Alkyladenine DNA Glycosylase (Aag)-Dependent
Cell-Specific Responses to Alkylating Agents
by
Carrie Marie Margulies
B.A. Chemistry Dartmouth College, 2008
Submitted to the Department of Biological Engineering in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2016
@ 2016 Massachusetts Institute of Technology. All rights reserved.
Signature of
Certified by:.
Sianature redacted
A u th o r:... ... ... ... . ... ...Department of Bioko'cal Engineering
Jnuary 8th 2016
U
~zignatu
re redactedi--j--_
.
...
...
...
...
...
...
Leona D. Samson Professor of Biological Engineering and Biology Thesis Supervisor
Accepted by:
Signature redacted
Chair, Biologic
... Forest White Professor of Biological Engineering al Engineering Graduate Committee
MASSACHUSETTS INSTITUTE OF
TECHNOLOGY-MAY 2
6
2015
LIBRARIES
Alkyladenine DNA Glycosylase (Aag)-Dependent Cell-Specific Responses to Alkylating Agents
By
Carrie Marie Margulies
Submitted to the Department of Biological Engineering on January 81h, 2016 in
Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biological Engineering
Abstract
Methylating agents are ubiquitous in our internal and external environments and can cause damage to all cellular components, including our DNA. If left unrepaired, methylated DNA can cause mutations, cell death, and disease, such as cancer and neurodegeneration. The majority of DNA lesions caused by methylating agents are repaired by the base excision repair (BER) pathway, which is initiated by the lesion-specific alkyladenine glycosylase (Aag). Loss of Aag in embryonic stem (ES) cells renders them sensitive to the methylating agent MMS (methyl methanesulfonate) compared to wild-type (WT). Surprisingly, this phenotype is reversed in hematopoietic myeloid progenitors and cerebellar granule neurons (CGNs) where Aag' cells are resistant to MMS induced killing compared to WT. In this study, we investigated how Aag can cause cell-specific
responses to alkylating agents.
We generated new WT, Aag', and Aag overexpressing (mAagTg) 129 and C57B1/6 ES cells and showed that inbred genetic background did not affect sensitivity to MMS, indicating this to be a cell-intrinsic response. Moreover, we found that cells overexpressing Aag were even more sensitive to MMS than
Aag' cells, suggesting that ES cells endure methylation treatment best when
they express Aag within an optimal range.
To study Aag-dependent neural sensitivity to methylating agents, we optimized protocols for the isolation and culture of primary cerebellar granule neurons and determination of cell death after drug treatment by high-throughput imaging. CGNs isolated from WT, Aag, and mAagTg mice exhibited cell sensitivity to MMS treatment that was dependent on Aag and Parp activity, thus recapitulating
in vivo results and proving that CGN death is cell-intrinsic. Cell death was
independent of caspases, mitochondrial depolarization, and AIF translocation. We did observe the formation of enlarged mitochondria and are investigating whether mitochondrial dynamics are causative of cell death in an Aag-dependent manner.
Finally, we used in vitro hematopoietic and neuronal differentiation to monitor cell responses to MMS as a function of cellular development. Three different methods all successfully generated mature neurons based on morphology,
immunochemical staining, and Aag expression. Though we successfully differentiated ES cells into cell types of interest, we are continuing to optimize methods for the assessment of alkylation sensitivity in the resulting heterogeneous populations.
Thesis Supervisor: Leona D. Samson
Acknowledgements
I would like to begin by thanking my advisor, Leona Samson, for her endless support and guidance throughout my graduate career. Through all the ups and down of research, she was a constant figure I could rely and depend on. I would also like to thank my committee members Bevin Engelward and Doug Lauffenburger for their assistance and contributions to my project.
I would not have been able to complete this without the assistance of all the members of the Samson lab, past and present. Whether during group meeting, over lunch/coffee, or outside of the lab, they have been wonderfully helpful on all aspects of my scientific and personal life. They made the lab an enjoyable place to work everyday and I look forward to staying friends with them for a long time.
Next, I want to acknowledge all my close friends who have been there for me throughout the past 6 years. To the BE Class of 2009, there is no way I would have survived at MIT without your assistance in the dungeon during the first year! In the years since then, each one of you is the reason MIT is exciting, invigorating, and full of laughter. To all my close friends in the Boston area, particularly all my Dartmouth hockey friends, thank you for reminding me to take breaks and relax to make cookies, dance, reminisce, and be 'me', no matter how crazy that is. To Sue, thank you for your 'wellness checks', Dan and I would not be the same without them.
Finally, I would like to thank my family. To my wonderful husband Dan, thank you for pushing me to become all that you know I can be and supporting me every step along the way. To my doggies Ted and Ruby, thank you for always being happy to see me, making me smile, and reminding me of the importance to relax and enjoy life. To my parents, Matt and Julie, thank you for your infinite support, financially and emotionally, and dedication toward all the endeavors I have undertaken, from hockey to academics. I would not have achieved everything I
have without your love and confidence in my abilities. To my brothers, Bud and Jake, thank you for your competitive spirits, without which I would have never learned how to push myself to the limits. To my aunt Trudy, though I have not always appreciated you as much as you deserve, you have been nothing but generous, dependable, and encouraging. To the rest of my enormous family, all the grandparents, aunts, uncles, cousins, etc., you have given me the love, support, and motivation I have needed to accomplish my goals and I hope I can offer you all the same. Finally, I'd like to thank my new family. Thank you to David, Else, Maja, and Nik for taking me in and providing me a family in Boston. In particular, thank you David for all of the scientific insight and support you have provided me in the past few years.
