau INP, qui regroupe essentiellement les grandes écoles d'ingénieurs de Bordeaux, Grenoble, Nancy et Toulouse (33 écoles). La Prépa des INP est présente dans 6 villes (Bordeaux, Grenoble, Nancy, Saint-Denis de la Réunion, Toulouse, et Valence). Le CPP permet donc d’intégrer des écoles aussi bien orientées « physique » que « chimie » ou « biologie ». Je suis responsable du module de cinétique pour lequel je réalise les cours, TD et TP.
2.3 Enseignement au Cycle Préparatoire Intégré de Strasbourg
Le CPI de Strasbourg est un des cycles préparatoires intégrés de la Fédération Gay-Lussac, qui regroupe 19 écoles de chimie et génie chimique. Le CPI est présent en France dans 5 villes (Clermont-Ferrand, Lille, Pau, Strasbourg et Rennes) et permet donc un accès aux écoles de chimie ou de génie chimique. Je dispense les cours de cinétique chimique.
2.4 Enseignement à l’École Nationale Supérieure en Génie des Systèmes et de l’Innovation
L’École Nationale Supérieure en Génie des Systèmes et de l’Innovation forme des ingénieurs destinés à piloter des projets d’innovation et de développement en entreprise. J’y encadre les TP de chimie-physique de première et deuxième année du cycle préparatoire intégré.
4
2.5 Enseignement en Formation Continue spécialisation Économie Circulaire
Cette formation de l’Université de Lorraine, était financée par la Région Lorraine et ouverte aux demandeurs d’emploi ayant au moins un niveau bac+2. Son objectif était de donner la capacité aux étudiants d’intervenir dans le cadre du recyclage industriel, en particulier en liaison avec ECOREVIA. J’y enseignais les procédés de recyclage en cours et TD. Cette formation n’est plus ouverte en 2020.
2.6 Récapitulatif horaire des enseignements sur les cinq dernières années
Cours TD TP Projet 2018-2019 38h 44h 158h 18h 2017-2018 38h 44h 194h 18h 2016-2017 38h 44h 194h 11h 2015-2016 38h 44h 194h 11h 2014-2015 44h 42h 194h 11h
3
Activités de recherche
De 2004 à 2014, j’ai contribué au LRGP à une activité de recherche centrée sur la cinétique des réactions thermiques en phase gazeuse mises en jeu dans des procédés industriels ou de potentiels procédés industriels. L’objectif des travaux était de développer les modèles cinétiques radicalaires détaillés des réactions mises en jeu dans ces procédés et de les valider à partir de pilotes expérimentaux de laboratoire ; l’objectif ultime de ces modèles étant de mieux comprendre les mécanismes cinétiques pour pouvoir optimiser les procédés. Dans la deuxième partie de ce manuscrit, trois problématiques seront abordées d’un point de vue cinétique : le procédé d’élaboration des matériaux composites C/C par dépôt chimique en phase vapeur, l’abattement des dioxines dans les procédés d’incinération des ordures ménagères et enfin le potentiel procédé de reformage des gaz issus de la gazéification de la biomasse.
Ensuite, à partir de 2014, j’ai rejoint le LERMAB et contribué aux activités de recherche sur les procédés de valorisation chimique de la biomasse et plus particulièrement sur les procédés de prétraitement de la biomasse. Dans la troisième partie de manuscrit, des travaux portant sur une étude cinétique du prétraitement de Saule contaminé par des éléments traces métalliques et de sa valorisation en vue d’une production de biocarburant seront présentés.
La suite de ce manuscrit sera composée de deux parties distinctes ayant pour fil conducteur commun « l’importance de la cinétique chimique dans les procédés ».
Le lecteur est donc invité à bien prendre en compte le fait que la seconde partie portera sur des problématiques de cinétique détaillée en phase gazeuse tandis que la troisième partie traitera de cinétique globale dans un procédé par voie humide.