Table of Contents Acknowledgements ... 4 Table of Contents ... 6 L ist o f F ig u re s ... . 9 L ist o f T a b le s ... . . 12 Chapter I: Introduction ... 16 Intro d u ctio n ... . . 16
Alkylating Agents in our Endogenous and Exogenous Environments Cause Cytotoxic DNA Damage ... 16
Methylating Agent Mechanism of Action ... 17
DNA Repair Pathways for Alkylation Damage ... 18
Alkyladenine Glycosylase (Aag) Initiates Repair of Methylated DNA Bases2l Cellular Consequences of Imbalanced BER ... 22
Alkylation Sensitivity of Cells with Altered Aag Expression...23
Poly (ADP-Ribose) Polymerase 1 Can Mediate Cell Death Caused by BER Im b a la n c e s ... 2 5 Model System to Study Aag-Dependent Alkylating Sensitivity...27
Overview of the Presented Study...27
F ig u re s ... . . 2 9 T a b le s ... . . 3 6 R e fe re n ce s ... . 3 7 Chapter II: Generation of WT, Aag' and mAagTg C57B1/6 and 129 Embryonic Stem Cells and Sensitivity to Alkylating Agents ... 51
In tro d u ctio n ... . . 5 1 Materials and Methods...54
R e s u lts ... . . 6 2 Generation of C57B1/6 and 129 Mouse Embryonic Stem Cells and Validation of Pluripotency... 62
BER Gene Expression and Aag Activity in WT, Aag-'- and mAagTg ES Cells ... 6 3
Sensitivity of ES Cell Lines to Alkylating Agents... 64
Discussion... 65
Figures ... 69
References... 78
Chapter III: In vitro Hematopoietic and Neuronal Differentiation of Mouse Em bryonic Stem Cells ... 86
Introduction ... 86
M aterials and Methods... 89
Results ... 95
In Vitro Hematopoietic Differentiation of 129 ES Cells... 95
In vitro Differentiation of Cerebellar Granule Neurons (CGNs) ... 96
In vitro Adherent Differentiation of Cortical Neurons... 98
In vitro Neural Differentiation Following 'Bibel' et al. (2007)...100
D is c u s s io n ... 1 0 1 F ig u re s ... 1 0 5 T a b le s ... 1 1 5 References...116
Chapter IV: Primary Mouse Cerebellar Granule Neuron Sensitivity to MMS is Dependent on Aag and Parp1 ... 123
In tro d u c tio n ... 1 2 3 M aterials and Methods...127
R e s u lts ... 1 3 3 Differential Expression of Aag in Primary Cerebellar Granule Neurons causes changes M MS Sensitivity ex vivo...133
Aag-Dependent CGN Sensitivity to MMS is mediated through Poly (ADP-Ribose) Polymerase Activity...134
Downstream Modulators of Neuron Sensitivity to M MS ... 136
Base Excision Repair Protein Expression in Primary CGNs...138 D is c u s s io n ... 1 3 9
F ig u re s ... 1 4 4 A p p e n d ix ... 1 6 4
CGN Sensitivity to Glutamate Excitotoxicity and Oxidative Stress is
Independent of Aag Activity...164 Genetic Deletion of Alkbh7 Reduces Female CGN Sensitivity to MMS...165 R e fe re n c e s ... 16 8 Chapter V: Discussion ... 180
D is c u s s io n ... 1 8 0 Imbalances in DNA Repair alter Embryonic Stem Cell Sensitivity to
A lky la tin g A g e nts ... 18 0 Aag and Parp1 Mediate Cerebellar Granule Neuron Sensitivity to MMS... 184 Aag-Dependent Cell-Specific Responses ... 187 R e fe re n c e s ... 18 9 Appendix: Development of a Method to Assay DNA Repair Capacity for
Mammalian Cells using High-Throughput Sequencing ... 196
In tro d u c tio n ... 1 9 6 Manuscript: Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. ... 197
List of Figures
Figure 1.1: Structures of Aag DNA Substrates...29
Figure 1.2: Base Excision Repair Pathway. ... 30
Figure 1.3: Alternate DNA Repair Pathways for Methylation Damage. ... 32
Figure 1.4: Parp1 utilizes NAD' to generate PAR polymers...33
Figure 1.5: Alternative mechanisms of Cell Death Caused during Base Excision R e p a ir (B E R ). ... . . 34
Figure 2.1: Generation of new Embryonic Stem (ES) Cells. ... 69
Figure 2.2: Karyotyping of C57B1/6 ES Cells... 70
Figure 2.3: Fluorescent Immunocytochemical Staining for Pluripotency Markers in C 57B1/6 and 129 ES C ells. ... 71
Figure 2.4: Aag Expression and Activity in C57BI/6 ES Cells. ... 72
Figure 2.5: Aag Expression and Activity in 129 ES Cells. ... 73
Figure 2.6: Expression of Base Excision Repair genes in ES cells...74
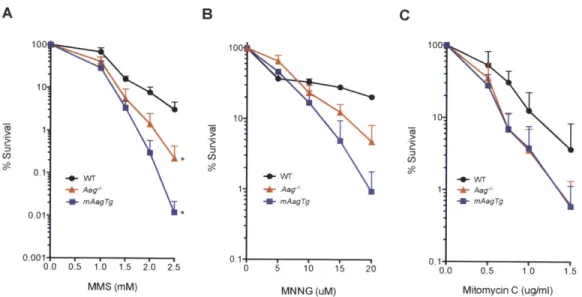
Figure 2.7: Sensitivity of C57B1/6 ES Cells to Alkylating Agents. ... 75
Figure 2.8: Sensitivity of 129 ES Cells to Alkylating Agents...76
Figure 2.9: MMS Sensitivity of 129 Hematopoietic Progenitors, Cerebellar Granule Neurons, and Retinal Photoreceptors is Aag-dependent. ... 77
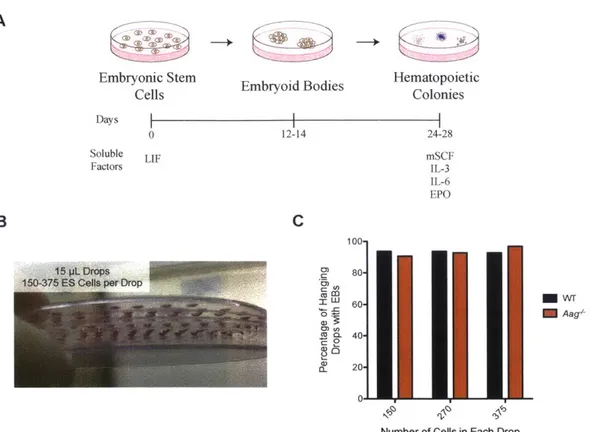
Figure 3.1: Mouse embryonic stem cells will be differentiated in vitro into hematopoietic progenitors and cerebellar granule neurons. ... 105
Figure 3.2: In vitro Hematopoietic Differentiation of 129 ES cells. ... 106
Figure 3.3: In vitro hematopoietic differentiation of VVT and Aag-'- 129 ES Cells a nd M M S se nsitiv ity ... 10 7 Figure 3.4: In vitro Differentiation of Cerebellar Granule Neurons. ... 108
Figure 3.6: Representative images of cells as they differentiate from ES cells into n e u ro n s ... 1 1 0 Figure 3.8: In vitro Neuronal Differentiation...112 Figure 3.9: Aag Expression Changes During In vitro Neuronal Differentiation.. 113
Figure 4.1: MMS Induced Cerebellar Degeneration in vivo is dependent on Aag a nd P a rp 1 exp re ssio n ... 14 4
Figure 4.2: Isolation of Primary Cerebellar Granule Neurons (CGNs). ... 145 Figure 4.3: Development of a High-Throughput Method to Determine Primary Neuron Sensitivity to Drug Treatment. ... 146 Figure 4.4: Sensitivity to MMS treatment ex vivo is dependent on Aag. ... 147 Figure 4.5: Aag is differentially expressed in primary CGNs from Aag', WT, and
m A a g Tg m ice . ... 14 8 Figure 4.6: Aag Glycosylase Activity is Significantly Different between Aag<, WT,
and m A ag Tg prim ary C G N s. ... 149 Figure 4.7: Primary CGN Sensitivity to MMS is dependent on Parp1 activity. .. 150 Figure 4.8: PAR formation post-MMS treatment (1 mM) is dependent on the expressio n leve l of A ag . ... 15 1 Figure 4.9: Parp inhibitor Veliparib inhibits PAR formation after MMS treatment. ... 1 5 2 Figure 4.10: Neither supplementation of NAD' nor pyruvate rescues WT or
mAagTg primary CGN sensitivity to MMS...153
Figure 4.11: Downstream molecular mechanisms of CGN Aag-dependent MMS s e n s itiv ity . ... 1 5 4 Figure 4.12: There is no loss of mitochondrial permeability in Aagt, WT, or
mAagTg neurons 1-7 hours after MMS treatment. ... 155 Figure 4.13: No evidence of AIF nuclear translocation after MMS treatment.... 156 Figure 4.14: Expression of BER genes in primary CGNs...157
Figure 4.15: Aag expression is not induced after MMS treatment in WT neurons. ... 1 5 8 Figure 4.16: Glycolysis and Tricarboxylic Acid (TCA) Cycle Schematic...159 Figure 4.17: Aag activity does not contribute to ex vivo sensitivity to glutamate e x c ito to x ic ity . ... 16 0 Figure 4.18: Aag does not contribute to ex vivo CGN sensitivity to treatment with hyd rogen pe roxide (H 20 2). ... 16 1
Figure 4.19: Female AlkbhT'CGNs are relatively resistant to MMS treatment ex
vivo compared to male AlkbhT', female WT and male WT neurons. ... 162 Figure 4.20: WT and Alkbh T' CGNs are rescued from MMS sensitivity by Parp inhibition but not by Caspase inhibition. ... 163
List of Tables
Table 1.1: Methylation patterns in single- and double-stranded DNA after
treatment with MMS represented as percentages of total methylation...36 Table 3.1: Attem pts at In vitro Cortical Neuronal...115
Table of Contents
C hapter 1: Introd uction ... . . 16 In tro d u ctio n ... . . 16
Alkylating Agents in our Endogenous and Exogenous Environments Cause Cytotoxic DNA Damage ... 16 Methylating Agent Mechanism of Action ... 17 DNA Repair Pathways for Alkylation Damage ... 18 Alkyladenine Glycosylase (Aag) Initiates Repair of Methylated DNA Bases2l Cellular Consequences of Imbalanced BER ... 22 Alkylation Sensitivity of Cells with Altered Aag Expression... 23
Poly (ADP-Ribose) Polymerase 1 Can Mediate Cell Death Caused by BER Im ba la nce s ... . . 2 5 Model System to Study Aag-Dependent Alkylating Sensitivity...27 Overview of the Presented Study... 27 F ig u re s ... . . 2 9 T a b le s ... . . 3 6 R e fe re n ce s ... . . 3 7
Chapter I: Introduction
Introduction
Alkylating Agents in our Endogenous and Exogenous Environments Cause Cytotoxic DNA Damage
The cells in our body are under constant attack from both endogenous and exogenous sources. At the genomic level, cells can accumulate around 100,000 DNA lesions per day (Ciccia, 2010). Fortunately, our cells have multiple repair mechanisms to restore DNA to its native sequence. Without repair, most of these events have the capacity to result in a heritable mutation that can contribute to cancer, degenerative conditions, and other maladies. Alkylating agents represent a broad category of DNA damaging agents that contribute to both the endogenous and exogenous sources of cellular damage.
Methylating agents are ubiquitous in our external and internal environments. Endogenous sources of alkylation damage include lipid peroxidation by-products and cellular cofactors. S-adenosyl-methionine is one potential source of aberrant DNA methylation, inducing 3-methyladenine (3MeA) and 7-methylguanine (7MeG) at rates of 600 and 4,000 lesions per cell per day, respectively (Fig. 1.1) (Fu et al., 2012b; Lindahl, 2000). The nitrosation of amines on amino acids, peptides, or polyamines is another potential source of methylating agents (Sedgwick, 1997). Moreover, the observation that alkylation DNA repair enzymes are conserved throughout evolution indicates that DNA alkylation must be naturally and continuously occurring. Exogenous alkylating agents include methyl halides derived from burning biomass and decaying vegetation, cigarette smoke, food, and other occupational exposures (Hamilton et al., 2003). Finally, patients are intentionally exposed to extremely high concentrations of alkylating agents
during chemotherapy for various cancers, including glioblastoma, lymphomas, and leukemias (Fu et al., 2012b).
Methylating Agent Mechanism of Action
Methylating agents act by covalently adding methyl (-CH3) groups to all
components of our cells, including proteins, lipids, and most importantly, our DNA. Methylation on DNA bases occurs on both the ring nitrogens and exocyclic oxygens. Methylating agents are categorized as either SN1 or SN2 agents based on their mode of chemical reaction and this classification is indicative of the proportion and type of DNA adducts they form. The predominant DNA adducts formed by all alkylating agents are on ring nitrogens. Methylation at the highly nucleophilic N7-position of guanine produces the most common methyl lesion, 7MeG (Table 1.1). 7MeG is neither mutagenic nor cytotoxic in itself; however, methylation of purines at the N7 position causes base destabilization, spontaneous depurination, and the formation of toxic and mutagenic abasic sites. The other primary N-methylation product in double-stranded (ds) DNA is 3MeA. Though 3MeA adducts are less common than 7MeG, they are toxic owing to their ability to block DNA replication by high fidelity replicative polymerases (Engelward et al., 1998; Groth et al., 2010; Johnson et al., 2007), although they can be bypassed by certain translesion DNA polymerases (Glassner et al., 1998; Johnson et al., 2007; Lange et al., 2011). When bypassed, 3MeA lesions are potentially mutagenic and can lead to A:T to T:A transversions (Fronza and Gold, 2004). The rarer 1-methyladenine (1MeA) lesion can be formed preferentially in single-stranded (ss)DNA and can block replication (Fu et al., 2012b). Methylation can also occur on exocyclic oxygen atoms. In addition to N-alkylation, SN
agents methylate the 06-position of guanine to generate 06-methylguanine
(06-meG). Even though these lesions are relatively rare, they are potentially more hazardous since O6meG can mispair with thymine during DNA replication leading
DNA Repair Pathways for Alkylation Damage
Fortunately, our cells have evolved multiple conserved DNA repair pathways through evolution to combat the mutagenic and cytotoxic effects of methylated bases. Below I discuss a few of the relevant pathways.
Base Excision Repair. The two most common methylation adducts, 7MeG and 3MeA, are repaired by the base excision repair (BER) pathway, a complex and multi-enzyme process (Fig. 1.2). This pathway is initiated by lesion-specific DNA glycosylases, which search the genome and recognize and excise damaged bases. DNA glycosylases are classified as either bi- or mono-function based upon their catalytic functions. Monofunctional glycosylases can remove damaged bases while bifunctional glycosylases can both remove a damaged base and cleave the DNA backbone. In mammalian cells, 11 different glycosylases have been identified, often with overlapping substrates for repair redundancy (Jacobs and Schar, 2012). However, there is only one glycosylase that acts on alkylated bases, the monofunctional alkyladenine glycosylase (Aag; also known as Mpg). Aag hydrolyzes the destabilized glycosyl bond between the DNA base and ribose in the sugar-phosphate backbone to generate an abasic site (AP site). The Aag glycosylase will be described in more detail below. In the next step of BER, the abasic site is processed by the mammalian endonuclease Apel to form a single strand break (SSB) with a 3'-OH and 5'-deoxyribosephosphate (5'-dRP). The bifunctional DNA polymerase
P
(Pol P) then carries out two important functions. First, PolP
removes the remaining sugar moiety left at the 5' end by APE1 with its 5'-dRP lyase activity (Sobol et al., 2000). Secondly, Pol P fills in the missing complementary nucleotides. BER is finalized by ligation of the single strand break by a DNA ligase. X-ray cross-complementing protein 1 (Xrccl) acts as a molecular scaffold during BER to recruit downstream enzymes and facilitate processing of repair intermediates. It should be noted that the pathway described above is the simplest form of BER and is termed short-patch (SP-) BER since only one nucleotide is replaced. In the alternative pathway, long-patch (LP-)BER, 2 to 12 nucleotides are replaced spanning the damaged DNA base (Kim and Wilson, 2012). Replication is thought to be mediated by replicative DNA polymerases Pol 6 and Pol E in combination with the accessory protein proliferating cell nuclear antigen (PCNA). The displaced DNA is then cleaved by flap endonuclease 1 (Feni) before the nick is sealed to complete repair (Fig.
1.2).