: Activités de recherche développées
au LRGP
5
Chapitre 1 : Objectifs et méthodologie
Les réactions thermiques en phase gazeuse (RTPG) ont une grande importance dans les procédés, que ce soit pour la production d’énergie mécanique et thermique par combustion ou même pour l’élaboration de molécules d’intérêt ou de vecteurs énergétiques par craquage, vapocraquage, oxydation ou pyrolyse. De ce fait, les RTPG ont également un fort impact sur l’environnement que ce soit lors de l’émission de gaz à effet de serre qui contribuent au réchauffement planétaire, de la production de composés halogénés qui engendrent la destruction de l’ozone stratosphérique ou même lors de la production de composés organiques volatils et de particules fines qui engendrent une pollution troposphérique croissante.
Une meilleure connaissance des réactions thermiques en phase gazeuse et de leur cinétique, et donc une meilleure maîtrise de cette dernière dans les procédés, ouvre des perspectives d’une part pour améliorer les procédés de production d’énergie en les rendant plus fiables, performants et propres mais également pour limiter et endiguer les causes de pollution atmosphérique.
À l’échelle du laboratoire, l’étude cinétique des réactions thermiques en phase gazeuse ayant lieu au cours d’un procédé se déroule en deux phases parallèles. La première partie du travail du cinéticien consiste à imaginer, dimensionner, construire puis valider un pilote expérimental représentatif du procédé à étudier et permettant de quantifier l’évolution des réactifs et produits de réaction au cours d’études paramétriques. Parallèlement une deuxième partie du travail consiste à élaborer un modèle de la réaction par le biais d’un mécanisme cinétique radicalaire détaillé décrivant à l’échelle élémentaire les processus impliqués dans les RTPG. Des simulations numériques à l’aide de logiciels de cinétique sont ensuite réalisées en reprenant les conditions expérimentales étudiées. La modélisation de ces résultats théoriques est ensuite confrontée aux résultats expérimentaux. L’objectif est d’atteindre la meilleure adéquation afin de valider le modèle qui a été développé. Une fois validé, le modèle permet par exemple de simuler le comportement d’une charge réactive dans différentes conditions de réaction et ainsi d’optimiser un point de fonctionnement. La figure 1 représente schématiquement la méthodologie utilisée.
Comme nous le verrons par la suite, la construction du pilote expérimental impose trois contraintes techniques majeures. Il faut au préalable définir et dimensionner le réacteur, de préférence idéal, utilisé pour l’étude, réfléchir à la solution technique d’introduction des réactifs dans le réacteur et enfin aux méthodes analytiques qui permettront la quantification des espèces intervenant dans la réaction. La réflexion sur la construction d’un pilote expérimental sera présentée au chapitre 3 de ce manuscrit.
La partie modélisation repose en général sur l’exploitation de bases de réactions radicalaires déjà existantes qui sont amenées à être complétées en fonction des conditions de l’étude et surtout de la nature des réactifs étudiés. Parfois, ce travail ne peut pas s’appuyer sur des travaux existants et nécessite la construction intégrale
6
Figure 1 : Méthodologie utilisée pour l’étude cinétique des réactions thermiques en phase gazeuse.
Etude expérimentale Modélisation homogène / hétérogène
Construction d’un pilote expérimental avec trois contraintes : ✓ Introduction des réactifs ✓ Choix d’un réacteur
✓ Choix des méthodes analytiques
Expérimentations
Etudes paramétriques de la réaction : ✓ Température
✓ Pression
✓ Composition d’entrée ✓ Temps de passage
Quantification :
Représentation des fractions molaires mesurées en fonction des paramètres
expérimentaux
Ecriture d’un mécanisme : ✓ Réactions ✓ Données cinétiques ✓ Données thermodynamiques Utilisation d’une base existante de réactions Simulations
Etudes paramétriques de la réaction : ✓ Température
✓ Pression
✓ Composition d’entrée ✓ Temps de passage
Modélisation :
Représentation des fractions molaires simulées en fonction des paramètres de simulation
Adéquation entre modélisation et expérimentation ?
OUI NON Validation du mécanisme Exploitation du mécanisme : ✓ Schéma de flux, ✓ Extrapolation, ✓ Optimisation… Mo d if ic a tio n / o p tim is a tio n d u m éc a n is m e
7 d’un modèle. La méthodologie de construction du modèle cinétique sera présentée au chapitre 4 de ce manuscrit.