Nucleotide Excision Repair. Nucleotide excision repair (NER) is another multi-enzyme and multi-step process generally reserved for the repair of bulky helix-distorting DNA lesions. NER is initiated via two sub-pathways: global genome repair (GGR) or transcription-coupled repair (TCR). The pathways vary in the way they detect DNA damage but converge to the same repair mechanism. After detection of a DNA lesion, NER proteins are recruited and the heterodimer Erccl-Xpf makes an incision 5' to the lesion. DNA replication is then initiated at the single-strand break thus displacing the lesion-containing DNA. Next, a second incision is created 3' to the lesion by Xpg, releasing the damaged DNA. Repair is finalized after all nucleotides are replaced and the DNA nick has been ligated (Fig. 1.3CB) (Scharer, 2013). Interestingly, NER has been shown to compensate in the repair of alkylated DNA in the absence of BER, primarily through the global genome repair subpathway (Huang et al., 1994; Memisoglu and Samson, 2000; Plosky et al., 2002; Samson et al., 1988).
Direct reversal proteins Mgmt and Alkbh2. The simplest repair strategy is to directly remove the methyl group from the DNA, thus repairing the lesion without affecting the overall DNA structure or sugar-phosphate backbone. Methylguanine methyl transferase (Mgmt) is one such 'suicide' protein that repairs toxic 06-meG lesions by transferring the methyl group to an internal cysteine residue, thus targeting the protein for ubiquitin-dependent degradation (Fig. 1.3A) (Gerson, 2004; Liu et al., 2002; Pegg et al., 1991; Xu-Welliver and Pegg, 2002). Given the highly cytotoxic and mutagenic nature of 06-meG lesions, Mgmt expression and promoter methylation is highly correlated to cell sensitivity after treatment with
SN1 methylating agents (Brandes et al., 2008; Gerson, 2004; Hegi et al., 2004; Maze et al., 1996). As mentioned above, 06-meG lesions readily mispair with thymine during DNA replication, thus creating a DNA mismatch that is recognized by the MutSa heterodimer, which initiates DNA mismatch repair (MMR). MMR removes the newly synthesized DNA base (T); however, polymerases repeatedly insert T opposite the 06-meG lesions, creating perpetual rounds of MMR and generating single strand DNA breaks as MMR intermediates (futile cycling) (Mojas et al., 2007); moreover, these single-strand DNA breaks can activate downstream DNA damage signaling responses (Hickman and Samson, 1999b). A second mechanism for direct DNA damage reversal is through the AlkB homologue (Alkbh) family of proteins, some of which can repair 1MeA and 3-methylcytosine (3MeC) in ss- and ds-DNA through an oxidative demethylation mechanism that results in the release of formaldehyde as a byproduct (Fig. 1.3A) (Begley and Samson, 2003; Falnes et al., 2002; Trewick et al., 2002).
Double Strand Break Repair. Double strand breaks (DSBs) can occur by a variety of mechanisms. Though treatment with alkylating agents does not cause DSBs directly, they can be generated by replication forks progressing past single-strand breaks that can be created during BER (Ma et al., 2011; Pascucci et al., 2005). Therefore, the ability of a cell to repair such DSBs is a determinate of sensitivity to alkylation DNA damage (Kondo et al., 2009; Roos et al., 2009). Two separate pathways can repair DSBs: error-prone non-homologous end joining (NHEJ) and the more accurate homologous recombination (HR). NHEJ is a relatively simple mechanism that ligates together double-stranded DNA ends with little to no homology (Fig. 1.3D). HR, on the other hand, functions primarily by using homologous DNA on sister chromatids as a template to resynthesize any missing DNA across the break, and is therefore only functional during S or
G2/M phases of the cell cycle (Fig. 1.3C). This cellular action results in the
appearance of sister chromatid exchanges (SCEs) which can be measured after alkylation treatment and serve as a measurement for HR activity. Additionally, some of the proteins involved in homologous recombination, most notably
Rad5l, have been shown to dually function in the regression of replication forks as a means to bypass replication-blocking lesions (Adelman et al., 2013; Hashimoto et al., 2010; Petermann et al., 2010; Schlacher et al., 2011; Shukla et al., 2005; Zellweger et al., 2015). Upon encountering replication stress, the parental DNA strand can re-anneal while the newly synthesized DNA strands unwind and eventually anneal to each other, forming a 4-way Holliday junction. The regression of the replication fork allows for DNA repair, replication past DNA-lesions by using an undamaged template, or filling in of single-strand DNA gaps (Neelsen and Lopes, 2015).
Alkyladenine Glycosylase (Aag) Initiates Repair of Methylated DNA Bases
Though Aag was first identified by its ability to remove alkylated DNA bases, it has since been demonstrated to initiate repair of a structurally diverse set of DNA lesions, including 3MeA, 7MeG, deaminated adenine (hypoxanthine), 1-methyladenine (1 MeA), 1-methylguanine (1MeG), 3-methylcytosine (3MeC), 8-oxoguanine (8-oxoG) and cyclic etheno adducts (EA; 1,2-EG) (Fig. 1.1) (Bessho et al., 1993; Lee et al., 2009a; Shrivastav et al., 2010b; Wyatt et al., 1999). The ability to effectively excise such a wide variety of DNA lesions while excluding normal bases makes Aag unique. Aag has been shown to employ both hopping and sliding mechanisms to move along double stranded DNA to search for rare DNA lesions among other normal undamaged bases. (Hedglin and O'Brien, 2008; Hedglin and O'Brien, 2010). During this search, Aag adopts a low-affinity conformation that is characterized by a disordered active site and non-specific hydrogen bonding to DNA (Setser et al., 2012). Upon binding an appropriate DNA lesion substrate, Aag assumes a higher-affinity conformation in which residue Tyr-162 is intercalated into the double helix causing the DNA lesion to be flipped into the glycosylase active site (Lau et al., 1998; Setser et al., 2012). A water molecule is appropriately placed to be activated by Asp-238 and act as a nucleophile to catalyze the hydrolysis of the N-glycosyl bond, causing the release of the damaged base (Lau et al., 1998). The active site is lined with hydrophobic,
electron-rich aromatic amino acid residues that effectively bind and stabilize positively charged alkylated bases (e.g. 3MeA, 7MeG), which additionally act as good leaving groups during nucleophilic elimination (Lau et al., 2000). However, not all of Aag's substrates are positively charged, such as the neutral lesions hypoxanthine and EA (Fig. 1.1). It seems a second requirement for base cleavage is dependent on the DNA lesion containing a hydrogen bond acceptor for residue His-136 (Lau et al., 2000). Moreover, release of neutral bases requires both a base to activate the water molecular and an acid to protonate the leaving group, while positively charged lesions only need a base for cleavage initiation (O'Brien and Ellenberger, 2003).
Cellular Consequences of Imbalanced BER
BER requires the tight coordination of multiple enzymes since the pathway intermediates have been shown to by cytotoxic if allowed to accumulate. AP sites and SSBs both inhibit transcription and replication machinery and can lead to the generation of DSBs (Fig. 1.5B) (Boiteux and Guillet, 2004). Though translesion polymerases exist to replicate past AP sites and avoid cytotoxic DSBs, this process often produces point mutations (Avkin et al., 2002; Pages et al., 2008; Schaaper et al., 1983; Weerasooriya et al., 2014). SSBs are rendered even more toxic during BER if the 5'-dRP termini is not cleared by the lyase activity of Pol
B.
Polfl' cells are hypersensitive to methylating agents, however reintroduction ofthe Pol P lyase domain, without an active polymerase domain, rescues sensitivity to near WT levels (Sobol et al., 2000). Moreover, Polf3' cells are only methylation sensitive if BER is initiated by Aag; Polf5'Aag-' cells exhibit the same sensitivity to MMS as WT cells (Sobol et al., 2003).
To help shuttle intermediates through repair to completion, Xrccl acts as a molecular scaffold to recruit and trigger the activity of BER proteins to avoid cellular accumulation of abasic sites or single-strand breaks. Xrccl is an essential protein has been shown to interact with glycosylases (including Aag)
(Campalans et al., 2005; Mutamba et al., 2011), Apel (Vidal et al., 2001), Pol P (Caldecott et al., 1996; Kubota et al., 1996), poly (ADP-ribose) polymerase (Parp1) (Caldecott et al., 1996; Masson et al., 1998), and DNA ligase III (Lig Ill) (Caldecott et al., 1994; Tebbs et al., 1999). The presence of XRCC1 in human cells was shown to facilitate AAGdependent excision of hypoxanthine and 1,N -ethenoadenine (sA) (Fig. 1.1) (Mutamba et al., 2011). Moreover, Xrccl competes with Apel and Parp1 to bind DNA repair intermediates themselves (Nazarkina et al., 2007). Knockout of Xrccl is embryonic lethal and knockdown of the protein leads to enhanced sensitivity to oxidative and alkylation treatment (Caldecott, 2003).
Even in the presence of Xrccl, BER pathway imbalances can occur due to increased pathway initiation via glycosylase activity or decreased activity of any downstream step. Decreases in Apel activity cause increases in AP sites and sensitivity to alkylation treatment (Ensminger et al., 2014; Silber et al., 2002). As mentioned above, downregulation of Pol P activity causes increases in SSBs, double-strand breaks, and sensitivity to methylating agents (Senejani et al., 2012; Sobol et al., 2000). Below, we will discuss in more detail the effects that altered Aag expression has on cell sensitivity to alkylating agents.
Alkylation Sensitivity of Cells with Altered Aag Expression
Given all that is known about BER and cellular responses, it is conceivable that
Aag-' cells could react in disparate ways to alkylating agents. Since BER is
initiated by the adduct-specific Aag DNA glycosylase, cells lacking Aag accumulate cytotoxic and mutagenic DNA methylation lesions 3MeA and 7MeG. Though 3MeA has been shown to block DNA polymerases, potentially leading to double strand breaks at collapsed replication forks that can be repaired by homologous recombination (Hendricks et al., 2002), translesion polymerases can bypass these lesions during DNA synthesis (Johnson et al., 2007; Lange et al., 2011). On the other hand, the loss of Aag could mask BER imbalances resulting
in accumulation of toxic intermediates that typically signal for cell death in wild-type (WT) cells.
In fact, there is a range of responses to alkylation damage in cells lacking the Aag DNA glycosylase. Aag' mouse embryonic stem ES cells are more sensitive to the SN2 alkylating agent methyl methanesulfonate and exhibit more alkylation induced sister chromatid exchanges, p53 stabilization, and apoptosis compared to wild-type ES cells (Engelward et al., 1998; Engelward et al., 1996). Primary mouse embryonic fibroblasts (MEFs) from Aag' mice are more sensitive than wild-type MEFs to Me-Lex, an agent that specifically induces 3MeA lesions, but exhibit no difference in sensitivity to MMS (Engelward et al., 1997; Sobol et al., 2003). Similarly, human cervical carcinoma (HeLa) cells and glioma cell lines with significantly reduced Aag protein expression are sensitized to chemotherapeutic methylating agents (Agnihotri et al., 2011; Paik, 2005).
Conversely, the increased sensitivity of Aag' ES cells does not extend to the adult Aag' mouse. Aag' mice are not more sensitive to alkylation-induced lethality compared to wild type nor do they exhibit significantly more spontaneous or alkylation induced cancers (Roth and Samson, 2002) (unpublished data). Surprisingly, the loss of Aag actually renders particular adult mouse tissues resistant to MMS induced cell death. Aag' myeloid progenitor cells of the hematopoietic lineage are resistant to MMS compared to WT progenitors in ex
vivo colony-forming assays (Meira et al., 2009a; Roth and Samson, 2002). Aag'
mice also exhibit hematopoietic resistance to MMS in vivo as measured by total bone marrow cell numbers and the micronucleus assay, a measure for large-scale erythropoietic chromosomal damage. A similar phenotype was observed in
Aag' retinal photoreceptors, which demonstrated remarkable resistance to
MMS-induced degeneration and blindness (Meira et al., 2009b). Finally, treatment of WT mice with methylating agents induces neurodegeneration of cerebellar granule neurons, yet this cell death is completely abolished in Aag-mice (Calvo et al., 2013b; Kisby et al., 2009).
These results suggest that initiation of BER in certain WT tissues causes cell death that is suppressed upon loss of Aag. Indeed, overexpression of Aag renders these tissues even more sensitive to MMS induced degeneration by creating a more severe imbalance in the BER pathway due to increased pathway initiation without concomitant increases in downstream enzymes (Calvo et al., 2013b). The Aag dependent sensitivity of hematopoietic progenitors, retinal photoreceptors, and cerebellar granule neurons is similarly completely suppressed in the absence of Parp1, independent of the level of Aag expressed, indicating that Parpi mediates cell death caused by BER imbalances in these tissues (Calvo et al., 2013b). All in all, these results support the notion that cell fate in response to methylating agents is both Aag-dependent and cell-specific.
Poly (ADP-Ribose) Polymerase I Can Mediate Cell Death Caused by BER Imbalances
Parp1 was first identified as a DNA repair protein with the ability to catalyze the formation of long poly (ADP-ribose) (PAR) polymers on itself and other proteins (Satoh and Lindahl, 1992). Parpl strongly binds single-strand breaks within minutes of their generation though two zinc-fingers and one zinc-binding domain that mediate DNA-dependent enzymatic activation (Langelier et al., 2008). PAR polymers are created through the hydrolysis of NAD' and transfer of the ADP-ribose moiety to protein acceptors (Fig. 1.4). The polymers are highly negatively charged and function both as posttranslation modifications as well as signaling molecules on their own right.
Upon binding to SSBs, Parp1 undergoes auto-PARylation that subsequently stimulates its release from DNA due to negatively charged interactions between the DNA and the PAR polymers. PARylation also occurs on multiple DNA damage repair and signaling proteins including Aag, Xrccl and the ataxia telangiectasia and Rad3 related kinase (ATR) (Jungmichel et al., 2013; Kedar et
al., 2008; Masson et al., 1998); however, the functional role of PARylation of Aag has not yet been explored. Though Parp1 is not required for accurate completion of BER, activation of Parp1 helps recruit Xrccl and can stimulate DNA repair (Dantzer et al., 1999; El-Khamisy et al., 2003; Prasad et al., 2015; Prasad et al., 2001). It should be noted that Parp1's cellular function extends beyond DNA repair as it has been found to play a role in transcription, metabolism, inflammation, cell differentiation, and regulation of chromatin structure (Bai and Canto, 2012; Ditsworth et al., 2007; Ji and Tulin, 2010; Krishnakumar and Kraus, 2010; Schreiber et al., 2006).