Lorsque les résultats obtenus par simulation ne présentent pas une bonne adéquation avec l’expérience, commence alors un long travail souvent chronophage (en partie en raison du temps de calcul lors des simulations) de correction, modification et optimisation du modèle, jusqu’à l’obtention d’un modèle final jugé acceptable.
Ainsi, dans ce travail cette méthodologie sera illustrée par l’étude de la cinétique de RTPG au travers de trois exemples de problématiques reposant sur des procédés existants ou potentiels, à savoir :
La production de matériaux composites C/C en vue d’une utilisation dans le domaine aérospatial. L’abattement des dioxines dans les procédés d’incinération des ordures ménagères.
La valorisation par reformage des gaz issus de la gazéification de biomasse.
Ces illustrations seront présentées au chapitre 5 de ce travail qui clôturera la deuxième partie de ce manuscrit. Enfin, avant de détailler ces différentes parties, le chapitre suivant propose de poser quelques rappels élémentaires de cinétique pour éclairer la lecture de la suite du manuscrit.
9
Chapitre 2 : Quelques rappels de cinétique
Souvent, les exemples d’études cinétiques rencontrés dans la littérature se limitent d’une part à décrire les vitesses expérimentales de réactions sous forme de dérivées par rapport au temps et d’autre part à décrire les vitesses de réaction avec des cinétiques d’ordre « un » sans prendre de précaution quant à la véracité de ces descriptions. La notion de « réacteur » y est trop souvent négligée.
Avant d’aborder, l’aspect cinétique radicalaire et détaillé des réactions thermiques en phase gazeuse, nous proposons donc de poser quelques rappels élémentaires de cinétique chimique dans le cas des réacteurs idéaux de laboratoire et isothermes dans le temps et dans le volume [Scacchi 2011].
En cinétique chimique, il existe deux manières d’exprimer la vitesse d’une réaction : la mesure de la vitesse et la loi de vitesse. La mesure de la vitesse d’une réaction est une expression expérimentale et donc, indissociable du réacteur utilisé pour étudier la réaction. Tandis que la loi de vitesse, est une expression mathématique, validée à partir de la mesure de vitesse et qui relie la vitesse aux paramètres qui peuvent l’influer.
1
Les réacteurs idéaux de laboratoire
Le réacteur se définit comme l’espace où peut se dérouler une transformation chimique. Parmi les différents types de réacteurs, on distingue :
Le réacteur fermé où les réactifs sont introduits en une seule fois dans le réacteur qui est vidangé en fin de
réaction. Dans ce réacteur, il n’y a pas d’échange de matière avec l’extérieur.
Le réacteur ouvert où il existe un courant total de matière qui traverse le réacteur : les réactifs sont
introduits en continu et les produits (et éventuels réactifs non consommés) sont extraits en continu.
Le réacteur semi-fermé, où il y a un échange de matière avec l’extérieur ; mais au moins un constituant n’est
ni apporté, ni extrait au cours de la réaction.
Ces réacteurs peuvent évoluer dans le temps soit :
En régime transitoire, où au moins un des paramètres de fonctionnement (composition, débit…) est
fonction du temps. Ainsi, les réacteurs fermés et semi-fermés fonctionnent en régime transitoire. L’utilisation de tels réacteurs est donc menée de manière discontinue.
En régime permanent, où aucun paramètre de fonctionnement n’est dépendant du temps. C’est le cas des
réacteurs ouverts qui, permettent un fonctionnement continu. Ils passent toutefois par une phase de régime transitoire à leur mise en service ou lors de leur arrêt.
Le degré de mélange à l’intérieur du réacteur est également un critère important ; on distingue deux cas limites :
10
Le réacteur parfaitement agité où l’agitation du milieu est suffisante pour assurer l’uniformité de la
composition dans tout le volume du réacteur. Ce réacteur peut être ouvert, fermé ou semi-fermé/ouvert. Dans ce cas le degré de mélange en retour1 est maximal.
Le réacteur ouvert à écoulement piston, où les réactifs et produits se déplacent le long du réacteur selon
des tranches qui n’échangent pas de matière entre elles, imposant ainsi que la composition du mélange réactionnel évolue tout au long de l’axe du réacteur. Le degré de mélange en retour y est donc nul.