While at moderate levels of DNA damage Parp1 activation can assist in DNA repair and cell survival, upon excessive levels of DNA damage and formation of SSBs Parp1 hyperactivation can cause cell death through caspase-independent programmed necrosis (Berghe et al., 2014). Historically, Parp1 mediated cell death has been hypothesized to be caused by cellular depletion of NAD+ through PARylation and subsequent ATP loss leading to bioenergetic failure (Fig. 1.5B). Indeed, in certain cases repletion of bioenergetic substrates such as NAD+ and pyruvate does rescue cell sensitivity to alkylating agents (Alano et al., 2010; Tang et al., 2010; Tang et a!., 2009). However, recent publications have challenged this hypothesis by demonstrating that ATP depletion precedes NAD+ loss. In fact, PAR polymers were shown to translocate from the nucleus to the mitochondria and bind directly to hexokinase (HK), the initiating enzyme of glycolysis, thus inhibiting its activity (Andrabi et al., 2014; Fouquerel et al., 2014). This glycolytic inhibition was independent of loss of NAD+ and glycolysis could be restored through the addition of tricarboxylic acid (TCA) cycle substrates pyruvate and glutamine (Andrabi et al., 2014). Furthermore, PAR translocation to the mitochondria has been shown to cause the mitochondrial release of apoptosis-inducing factor (AIF) (Hong and Dawson, 2004; Yu et al., 2006). AIF translocates to the nucleus where it interacts with histone H2AX, leading to chromatinolysis through interaction with cyclophilin A (Artus et al., 2010). It
should be noted, however, that AIF translocation is not a necessary component of Parp1-mediated programmed necrosis (Tang et al., 2010).
Model System to Study Aag-Dependent Alkylating Sensitivity
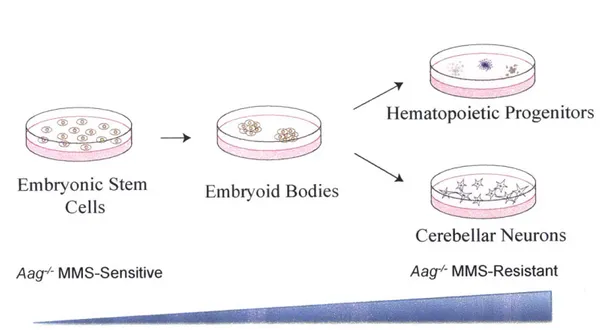
Despite the known and obvious cell-specific differences in Aag-dependent sensitivity to methylating agents, no attempts to understand the underlying biology have been made. One option is to study pure populations of cells exhibiting varied Aag-dependent alkylation responses, for example, mouse embryonic stem (ES) cell lines and primary neurons. Another possibility, however, is to utilize the pluripotency of one of the cell types of interest - mouse ES cells. ES cells have the capacity to differentiate in vitro into any cell type of interest. Moreover, if in vitro differentiation recapitulates in vivo development then
Aag* MMS-sensitive ES cells will transform to become Aag4 MMS-resistant
hematopoietic progenitors and cerebellar neurons. Benefits of this method include a shorter timeline of differentiation in vitro compared to in vivo, abrogating the need for maintaining mouse colonies, and offering the option of assessing
Aag' sensitivity as a function of development. Limitations of this method include
heterogenous populations of cells after differentiation and the inability to know forthright whether in vitro differentiation will reflect in vivo work.
Overview of the Presented Study
In the presented work, we explore the Aag dependent DNA-damage responses to methylating agents in embryonic stem cells compared to primary neurons. To this end, new ES cells have been generated and validated for pluripotency. We show that Aag' sensitivity to MMS compared to WT is independent of inbred genetic background. Surprisingly, we find that overexpression of Aag in ES cells renders them even more sensitive to MMS than loss of Aag, indicating that methylation sensitivity in ES cells is not Aag gene dose dependent as seen in
primary neurons or retinal photoreceptors. Additionally, we studied in detail the Aag- and Parp1-dependent cell sensitivity of mouse cerebellar granule neurons in ex vivo conditions, demonstrating that methylation responses are cell-intrinsic phenomena that can be studied outside of brain tissue. Neurons undergo caspase-independent cell death after MMS treatment and do not exhibit mitochondrial permeabilization or AIF nuclear translocation. Finally, we employed
in vitro differentiation of hematopoietic progenitors and cerebellar neurons in an
attempt to recapitulate the transition in Aag-' MMS-responses from sensitivity in ES cells to resistance in differentiated cells.
0 (D N </ NH NN N NH2 7meG N NH N Hypoxanth ine 0 H N NNH N NH 2 8-oxoG NH2 N NN 3meA 0 N NN NH-2 ImeG N N N 1,N6-E A NH 2 N NN NN lmeA NH N 3meC 0 N / N N N 1,N2-EG
Figure 1.1: Structures of Aag DNA Substrates.
7MeG, 7-methylguanine; 3MeA, 3-methyladenine; 1MeA, 1-methyladenine; 1MeG, 1methylguanine; 3MeC, 3methylcytosine; 8oxoG, 8oxoguanine; 1,N -EA, 1,N6-ethenoadenine; 1,N2-G, 1,N2-ethenoguanine.
I'I
DNA Base Damage 4 Mono-functional UNG) YI ISMLJ61 MBD4) AAGIMPGj 3' A G T T C A T A C A A G T 3 I 1 , 1 5 T--- 3 A G T 5 - -T 3' A'7 G T T C A T A C A A G T yj __ T A AG G 3 5 A G T T C A T A C A L G la/XRCC1A G T 5 -111 3 SP-BER Bi-functional (P) (OGG1 NTH1 Bi-functional (0,6) riNE1L I 2 1 1 1 1 35'--T~~ A G T T C A A G T T C A T A C A A G T T A C A A GT 3 5 3' 5 APE1' PNKP 5' - -T 1 3 5 A C T T C A T A C A A G T 3~~~~ A . 1~..L I.. 5' PCNA Po6 PoIE C 5 -- T - 3 33 A ' ~ ' C A T A C A A G T 3' 1 1 1 5' FENI A G C A TA C A AG T LIGI ---- - --, -r---'r 3' A ; ' C A T A C A A GI LP-BER
Figure 1.2: Base Excision Repair Pathway.
Base excision repair (BER) pathway is initiated by the mono-functional Aag glycosylase that remove a methylated base, leaving an abasic site. The abasic site is cleaved by AP endonuclease 1 (Apel) resulting in a 5'-deoxyribosephosphase (5'-dRP) that is cleared by the lyase activity of Pol 1, which also synthesizes any missing DNA nucleotides. Finally the single-strand break is sealed together by a ligase (Lig). The whole process is coordinated by Xrccl. LP-BER is initiated by bi-functional DNA glycosylases that remove a damaged base and also cleave the DNA backbone. Inhibitory DNA termini are removed by either Apel or Pnkp and DNA replication is completed by either Pol
P, Pol 6, or Pol E. The displaced DNA is removed by the flap endonuclease 1 (Fen1) and the nick is sealed. Figure adapted from (Kim and Wilson, 2012).
A Direct repair (MGMT) CH1 MGMT . CysCH, MGMT - Cys-CH,
IIIIIIIIII
CDirect repair (ALKBH) B NER
CH XPC-RAD23B mifhrm17l ALKBH OH H , LK -HCOH 0 HR XPA RPA 5 fnc sion 3' incsion XPA TFIH ERCCXG XPE Repair synthesis Pol 6. or L
D=I|
PCNA LigationniwiQItTIi
NHEJ DNA-PK binding and synapsis Processing by Artemis, PNK and other proteinsRecruitment of LIG4-XRCC4 and rejoining
Figure 1.3: Alternate DNA Repair Pathways for Methylation Damage.
(A) Direct Reversal repair mechanisms include MGMT and the AlkBH proteins. MGMT directly removes methyl groups from the 06-position of guanine to an internal cysteine residue. AlkBH proteins remove methyl lesions through an oxidative demethylation mechanism that results in the release of formaldehyde.
(B) Nucleotide excision repair (NER) pathway identifies DNA damage through global genome repair subpathway. Single-strand nicks are created 5' and 3' of the damage so that a fragment of lesion-containing DNA can be removed. Polymerases (Pol 6 or Pol E) fill in the empty segment before ligation. TFIIH, Transcription factor IIH; XP, xerodera pigmentosum proteins.
(C)A replication fork passing a single-strand DNA break will generated a one ended double-strand break. The 3' overhang of the double-strand break will invade the complementary sequence on the sister chromatid to generate a D-loop and DNA is synthesized past the single-strand break. The resulting Holliday junction will be cleaved, thus restoring the replication forks.
( n- g IIU JUIIIIId jInI I I J) Is Iniliated by the binding of Ku
proteins (green circles) to double-stranded DNA ends, recruitment of DNA-PKcs (taupe cylinder), processing of the ends by Artemis (red triangle), and eventual ligation by Lig4-XRCC4 complex (orange ovals). DNA-PKcs, DNA-dependent protein kinase catalytic subunit; Lig4, DNA ligase 4; XRCC4, X-ray repair cross-complementing protein 4.
Figures adapted from (Fu et al., 2012b; O'Driscoll and Jeggo, 2006, Helleday, et al., 2007)
0 0 NHH N H N A D NH H NAD Nicotinamide OHN O
Parp1l
Acceptor Acceptor Protein Protein SE. D, or K NHC=O <N N IUHydrohise W NH O OH NN 0 OH NHARG AR NHO N OHOHI OOH OH OHFigure 1.4: Parp1 utilizes NAD* to generate PAR polymers.
Parp1 catalyzes the addition of the ADP-ribose portion of NAD* to an acceptor protein, releasing nicotinamine in the process. Long polymers of ADP-ribose are called poly (ADP-ribose) or PAR polymers. Polymers can be either branched or linear and are primarily hydrolyzed by poly (ADP-ribose) glycohydrolase (PARG). Figure adapted from (Krishnakumar and Kraus, 2010)
Alkylating Agent AAG APE - ~ OH jG Alkylating Agent .3'OH .3'OH
(
LID
DNA Survival replication Mutation block DNA replication DSB and fork collapseSurvival Mutation NHEJ HR Survival SCE TLS Surviva Mutation Parpi Parp1 Activation Hyperactivation DNA NAD'/ATP Repair depletion
4.riv
C
4lDe
Figure 1.5: Alternative mechanisms of Cell Death Caused during Base Excision Repair (BER).
Alkylating agents cause the formation of DNA damage that is repair by the BER pathway (described in the text). DNA lesions and abasic sites (AP sites) inhibit
34
_____________I
A
%A 0 1A E 4, 12 Apoptosis Necrosis Cell deathDNA replication but can be bypassed by translesion synthesis (TLS), resulting in survival albeit with mutations. Single-strand breaks, however, can cause death through two different mechanisms. (A) SSBs strong inhibit replication fork progress leading to cytotoxic double-strand breaks which can be repaired by the error-prone NHEJ or more accurate HR. DSBs can also cause cell death, often through apoptotic mechanisms. (B) Alternatively, SSBs can be bound by Parp1, leading to activation. At low levels of DNA damage, Parp activation stimulates DNA repair; however, Parp hyperactivation can cause cell death through energetic failure and programmed necrosis. Figures adapted from (Calvo et al., 2013b; Fu et al., 2012b).
Tables
Table 1.1: Methylation patterns in single- and double-stranded DNA after treatment with MMS represented as percentages of total methylation.
nd = not detected. Data adapted from (Beranek, 1990; Drablos et al., 2004).
Site of Methylation ssDNA/RNA Adenine N1- 18 3.8 N3- 1.4 10.4 N7- 3.8 1.8 Guanine N3- -1 0.6 o6- - 0.3 N7- 68 83 Cytosine 2 - N3-nd 10 Diester 2 nd <1 0.8 dsDNA
References
Adelman, C.A., Lolo, R.L., Birkbak, N.J., Murina, 0., Matsuzaki, K., Horejsi, Z., Parmar, K., Borel, V., Skehel, J.M., Stamp, G., et al. (2013). HELQ promotes RAD51 paralogue-dependent repair to avert germ cell loss and tumorigenesis. Nature 502, 381-384.
Agnihotri, S., Gajadhar, A.S., Ternamian, C., Gorlia, T., Diefes, K.L., Mischel, P.S., Kelly, J., McGown, G., Thorncroft, M., Carlson, B.L., et al. (2011). Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. In The Journal of clinical investigation.
Alano, C.C., Garnier, P., Ying, W., Higashi, Y., Kauppinen, T.M., and Swanson, R.A. (2010). NAD+ Depletion Is Necessary and Sufficient forPoly(ADP-Ribose) Polymerase-1-Mediated Neuronal Death. In The Journal of
Andrabi, S.A., Umanah, G.K.E., Chang, C., Stevens, D.A., Karuppagounder, S.S., Gagne, J.-P., Poirier, G.G., Dawson, V.L., and Dawson, T.M. (2014). Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. In Proceedings of the National Academy of Sciences, pp.
10209-10214.
Artus, C., Boujrad, H., Bouharrour, A., Brunelle, M.N., Hoos, S., Yuste, V.J., Lenormand, P., Rousselle, J.C., Namane, A., England, P., et al. (2010). AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. The EMBO journal 29, 1585-1599.
Avkin, S., Adar, S., Blander, G., and Livneh, Z. (2002). Quantitative measurement of translesion replication in human cells: evidence for bypass of abasic sites by a replicative DNA polymerase. Proceedings of the National Academy of Sciences of the United States of America 99, 3764-3769.
Bai, P., and Canto, C. (2012). The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell metabolism 16, 290-295.
Begley, T.J., and Samson, L.D. (2003). AlkB mystery solved: oxidative demethylation of N1-methyladenine and N3-methylcytosine adducts by a direct reversal mechanism. Trends in biochemical sciences 28, 2-5.
Beranek, D.T. (1990). Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. In Mutat Res, pp. 11-30.
Berghe, T.V., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., and Vandenabeele, P. (2014). Regulated necrosis: the expanding network of non-apoptotic cell death pathways. In Nat Rev Mol Cell Biol (Nature Publishing Group), pp. 135-147.
Bessho, T., Roy, R., Yamamoto, K., Kasai, H., Nishimura, S., Tano, K., and Mitra, S. (1993). Repair of 8-hydroxyguanine in DNA by mammalian N-methylpurine-DNA glycosylase. Proceedings of the National Academy of Sciences of the United States of America 90, 8901-8904.
Boiteux, S., and Guillet, M. (2004). Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA repair 3, 1-12.
Brandes, A.A., Brandes, A.A., Franceschi, E., Franceschi, E., Tosoni, A., Tosoni, A., Blatt, V., Blatt, V., Pession, A., Pession, A., et al. (2008). MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. In J Clin Oncol, pp. 2192-2197.
Caldecott, K.W. (2003). XRCC1 and DNA strand break repair. DNA repair 2, 955-969.
Caldecott, K.W., Aoufouchi, S., Johnson, P., and Shall, S. (1996). XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase IlIl is a novel molecular 'nick-sensor' in vitro. Nucleic Acids Res 24, 4387-4394.
Caldecott, K.W., McKeown, C.K., Tucker, J.D., Ljungquist, S., and Thompson, L.H. (1994). An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase Ill. Mol Cell Biol 14, 68-76.
Calvo, J.A., Moroski-Erkul, C.A., Lake, A., Eichinger, L.W., Shah, D., Jhun, I., Limsirichai, P., Bronson, R.T., Christiani, D.C., Meira, L.B., et al. (2013). Aag
DNA Glycosylase Promotes Alkylation-Induced Tissue Damage Mediated by Parp1. In PLoS Genet (Public Library of Science), pp. e1003413.