Même si cela semble évident, il faut également rappeler que le contrôle de la température du réacteur est également une donnée cruciale. Avoir une température uniforme dans le temps et dans le volume du réacteur (réacteur dit isotherme) est un avantage dans les études cinétiques, car cela permet de traiter la température comme une variable indépendante.
Les critères d’introduction d’entrée et de sortie des composants chimiques, de mélange, d’évolution temporelle et de température du réacteur permettent de poser les conditions limites de fonctionnement des réacteurs. Lorsqu’un réacteur peut être décrit à l’aide de ces conditions limites, on parle alors de réacteur idéal de laboratoire. Un réacteur idéal de laboratoire est bien entendu un modèle simple qui permet de mener des études cinétiques mais il diffère du réacteur réel car il ne prend pas en compte les phénomènes d’échange de chaleur et de transport de matière qui ont lieu au cours de la réaction.
2
La mesure de la vitesse
Comme son nom l’indique, la mesure de la vitesse correspond à la « quantification » expérimentale de la vitesse d’une réaction. Elle s’obtient en écrivant l’équation de conservation de la matière dans le réacteur où se déroule la réaction étudiée, sous forme d’un bilan instantané en termes de débits molaires sur un des constituants chimiques de la réaction (Fig. 2).
FR,e : débit molaire d’entrée du réactif R dans le réacteur
FP,e : débit molaire d’entrée du produit P (en général nul)
FR,s : débit molaire de sortie du réactif R dans le réacteur
FP,s : débit molaire de sortie du produit P dans le réacteur
nR : quantité instantanée du réactif R dans le réacteur
nP : quantité instantanée du produit P dans le réacteur
rcR : vitesse locale instantanée de consommation du réactif R
rFP : vitesse locale instantanée de formation de du produit P
V : volume du réacteur
dV : élément de volume du réacteur Figure 2 : Schéma d’un réacteur quelconque.
1 Mélange entre réactifs entrant et produits de la réaction.
Réacteur quelconque de volume V
Elément du réacteur de volume dV Vitesse dans dV : rcR et rFP FR,e FP,e souevnt FR,s FP,s nR nP
11 Ainsi pour un réactif quelconque « R » entrant dans un réacteur quelconque, l’équation de conservation de la matière s’exprime sous la forme :
FR,e= FR,s+ dnR dt + vrdV c R
Pour un produit quelconque « P », il s’exprimera par :
FP,e+vrPFdV= FP,s+ dnP
dt
Ces bilans de conservation de la matière vont pouvoir se simplifier en fonction des conditions limites de fonctionnement du réacteur. On obtiendra ainsi des expressions différentes de la mesure de la vitesse s’il s’agit d’une réaction étudiée en réacteur fermé, réacteur ouvert à écoulement piston, réacteur ouvert parfaitement agité, si le régime de l’écoulement est transitoire ou permanent…. La mesure de la vitesse est donc expérimentale et dépend du réacteur utilisé.
Il n’est donc pas utile de connaître la ou les stœchiométries de la réaction étudiée pour connaître la mesure de la vitesse de formation ou de consommation d’un constituant chimique. Cependant, si l’on souhaite connaître la vitesse globale d’une réaction, il est nécessaire de connaître sa stœchiométrie. Par exemple, dans le cas d’une stœchiométrie unique la vitesse globale de la réaction « r » s’obtient selon la relation suivante :
Stœchiométrie : νR1R1 + νR2R2 + …. + νRnRn = νP1P1 + νP2P2 + … + νPnPn r =rR1 νR1 =rR2 νR2 =…=rRn νRn = rP1 νP1 =rP2 νP2 =…=rPn νPn
Où νRj est le coefficient stœchiométrique du réactif Rj
rRj la vitesse de consommation du réactif Rj
νPj le coefficient stœchiométrique du produit Pj
rPj la vitesse de formation du produit Pj
3
La loi de vitesse
La loi de vitesse s’obtient après avoir mesuré la vitesse. C’est une expression mathématique qui va relier la vitesse aux paramètres qui l’influent ; à savoir dans le cas des réactions thermiques en phase gazeuse : la température et les concentrations des réactifs et éventuellement des produits. La loi de vitesse peut être ainsi décrite comme le produit de deux fonctions indépendantes, l’une dépendant uniquement de la température appelée constante de vitesse et l’autre dépendant des concentrations :
12 La loi de vitesse est indépendante du type de réacteur utilisé. On distingue la loi de vitesse initiale de la loi de vitesse courante :
La loi de vitesse initiale décrit la vitesse aux premiers instants de la réaction dans un milieu quasiment exempt
de produits. Par exemple pour une réaction générique :
νa A + νb B νb C + νd D
Où A et B sont des réactifs quelconques - C et D sont des produits quelconques
νa, νb, νc νd les coefficients stœchiométriques
La loi de vitesse initiale, si elle existe, s’écrira :
r0 = k0 [A]0 nA0
[B]0nB0
Où r0 est la vitesse initiale
[A]0 et [B]0 les concentrations initiales de A et B
k0 la constante de vitesse initiale de la réaction
0 A
n et nB0les ordres initiaux partiels par rapport à A et B
La détermination des ordres initiaux d’une réaction constitue un important point de départ pour aborder l’écriture d’un mécanisme réactionnel détaillé.
La loi de vitesse courante décrit la vitesse à un avancement quelconque en présence de produits de réaction.
Ces produits peuvent avoir une influence cinétique sur la réaction, ce qui complexifie la recherche des lois de vitesse. Par exemple pour la même réaction générique que précédemment et si les produits de réaction n’ont pas d’influence cinétique, la loi de vitesse courante si elle existe s’écrira :
r = k [A]nA[B]nB
Où r est la vitesse courante de la réaction. k la constante de vitesse de la réaction
[A] et [B] les concentrations de A et B à avancement non nul
nA et nB les ordres courants partiels par rapport à A et B ; ils ont les mêmes valeurs respectivement que les ordres initiaux partiels
Lorsque les produits ont une influence cinétique, on cherche plutôt à écrire la vitesse sous la forme :
r = rfictive x ϕ
Où r est la vitesse de la réaction
rfictive la vitesse fictive de la réaction ; c’est à dire la vitesse que la réaction aurait à même avancement si les produits
n’intervenaient pas
13 À partir de valeurs expérimentales de la mesure de la vitesse et des concentrations des réactifs, il est donc possible de déterminer, s’ils existent, les valeurs des ordres partiels expérimentaux et la constante de vitesse expérimentale de la réaction étudiée ; pour cela, on linéarise l’expression de la loi de vitesse (méthode dite différentielle). Les ordres sont donc des grandeurs expérimentales qui caractérisent l’influence de la concentration des réactifs sur la vitesse d’une réaction.
4
La loi d’Arrhenius
La constante de vitesse d’une réaction varie avec la température absolue, sur une plage restreinte de de température, en suivant la relation d’Arrhenius :
k = A exp - E RT
Où E est l’énergie d’activation de la réaction en J.mol-1
A le facteur préexponentiel (il a la même unité que k) R la constante des gaz parfait (8,314 J.mol-1.K-1)
T la température (K)
Dans certains cas, on utilise également la loi d’Arrhenius-Kooij, qui prend en compte la dépendance du facteur préexponentiel à la température (Tn) :
k = A Tn exp - E RT
Où E est l’énergie d’activation de la réaction en J.mol-1
A le facteur préexponentiel (il a la même unité que k) R la constante des gaz parfait (8,314 J.mol-1.K-1)
T la température (K) n l’exposant de correction
Remarque : dans ce travail l’énergie d’activation sera également parfois exprimée en calories par mole.
5
Les processus élémentaires
Une réaction peut être considérée comme un processus élémentaire, si elle se produit en un acte irréductible à l’échelle moléculaire. C’est-à-dire lorsqu’on ne peut pas détecter d’intermédiaires réactionnels entre les réactifs et les produits à l’aide d’une technique analytique quelconque.
La molécularité d’un processus élémentaire correspond aux nombres de particules de réactifs, qui peuvent être des molécules, des ions ou des radicaux, participant à cet acte. Dans l’écriture de la loi de vitesse d’un processus élémentaire, l’ordre et la molécularité se confondent.
14 On distingue trois types de processus élémentaires :
Les processus bimoléculaires, les plus fréquents, qui résultent de la collision entre deux particules A et B
identiques ou non :
A + B → produits et dont la loi de vitesse s’écrit r = k [A] [B]
Les processus unimoléculaires, où une particule A subit isolément :
- soit une isomérisation
A → B et dont la loi de vitesse s’écrit r = k [A] - soit une décomposition
A → B1 + B2 et dont la loi de vitesse s’écrit r = k [A]
Les processus trimoléculaires, les plus rares, qui nécessitent le choc simultané de trois particules :
A + B + M → produits et dont la loi de vitesse s’écrit r = k [A] [B] [M]
Dans ce cas, en général « M » désigne un partenaire de collision, c’est-à-dire une espèce quelconque du milieu réactionnel qui ne participe pas à la réaction mais qui a le rôle d’un tampon énergétique en apportant ou en absorbant de l’énergie lors du choc trimoléculaire.
6
Les réactions complexes
La plupart des réactions chimiques mettent en fait en œuvre plusieurs processus élémentaires concomitants. On parle alors de réactions complexes. La compréhension et la description de ces réactions complexes sous forme de mécanismes réactionnels composés de processus élémentaires dont les lois de vitesses sont connues est donc l’objectif final du cinéticien, comme on pourra le voir dans la suite de ce manuscrit.
15
Chapitre 3 : Les montages expérimentaux développés
La compréhension et la description sous forme mécanistique des réactions thermiques en phase gazeuse impliquées dans un procédé requièrent l’obtention de données expérimentales fiables sur ces réactions. Partant des informations relatives au procédé industriel étudié, il faut être capable de construire un pilote à l’échelle du laboratoire permettant d’étudier les réactions mises en jeu. Pour cela, il faut :
Mettre au point des solutions techniques pour entraîner les réactifs dans le réacteur. Choisir et dimensionner un réacteur et son mode de chauffage.
Mettre au point des solutions techniques pour caractériser et quantifier les produits issus des RTPG. De plus, dans la mesure où les études se font essentiellement en réacteur ouvert, il faut pouvoir faire fonctionner le pilote en régime permanent.
Schématiquement (Fig. 3) on peut donc distinguer trois zones essentielles dans chacun des pilotes expérimentaux développés pour les études cinétiques des réactions thermiques en phase gazeuse :
La zone réactionnelle constituée du réacteur et de son moyen de chauffage
La zone en amont du réacteur qui permet de générer et réguler les débits de gaz entrant dans le réacteur. La zone en aval du réacteur qui se compose des outils analytiques permettant de déterminer la composition de gaz en sortie de réacteur.
Figure 3 : Schéma de principe des montages réactionnels développés.
Les solutions techniques sont choisies en fonction de l’état des réactifs en condition ambiante de température et de pression (liquide, solide, gazeux), de la température de l’étude, de l’avancement des réactions…
1
La zone réactionnelle : les réacteurs utilisés et leur mode de chauffage
La zone réactionnelle contient deux éléments essentiels du montage ; à savoir le réacteur utilisé et son mode de chauffage.
1.1 Les réacteurs
Le choix du réacteur est primordial dans la construction d’un pilote expérimental destiné à l’étude des réactions thermiques en phase gazeuse. Comme nous l’avons décrit précédemment, trois réacteurs idéaux de
ZONE PRE-REACTIONNELLE : Génération et régulation
des débits des réactifs gazeux ZONE REACTIONNELLE : Réacteur chauffé Mesure et contrôle de la pression ZONE POST-REACTIONNELLE : Analyses des produits
![Figure 5 : Schéma de principe du réacteur auto-agité par jets gazeux [Tritz 2014a].](https://thumb-eu.123doks.com/thumbv2/123doknet/15048349.693829/42.892.229.699.507.790/figure-schéma-principe-réacteur-auto-agité-gazeux-tritz.webp)
![Figure 19 : Classement des fibres de carbone en fonction de leurs propriétés mécaniques [Camus 2016]](https://thumb-eu.123doks.com/thumbv2/123doknet/15048349.693829/64.892.197.691.123.446/figure-classement-fibres-carbone-fonction-propriétés-mécaniques-camus.webp)