Campalans, A., Marsin, S., Nakabeppu, Y., O'Connor, T.R., Boiteux, S., and Radicella, J.P. (2005). XRCC1 interactions with multiple DNA glycosylases: A model for its recruitment to base excision repair. In DNA repair, pp. 826-835.
Ciccia, A. (2010). The DNA damage response: making it safe to play with knives. In Molecular cell.
Dantzer, F., Schreiber, V., Niedergang, C., Trucco, C., Flatter, E., De La Rubia, G., Oliver, J., Rolli, V., Menissier-de Murcia, J., and de Murcia, G. (1999). Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie
81, 69-75.
Ditsworth, D., Zong, W.X., and Thompson, C.B. (2007). Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. The Journal of biological chemistry 282,
17845-17854.
Drablos, F., Feyzi, E., Aas, P.A., Vaagbo, C.B., Kavli, B., Bratlie, M.S., Pena-Diaz, J., Otterlei, M., Slupphaug, G., and Krokan, H.E. (2004). Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA repair 3, 1389-1407.
EI-Khamisy, S.F., Masutani, M., Suzuki, H., and Caldecott, K.W. (2003). A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res 31, 5526-5533.
Engelward, B.P., Allan, J.M., Dreslin, A.J., Kelly, J.D., Wu, M.M., Gold, B., and Samson, L.D. (1998). A chemical and genetic approach together define the biological consequences of 3-methyladenine lesions in the mammalian genome. In The Journal of biological chemistry, pp. 5412-5418.
Engelward, B.P., Dreslin, A., Christensen, J., Huszar, D., Kurahara, C., and Samson, L.D. (1996). Repair-deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation-induced chromosome damage and cell killing. In The EMBO journal (Nature Publishing Group), pp. 945.
Engelward, B.P., Weeda, G., Wyatt, M.D., Broekhof, J.L., de Wit, J., Donker, I., Allan, J.M., Gold, B., Hoeijmakers, J.H., and Samson, L.D. (1997). Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proceedings of the National Academy of Sciences of the United States of America 94,13087-13092.
Ensminger, M., lloff, L., Ebel, C., Nikolova, T., Kaina, B., and Lbrich, M. (2014). DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J Cell Biol 206, 29-43.
Falnes, P.O., Johansen, R.F., and Seeberg, E. (2002). AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 419, 178-182.
Fouquerel, E., Goeliner, E.M., Yu, Z., Gagne, J.P., Barbi de Moura, M., Feinstein, T., Wheeler, D., Redpath, P., Li, J., Romero, G., et al. (2014). ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep 8, 1819-1831.
Fronza, G., and Gold, B. (2004). The biological effects of N3-methyladenine. Journal of cellular biochemistry 91, 250-257.
Fu, D., Calvo, J.A., and Samson, L.D. (2012). Balancing repair and tolerance
f DA. ~dLmag I used by clkMy ICiLng JeIILs. In Nature Reviews Cancer, pp.
-18.
Gerson, S.L. (2004). MGMT: its role in cancer aetiology and cancer therapeutics. In Nature Reviews Cancer, pp. 296-307.
Glassner, B.J., Rasmussen, L.J., Najarian, M.T., Posnick, L.M., and Samson, L.D. (1998). Generation of a strong mutator phenotype in yeast by imbalanced base excision repair. In Proc Natl Acad Sci USA, pp. 9997-10002.
Groth, P., Auslander, S., Majumder, M.M., Schultz, N., Johansson, F., Petermann, E., and Helleday, T. (2010). Methylated DNA causes a physical block to replication forks independently of damage signalling, 0(6)-methylguanine or DNA single-strand breaks and results in DNA damage. Journal of molecular biology 402, 70-82.
Hamilton, J.T.G., Hamilton, J.T.G., McRoberts, W.C., McRoberts, W.C., Keppler, F., Keppler, F., Kalin, R.M., Kalin, R.M., Harper, D.B., and Harper, D.B. (2003). Chloride methylation by plant pectin: an efficient environmentally significant process. In Science, pp. 206-209.
Hashimoto, Y., Ray Chaudhuri, A., Lopes, M., and Costanzo, V. (2010). Rad5l protects nascent DNA from Mrel 1-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305-1311.
Hedglin, M., and O'Brien, P.J. (2008). Human alkyladenine DNA glycosylase employs a processive search for DNA damage. Biochemistry 47, 11434-11445.
Hedglin, M., and O'Brien, P.J. (2010). Hopping enables a DNA repair glycosylase to search both strands and bypass a bound protein. ACS chemical
biology 5, 427-436.
Hegi, M.E., Hegi, M.E., Diserens, A.-C., Diserens, A.-C., Godard, S., Godard, S., Dietrich, P.-Y., Dietrich, P.-Y., Regli, L., Regli, L., et aL. (2004). Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. In Clin Cancer Res, pp. 1871-1874.
Hickman, M.J., and Samson, L.D. (1999). Role of DNA mismatch repair and p53 in signaling induction of apoptosis by alkylating agents. Proceedings of the National Academy of Sciences of the United States of America 96, 10764-10769.
Hong, S.J., and Dawson, T.M. (2004). Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. In Trends in ....
Huang, J.C., Hsu, D.S., Kazantsev, A., and Sancar, A. (1994). Substrate spectrum of human excinuclease: repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proceedings of the National Academy of Sciences of the United States of America 91, 12213-12217.
Jacobs, A.L., and Schar, P. (2012). DNA glycosylases: in DNA repair and beyond. Chromosoma 121, 1-20.
Ji, Y., and Tulin, A.V. (2010). The roles of PARP1 in gene control and cell differentiation. In Current Opinion in Genetics & Development, pp. 512-518.
Johnson, R., Yu, S., and Prakash, S. (2007). A Role for Yeast and Human Translesion Synthesis DNA Polymerases in Promoting Replication through 3-Methyl Adenine. In Molecular & Cellular ....
Jungmichel, S., Rosenthal, F., Altmeyer, M., Lukas, J., Hottiger, M.O., and Nielsen, M.L. (2013). Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. In MOLCEL (Elsevier), pp. 272-285.
Kedar, P.S., Stefanick, D.F., Horton, J.K., and Wilson, S.H. (2008). Interaction between PARP-1 and ATR in mouse fibroblasts is blocked by PARP inhibition. DNA repair 7, 1787-1798.
Kim, Y.J., and Wilson, D.M., 3rd (2012). Overview of base excision repair biochemistry. Current molecular pharmacology 5, 3-13.
Kisby, G.E., Kisby, G.E., Olivas, A., Olivas, A., Park, T., Park, T., Churchwell, M., Churchwell, M., Doerge, D., Doerge, D., et al. (2009). DNA repair modulates the vulnerability of the developing brain to alkylating agents. In DNA repair, pp. 400-412.
Kondo, N., Takahashi, A., Mori, E., Ohnishi, K., McKinnon, P.J., Sakaki, T., Nakase, H., and Ohnishi, T. (2009). DNA ligase IV as a new molecular target for temozolomide. Biochemical and biophysical research communications 387, 656-660.
Krishnakumar, R., and Kraus, W.L. (2010). The PARP Side of the Nucleus: Molecular Actions, Physiological Outcomes, and Clinical Targets. In Molecular cell, pp. 8-24.
Kubota, Y., Nash, R.A., Klungland, A., Schar, P., Barnes, D.E., and Lindahl, T. (1996). Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. The EMBO journal 15, 6662-6670.
Lange, S.S., Takata, K.-i., and Wood, R.D. (2011). DNA polymerases and cancer. In Nature Reviews Cancer, pp. 96-110.
Langelier, M.F., Servent, K.M., Rogers, E.E., and Pascal, J.M. (2008). A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates