HAL Id: tel-01279986
https://tel.archives-ouvertes.fr/tel-01279986
Submitted on 2 Mar 2016
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License
NOUVEAUX SUPPORTS POUR MICROSYSTEMES
INTEGRES DE CHROMATOGRAPHIE EN PHASE
GAZEUSE
Imadeddine Azzouz
To cite this version:
Imadeddine Azzouz. ETUDE, REALISATION ET CARACTERISATION DE NOUVEAUX
SUPPORTS POUR MICROSYSTEMES INTEGRES DE CHROMATOGRAPHIE EN PHASE GAZEUSE. Chimie analytique. Université Paris 6, 2013. Français. �tel-01279986�
THÈSE
PRÉSENTÉE A
L’UNIVERSITÉ PIERRE ET MARIE CURIE
ÉCOLE DOCTORALE : 388
Par Imadeddine AZZOUZ
POUR OBTENIR LE GRADE DE DOCTEUR
SPÉCIALITÉ : Chimie Analytique
ETUDE, REALISATION ET CARACTERISATION DE
NOUVEAUX SUPPORTS POUR MICROSYSTEMES
INTEGRES DE CHROMATOGRAPHIE EN PHASE GAZEUSE
Soutenue publiquement le : 29 Octobre 2013 Devant le jury formé de :
Pr.Benamar DAHMANI Université de Tlemcen, Algérie Rapporteur
Pr.Pascal CARDINAEL Université de Rouen Rapporteur
Dr.Jérôme VIAL ESPCI Directeur de thèse
Dr.Didier THIEBAUT ESPCI Co-encadrant de thèse
M.Jérôme BREVIERE Géoservices Invité
Pr.Franck LAUNAY UPMC Examinateur
Dr.Simion BELDEAN-GALEA Université Babes-Bolyai, Roumanie Examinateur
I
Remerciements :
Je tiens tout d’abord à remercier Marie-Claire HENNION puis Valérie PICHON qui se sont su ccédé à l a d irection d u l aboratoire sci ences an alytiques, b ioanalytiques e t miniaturisation (LSABM-ESPCI), de m’avoir accueilli.
Je remercie l’ensemble des membres du jury de thèse pour le temps qu’ils ont consacré à l’examen de ca manuscrit.
Je remercie mon directeur de thèse Jérôme VIAL, pour m’avoir proposé un s ujet de thèse riche et intéressant. Je souhaiterai le remercier pour la confiance et la liberté qu’il a su m’accorder p our effectuer ce t ravail. C onfiance q u’il a su au ssi m ’accorder p our l’encadrement des étudiants.
Je ser ai i ngrat s i j e n e r emercie p ar m on co -encadrant D idier T HIEBAUT. Je l e remercie sincèrement de ses éch anges, orientations et deson apport au projet. Je le remercie pour m’avoir fait bénéficier de son regard critique et de sa grande culture scientifique.
Je so uhaite également t émoigner t oute m a r econnaissance à m es st agiaires A nouar ESSOUSSI et Joachim FLEURY avec lesquels j’ai eu le plaisir d’encadrer et d’interagir
Ce travail a été réalisé avec la contribution de nombreuses personnes extérieures au laboratoire et j e t iens à l es r emercier : Jean-Michel PEREIRA d u l aboratoire NAVI ER d e l’école nationale des ponts et chaussées pour les mesures de porosité. Mohamed HANAFI du laboratoire P PMD-EPSCI p our les an alyses thermiques g ravimétriques et d ifférentielles. Remerciement au x membres d u l aboratoire LMEP-ESPCI q ui n ous f aciliter l ’accès e t l’utilisation du microscope MEB et à Patrice pour la formation.
Plus pr ès d e moi, un g rand m erci à t ous l es membres de not re é quipe, r etrouvées impliquées dans ce travail: Patrick, Raphael, Kamran, Zineb et Emna
Je r emercie t ous les m embres d u l aboratoire L SABM et c ollègues q ui m ’ont acco mpagné durant ces an nées de thèse. La liste e st t rès l ongue... je cite à tit re d’exemple : W assim, Valérie, R amia, Youcef, Manel, Aurélien... et tous l es doctorants, pos t-doctorants e t les dizaines de stagiaires... j’ai eu vraiment le plaisir de les connaitre
Enfin, je voudrais remercier tous les membres de ma famille pour leur aide et leur soutien indispensable d urant ce s an nées d e t hèse... « ma b kach b ezzef w t kammal » Je r emercie particulièrement mes parents pour l‘amour et la confiance qu’ils m’ont toujours témoignés.
III
Liste des publications, communications orales et écrites :
Publications dans des revues nationales et internationales à comité de lecture :
2011 A zzouz I , V ial J , T hiébaut D , S assiat P, M arty F , D anaie K , B ockrath M , Wong J , Haudebourg R , B ourlon B ( 2011) « Comparaison de phases stationnaires originales adaptées aux colonnes de CPG sur puces ». Spectra Analyse 82:46-51
2013 Haudebourg R, Vial J, Thiebaut D, Danaie K, Breviere J, Sassiat P, Azzouz I, Bourlon B (2012) “Temperature-Programmed Sputtered Micromachined Gas Chromatography Columns: An Approach to Fast Separations in Oilfield Applications” . A nalytical Chemistry 85 (1):114-120
2013 H audebourg, R aphael; E Matouk, Z ineb; Zoghlami, E mna;Danaie, K amran;Azzouz, Imadeddine; E Sassiat, P atrick; E Thiébaut, D idier; V ial, Jérôme; “Sputtered alumina as a novel stationary phase for micro machined gas chromatography columns” DOI: 10.1007/s00216-013-7289-z
2013 I. Azzouz, J. Vial, D. Thiébaut, R. Haudebourg, K. Danaie, P. Sassiat, J. Breviere Analytical a nd B ioanalytical C hemistry, A ugust 2013,” Review of stationary phases for microelectromechanical systems in gas chromatography: feasibility and separations”, DOI: 10.1007/s00216-013-7168-7
IV Autres publications:
2011 CJ MAG N°3, journal semestriel du club jeunes de l’Afsep (association francophone des sciences sé paratives): « Comparaison chromatographique de phases stationnaires originales adaptées aux GC sur puce: faisabilité et premières séparations ».
Posters
1. 9ème congrès francophone de l'AfSep SEP 2011, 23-25 Mars 2011, Toulouse, France. « Comparaison chromatographique de phases stationnaires originales adaptées aux colonnes de CPG sur puces ». Prix Poster.
2. 36th I nternational S ymposium on C apillary C hromatography- 9th GCx GC Symposium, Riva del Garda, Italy May 27 – June 1, 2012 “ silica-based monoliths a novel stationary phase for light hydrocarbons gas chromatography separation”
Orales
1. 4ème Journée scientifique du club jeunes Afsep , 15 et 16 Novembre 2011 à Lyon 2. 5ème Journée scientifique du club jeunes Afsep, 26 octobre 2012, Paris
V
Table des matières
Introduction générale...1
Chapitre 1: Etude bibliographique
1. Introduction:... .. 42. Les différents types de colonnes utilisées en CPG :... 5
2.1. Les colonnes remplies ... 5
2.2. Les colonnes ouvertes ... 7
2.2.1. Les films de phases stationnaires ... 7
2.2.2. Méthodes de dépôt du film de phase stationnaire ... 9
2.2.3. Epaisseur du film et incidences sur les propriétés de séparation ... 10
2.3. Colonnes PLOT ... 11
3. Miniaturisation en CPG :... 12
3.1. Intérêt de la miniaturisation ... 12
3.2. Méthode de fabrication des dispositifs miniaturisés ... 13
3.2.1. Méthodes conventionnelles ... 13
3.2.2. Méthode non conventionnelle : La lithographie molle ... 14
3.3. Phases stationnaires utilisées pour les MEMS en CPG ... 15
3.4. Programmation de température en chromatographie en phase gazeuse ... 30
3.5. Techniques de chauffage pour la chromatographie gazeuse sur puce ... 31
4. Les phases stationnaires monolithiques:... 32
4.1. Les monolithes inorganiques ... 33
4.2. Synthèse et caractérisation des monolithes de silice ... 34
4.3. Généralités sur le procédé sol-gel ... 35
4.4. Procédé sol-gel dédié à la chromatographie ... 36
4.4.1. Hydrolyse ... 37
4.4.2. Condensation et gélification ... 37
4.4.3. Restructuration de la microporosité ... 39
4.4.4. Elimination du porogène ... 40
5. Influence des différents paramètres sur la morphologie du monolithe de silice : 40 5.1. Nature et quantité du précurseur ... 41
5.2. Nature et quantité de porogène ... 42
5.3. Influence de la masse moléculaire du porogène ... 43
5.4. Nature du catalyseur et pH initial du milieu ... 44
5.5. Nature du catalyseur ... 45
6. Conclusion :...4 6 7. Références bibliographiques……….………47
VI
Chapitre 2: Matériels et méthodes
1. Introduction : 53
2. Procédé de sythèse sol-gel : 53
2.1. Prélèvement des réactifs: 53
2.2. Préparation des mélanges de polymérisation 53
2.3. Activation des parois du capillaire 53
2.4. Remplissage des tubes capillaires 54
2.5. Etape de gélification vieillissement 54
2.6. Echange de solvant (rinçage) 55
2.7. Séchage et calcination 55
3. Procédé de dépôt de film sur puces : 55
3.1. Système de remplissage des puces 55
4. Appareillage : 56
4.1. Test des puces de PDMS 56
4.2. Echantillons de gaz 56
4.3. Système d’acquisition 57
4.4. Test des colonnes et puces de monolithes 57
5. Caractérisation morphologique des gels : 58
5.1. Porosité et surface spécifique-Adsorption d’azote 58
5.2. Porosimétrie par intrusion de mercure 58
5.3. Microscopie électronique à balayage 59
5.4. Analyse thermique 60
6. Rappel des définitions des grandeurs chromatographiques mesurées : 61
6.1. Nombre de plateaux 61
6.2. Hauteur équivalente à un plateau théorique 62
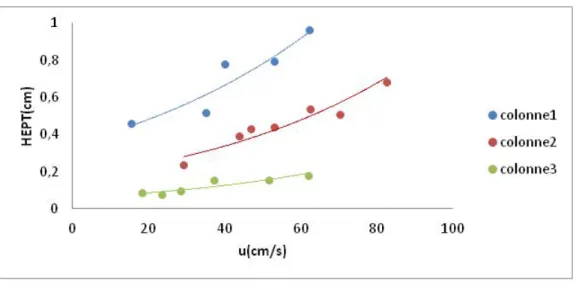
6.3. Courbes de Van Deemter 62
6.4. Facteur de rétention 63
6.5. Sélectivité 64
6.6. Résolution 64
6.7. Asymétrie d’un pic chromatographique 64
6.8. Ecart-type 65
VII
Chapitre 3: Expériences préliminaires
1.Contexte : 66
2. Comparaison de phases stationnaires originales adaptées aux colonnes CPG sur
puces 67
3. Conclusion : 72
Chapitre 4: Etude physico-chimique et morphologique des monolithes de
silice
1. Introduction : 73
2. Conditions de départ 74
2.1. Répétabilité de la synthèse 74
2.2. Effet de la température 76
2.3. Effet du temps de mélange 77
2.4. Effet de la réduction du ratio molaire TMOS/PEG 79
3. Limitations et perspectives d’optimisation: 80
4. Elaboration du nouveau procédé : 80
5. Légères variations autour du point zéro: 81
5.1. Analyse thermique des échantillons 83
6. Optimisation du mélange réactionnel : 85
6.1. Détermination des limites du domaine d’obtention de monolithes 85 6.2. Etude fine à l’intérieur du domaine d’obtention de monolithes 87 7. Effet de la concentration de l’hydroxyde d’ammonium : 89
8. Conclusion : 91
VIII
Chapitre 5: Synthèse dans les tubes capillaires
1. Introduction 93
2. Essais préliminaires 94
2.1. Synthèse des monolithes (point de départ 94
1.1. Réduction des inhomogénéités longitudinales 98
1.2. Réduction des inhomogénéités radiales 98
1.2.1. Activation 98
1.2.2. Influence de l’activation des parois du capillaire (75µm 100
1.3. Conclusion 100
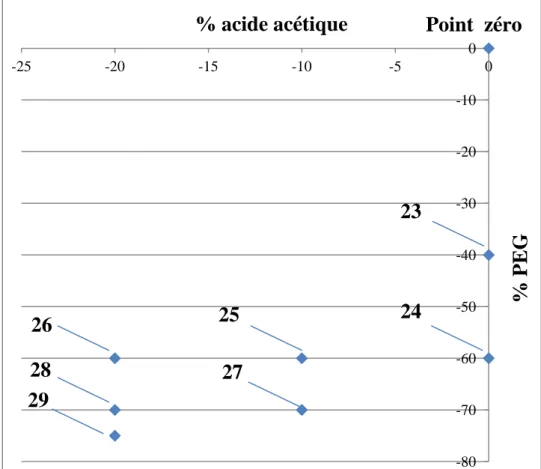
2. Modification du procédé d’Ishizuka au point zéro 101
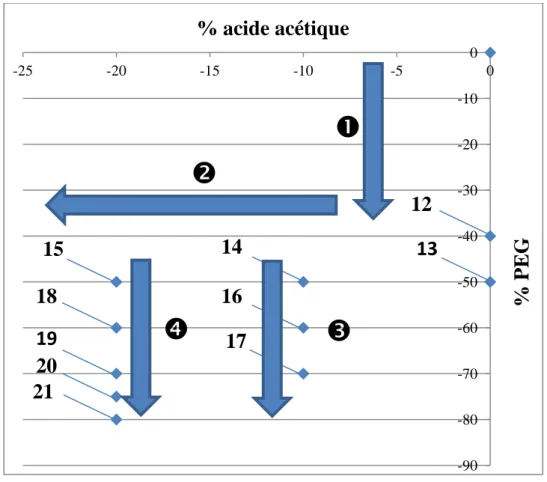
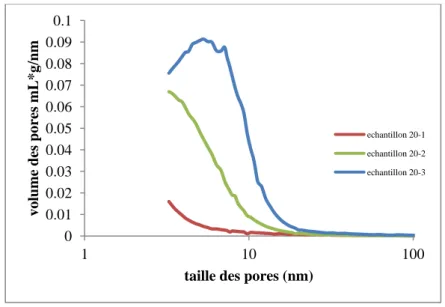
3. Optimisation de la synthèse des monolithes dans les tubes capillaires 104 3.1. Synthèse 23 (diminution de 40% de la quantité du PEG 105 3.2. Synthèse 24 (diminution de 60% de la quantité du PEG 109 3.3. Synthèse 25 (-60% de la quantité du PEG et -10% la quantité de l’acide 113 3.4. Synthèse 26 (-60% de la quantité du PEG et -20% la quantité de l’acide 116 3.5. Synthèse 27 (-70% de la quantité du PEG et -10% la quantité de l’acide 120 3.6. Synthèse 28 (-70% de la quantité du PEG et -20% la quantité de l’acide 124 3.7. Synthèse 29 (-75% de la quantité du PEG et -20% la quantité de l’acide 128
3.8. Amélioration de la perméabilité 130
4. Conclusion 133
5. Références bibliographiques 134
Chapitre 6: Transfert de la synthèse dans des puces
1. Introduction 135
2. Géométrie du microsystème 136
3. Mise en œuvre et adaptation du procédé de synthèse 136
3.1. La pulvérisation cathodique 137
3.2. Synthèse des monolithes 138
3.2.1. Activation de la couche de silice 138
3.2.2. Transposition de la synthèse 28 138
3.3. Amélioration du procédé 145
3.3.1. Elimination du porogène 145
IX
4. Conclusion : 150
5. Références bibliographiques 151
Chapitre 7: utilisation de routine des monolithes de silice en CPG : mythe
ou réalité
1. Contexte 152
2. Applications : 152
2.1. Séparations rapides d’hydrocarbures linéaires sur puce 153 2.2. Séparation d’hydrocarbures cycliques sur puce 154 2.3. Séparation de dérivés halogénés légers sur puce 154 2.4. Séparation d’hydrocarbures linéaires sur tube capillaire 155
3. Fidélité : 156
Conclusion générale 159
Perspectives 161
Annexe 162
XI
Abréviations
As Asymétrie d'un pic
ATD Analyse thermique
différentielle
ATG Analyse thermique
gravimétrique
CEC Capillary electrokinetic
chromatography
CPG Chromatographie en phase
gazeuse
R2 Coefficient de corrélation
DRIE Deep reactive ion etching
RSD Ecart-type relatif k facteur de rétention GC Gas chromatography h Hauteur du pic chromatographique H Hauteur équivalente à un plateau théorique [cm]
HPLC High performance liquid
chromatography
IUPAC International union of pure
and applied chemistry
MEB Microscopie électronique à
balayage MEMS Microelectromechanical systems N Nombre de plateaux théoriques KD Perméabilité [m2] PDMS Polydiméthylsiloxane
XII
PEG Polyéthylène glycol
Rs Résolution
tr Temps de rétention du
composé
TEOS Tetraéthyle orthosilicate
TMOS Tetraméthyle orthosilicate
TCD Thermal conductivity
detector
TAS Total analysis systems
UV Ultraviolet
u Vitesse de la phase mobile
(cm/s)
𝜔 Largeur à la base du pic
chromatographique
𝛿 Largeur à mi-hateur du pic
chromatographique
1 | P a g e
Introduction générale
Au co urs d es d ernières an nées, les exigences et l es b esoins d ans l e domaine d e l ’analyse chimique, qui se traduisent notamment par la demande toujours plus pressante de l’industrie pour réduire à l a fois les coûts et les temps d’analyse font de la miniaturisation un des défis majeurs pour la conception de systèmes toujours plus performants. Ces systèmes miniaturisés (micro-TAS pour micro-Total Analysis System), doivent intégrer les différents étapes de la chaîne a nalytique, de puis la p réparation de l ’échantillon jusqu’à la détection, et re quièrent pour l eur d éveloppement u ne ap proche co ncertée n écessitant d es co mpétences dans d es domaines v ariés q ue ce so it l a ch imie, les m atériaux, la micro f abrication ou l a microfluidique.
Dans ce contexte scientifique pluridisciplinaire, l’objectif de ce travail de thèse s’inscrit dans l’axe d édié au dé veloppement d’un c hromatographe en p hase gazeuse transportable, indépendant et aussi efficace que les chromatographes gaz conventionnels.
L’originalité d e n otre tr avail réside d ans l e ch oix d’un m onolithe de s ilice comme phase stationnaire en chromatographie en phase gazeuse. Constituant le cœur du système séparatif, cette phase stationnaire doit répondre au cahier des charges imposé par le format réduit de ces systèmes analytiques et des contraintes de la chromatographie en phase gazeuse notamment en pression.
Les monolithes à base de polymère inorganique ont été proposés dès le début des années 1990 par l e g roupe d e T anaka co mme matériau h autement p erméable p our la p réparation d e matrices d’extractions et de séparation pour la chromatographie liquide. L’attrait principal des monolithes i norganiques r éside cer tainement d ans l eur f acilité r elative de p réparation et d e mise en œ uvre. Les m onolithes p résentent u n in térêt m ajeur d ans la miniaturisation d es techniques séparatives puisqu’ils peuvent être synthétisés in-situ directement à l’intérieur des capillaires ou m icrosystèmes. I ls p ermettent ai nsi d e s’ affranchir d es contraintes technologiques liées à l’utilisation de particules qui nécessitent le remplissage homogène des colonnes capillaires.
Ces matériaux so nt donc préparé en u ne ét ape p ar p olymérisation in-situ d’un m élange composé d’un monomère qui peut être fonctionnalisé par la suite, d’un agent porogène qui
2 | P a g e après extraction donnera lieu à une porosité macroscopique indispensable à l’écoulement sans pertes de charge élevées du gaz vecteur (ou de la phase mobile) et d’un catalyseur qui peut être acide ou b asique pour h ydrolyser l e p récurseur en en tité f acilement p olymérisable. C e mélange multi-composant est introduit sous forme liquide dans un capillaire ou dans une puce chromatographique. La réaction de polymérisation est généralement initiée par voie thermique et ne nécessite aucun dispositif particulier. Les difficultés rencontrées pour le remplissage et la fabrication des frittés dans le cas des colonnes particulaires sont ainsi évitées. Le contrôle des c onditions d e p olymérisation co nstitue le p aramètre cl é p ermettant d ’optimiser la morphologie du m onolithe f inal qui est é troitement r eliée a ux pe rformances chromatographiques.
La di fficulté e t l ’aspect i nnovant de not re travail r epose s ur l a pr éparation d’ un monolithe compatible avec les contraintes de la chromatographie en phase gazeuse notamment en termes de pression et de stabilité thermique. Ainsi, le procédé de synthèse va devoir être adapté à une synthèse in-situ dans des colonnes de faible diamètre et/ou dans des canaux micrométriques de microsystèmes analytiques de type silicium-pyrex.
Le premier chapitre de ce manuscrit est une étude bibliographique et qui est divisée en deux parties :
La première partie de type « revue » traite les différentes phases stationnaires utilisées dans les sy stèmes d e CPG sur p uces. Une l iste la pl us exhaustive que possible de s phases stationnaires utilisées a été dressée et les différentes applications ont été revues en détail. Les performances de ces p hases stationnaires ont été présentées pour la séparation de différentes classes de produits.
La seconde illustre les atouts des phases stationnaires monolithiques de silice dans le contexte de l ’évolution d es t echniques sép aratives, au t ravers d e l eurs p erformances et de leurs multiples domaines d’applications. Un descriptif détaillé du procédé sol-gel employé pour la synthèse de ces monolithes, de l’ensemble des paramètres influents ce procédé, ainsi que des méthodes de caractérisation de ces phases stationnaires est ensuite présenté.
Le seco nd chapitre es t en tièrement co nsacré à l a d escription d es d ifférents p rotocoles d e synthèse ainsi que des dispositifs expérimentaux utilisés pour la caractérisation de ces phases stationnaires monolithiques de silice synthétisés en bloc dans des flacons.
3 | P a g e Le troisième chapitre c onsiste en u ne co mparaison ex périmentale d e p hases st ationnaires décrites pour la CPG sur puce. Les puces obtenues ont été évaluées vis-à-vis de leur aptitude à séparer les hydrocarbures légers.
Le quatrième chapitre est dédié au développement de monolithes synthétisés dans des flacons. Le but est d’évaluer morphologiquement la synthèse des monolithes avant de passer aux tubes capillaires e t a ux puc es de d imensions trop réduites. En a bsence de travaux t raitant de l’utilisation des monolithes de silice en CPG, nous sommes partis d’une synthèse décrite pour la chromatographie liquide. Etant donnée la multitude de paramètres à optimiser, nous nous sommes focalisés sur les paramètres les plus critiques du procédé sol-gel, citons le temps de mélange du mélange initial, la constitution de ce mélange, la température de vieillissement... Le c inquième c hapitre décrit le transfert d e l a sy nthèse dans des flacons vers l es t ubes capillaires. Si les synthèses de monolithes de silice dans des capillaires sont l’objet de très nombreuses recherches et publications, les exemples de mise en œuvre de ces monolithes en adéquation avec les contraintes de la chromatographie en phase gazeuse restent encore très marginaux. Les performances des capillaires remplis avec ces phases seront évaluées du point de vue c inétique e t thermodynamique en prenant un mélange d’alcanes co mme éch antillon test. . A notre c onnaissance, il n ’y a a ucun tr avail p ublié tra itant de l a mise en œ uvre en microsystèmes de monolithes de silice en chromatographie en phase gazeuse.
Le si xième ch apitre t raitera donc du transfert d e l a sy nthèse d e monolithes des t ubes capillaires v ers l es microsystèmes séparatifs. Plusieurs s olutions ont été p roposées p our s’affranchir aux différents leviers rencontrés.
Enfin, l e d ernier ch apitre m et l ’accent sur la p ossibilité d e l ’utilisation d e c es p hases stationnaires en CPG de routine. En effet, plusieurs applications de séparations rapides seront présentées. De pl us, une é tude de fidélité a é té menée su r l a sy nthèse la p lus ef ficace de manière à sa faire une idée de la variabilité synthèse à synthèse et colonne à colonne pour une même sy nthèse. P our t erminer, u ne b rève ét ude d e v ieillissement a ét é menée d ans l e b ut d’étudier le comportement de ces phases stationnaires après plusieurs mois d’utilisation.
Chapitre 1 ETUDE
4 | P a g e
1. Introduction:
Après une description générale des différentes classes de colonnes utilisées en chromatographie en phase gazeuse, une partie de ce chapitre bibliographique est consacrée aux différentes phases stationnaires utilisées dans des puces dédiées à la chromatographie en phase gazeuse. Cette partie correspond à une revue aussi exhaustive que possible des phases stationnaires adaptées aux puces de chromatographie en phase gazeuse allant des films de polydimethylsiloxane aux dépôt moins conventionnels (tels que la pulvérisation cathodique par exemple). Les différents types de phases stationnaires, leurs performances, leurs avantages et limitations sont largement discutées.
Ensuite, les solutions employées pour la programmation de température en chromatographie en phase gazeuse seront décrites. Les différentes techniques de chauffe appliquées sur les appareils conventionnels et sur puce seront détaillées.
Ce chapitre se termine enfin par une partie consacrée aux monolithes de silice : leur synthèse et caractérisation. Ainsi, après quelques rappels sur le procédé sol-gel classique, une description des adaptations de ce procédé afin de répondre aux exigences morphologiques et texturales liées à la chromatographie est effectuée. Une étude détaillée de l’influence des différents paramètres de synthèse sur les propriétés du matériau obtenu accompagne cette description.
5 | P a g e
2. Les différents types de colonnes utilisées en CPG :
La colonne est le cœur du chromatographe et c’est d’elle que dépend principalement le succès des séparations. On distingue deux types de colonnes (Figure I.1):
• Les colonnes remplies
• Les colonnes capillaires à films (épais ou mince) Chacun de ces types de colonnes sera décrit dans ce chapitre.
Figure I.1 : Comparaison des coupes d’une colonne remplie (3,2 mm ID) et d’une colonne capillaire (0,32 mm ID).
2.1. Les colonnes remplies
Les colonnes remplies (en anglais micropacked columns) représentent la première technologie de phases stationnaires développées, utilisées, et étudiées en chromatographie en phase gazeuse [1]. La Figure I.2 illustre l’agencement d’une colonne remplie de phase stationnaire particulaire. L’obtention de performances séparatives stables et reproductibles dépend de la qualité du remplissage et de l’immobilisation des particules.
Figure I.2 : Schéma d’une colonne remplie pour la chromatographie en phase gazeuse.
Ces colonnes remplies fonctionnent selon un mode de chromatographie dit « gaz-liquide »: le remplissage est constitué de particules d’un support inactif (particules) imprégnées de phase stationnaire liquide ou gomme. Cette imprégnation est obtenue en immergeant ces grains dans une solution de phase stationnaire dissoute dans un solvant, puis en évaporant le solvant. On distingue les supports siliceux poreux d’origine naturelle à base de diatomite (type
6 | P a g e
Chromosorb), les supports siliceux artificiels (type Sphérosil) et les supports non siliceux
(type carbone graphitisé).
La perméabilité étant proportionnelle au carré du diamètre des particules, l’utilisation de particules plus fines conduit vite à de faibles valeurs de perméabilité et donc à de fortes pertes de charge. Le Tableau I.1 rassemble quelques propriétés et domaines d’applications de plusieurs types d’adsorbants.
Adsorbant Matériau Tmax °C Applications
HayeSep A DVB-EGDMA 165 Gaz permanents, hydrogène, azote, Oxygène, NO, méthane, éthane HayeSep B DVB-PEI 190 Amines en C1 et C2, traces d’ammoniac
et d’eau
HayeSep C ACN-DVB 250 Analyse de gaz polaires (HCN, ammoniac, sulfure d’hydrogène
Porapak T EDGMA (PM) 190 Pour les composés très polaires, détermination de formaldéhyde dans l’eau
Porapak R Vinyl
pyrollidone
250 Séparation d’alcools linéaires et ramifiés Porapak P Styrene-DVB 250 Séparation d’une large gamme d’alcools,
glycols et carbonyles Chromosorb
101
Styrene-DVB 275 Acides, alcools, glycols, esters, cétones, Aldéhydes, éthers
Chromosorb 102
Styrene-DVB 250 Composés organiques volatils, gaz permanant, Alcools et eau
Chromosorb 104
ACN-DVB 250 Nitriles, nitroparafines, ammoniac, SO2,
Dioxyde de carbone, traces d’eau
Tableau I.1 : Quelques polymères adsorbants : DVB : divinylbenzène, EGDMA : éthylène glycol diméthacrylate,
PEI : polyéthylèneimine, ACN : acétonitrile.
Le coût des colonnes remplies est réduit et l’appareillage nécessaire, simple, peut même être rudimentaire. Ce type de colonne présente une grande surface spécifique comparée aux colonnes non-remplies, permettant une capacité d’injection plus élevée. Par ailleurs, ces colonnes permettent de bonnes efficacités en réduisant les dimensions des particules [2]. La possibilité d’utiliser de petites particules permet un transfert de masse rapide au travers de la phase stationnaire. Certaines séparations nécessitent l’emploi de phases stationnaires très spécifiques : gaz permanents, isomères insaturés d’hydrocarbures légers…, et des méthodes normalisées (ASTM E260, NF ISO 17494…), font encore appel aux colonnes remplies.
7 | P a g e Cependant, la mise en œuvre laborieuse du remplissage doublée de la difficulté à obtenir des frittés de bonne qualité et reproductibles limitent les performances de ce type de colonne. En effet, la présence de frittés peut également engendrer des contaminations. Une autre limitation de cette méthode est due à la forte pression nécessaire en tête de colonne. Il est donc difficile de remplir de longues colonnes ou d’utiliser des particules de faibles dimensions sans un dispositif adapté à de très hautes pressions [3]. De plus, leur efficacité est plus faible que celle des colonnes capillaires et leur adsorption résiduelle est très importante.
2.2. Les colonnes ouvertes
Appelées en anglais « Open tubular columns » ou « colonnes ouvertes » en français, les colonnes capillaires ouvertes regroupent toutes les colonnes ne contenant pas de remplissage granulé. Proposées comme une alternative aux colonnes remplies, les colonnes capillaires non-remplies se présentent sous la forme d’un tube capillaire ouvert de faible diamètre dont la paroi interne a été tapissée par une phase stationnaire, greffée ou adsorbée, d’épaisseur variable. On trouve plusieurs appellations comme WCOT pour « Wall Coated Open tubular », SCOT pour « Support Coated Open Tubular » ou PLOT pour « Porous Layer Open Tubular ». L’épaisseur du film de phase stationnaire et le diamètre de colonne jouent un rôle considérable sur la rétention, l’efficacité, la vitesse d’analyse et la stabilité thermique de la colonne. Les colonnes capillaires ouvertes offrent des solutions de séparation parmi les plus efficaces et les plus rapides. On distingue la chromatographie gaz-liquide dans le cas où la phase stationnaire est obtenue par évaporation d’une solution, de la chromatographie gaz- solide quand le tube est recouvert d’une fine couche d’adsorbant. Plusieurs méthodes ont été décrites pour attacher une phase stationnaire aux parois des tubes et chacune présente un grand choix de fonctionnalités disponibles ce qui permet de couvrir une large classe de produits à analyser.
Nous discuterons dans cette partie les différentes colonnes ainsi que leurs avantages et inconvénients.
2.2.1. Les films de phases stationnaires
Les films de phases stationnaires (Figure I.3) peuvent être des liquides, des polymères réticulés ou même des cristaux liquides, répartis en films minces, sur la paroi interne d’un tube capillaire. Ainsi, ce genre de phase stationnaire doit satisfaire à plusieurs conditions:
8 | P a g e • Inertie chimique : la phase stationnaire ne doit pas réagir chimiquement avec les
substances analysées. Dans certains cas l’interaction phase stationnaire-solutés se traduit par une disparition des pics, un dédoublement ou par l’apparition épaulements. • Stabilité thermique : une bonne stabilité de la phase stationnaire s’impose dans la
mesure où les séparations rapides sont souvent réalisées en gradient de température. Les signes de décomposition « bleeding » en anglais et ressuage en français, se traduisent par une dérive de la ligne de base, d’autant plus prononcée que l’on se rapproche de la température limite d’emploi de la colonne.
Dans une colonne ouverte, le terme appelé β, représente le rapport des volumes de la phase gazeuse et la phase liquide en fonction du diamètre de la colonne et de l’épaisseur du film (équation 1) 𝛃 =𝐕𝐆 𝐕𝐋 = 𝛑𝐫𝟐𝐋𝐜 𝟐𝛑𝐫𝐞𝐟𝐋𝐜 = 𝐝𝐜 𝟒𝐞𝐟 Équation I.1
Figure I.3: Colonne ouverte ayant une épaisseur de film ef et diamètre d.
Pour améliorer l’immobilisation de la phase stationnaire à la surface interne du capillaire, des fines particules adhérentes peuvent y être déposées, ce qui donne les colonnes SCOT pour Support Coated Open Tubular. Cependant, ce type de colonne est de moins en moins utilisé étant donné l’amélioration des techniques de dépôt et de greffage des phases stationnaires directement sur la paroi. Les propriétés ainsi que les domaines d’applications des principales phases stationnaires à base de films sont décrits dans le Tableau I.2.
9 | P a g e Nom de la phase composition Polarité Tmax °C Applications
SE-30, OV-1, OV-101, DB-1, SPB-1, BP-1,HP-1, ULTRA-1, RTx-1, AT-1, CH3 dimethyl apolaire 325 Universelle, non spécifique : amines, thiols, hydrocarbures SE-54, OV-23, DB-5, SPB-5, BP-5, HP-5, ULTRA 2, RTx-5, 5% phényl
95% dimethyl Peu polaire 325
Pesticides, composés semi-volatils, hydrocarbures halogénés OV-17, DB-17, SPB-7, BP-10, HP-17, RTx-17, AT-50 50% phényl 50% dimethyl Moyennement polaire 325 HAP, sucres, stéroïdes, médicaments, glycols, cholestérols DB-Wax, Supelsowax 10, Super-ox, CPWax-52, Stabilwax, BP-20, HP-20M, AT-Wax HO(CH2)nOH (PEG) Très polaire 260 Alcools, solvants très polaires, aldéhydes, glycols, aromatiques, huiles essentielles, composés basiques
Tableau I.2 : Propriétés des principales phases stationnaires commerciales.
2.2.2. Méthodes de dépôt du film de phase stationnaire
La formation du film de phase stationnaire sur la paroi interne du tube capillaire est obtenue par évaporation d’une solution. L’épaisseur de ce film, comprise entre quelques dixièmes de µm et quelques µm, doit être constante tout le long de la colonne. Ceci peut être obtenu, soit par la méthode statique, soit par la méthode dynamique.
La méthode dynamique simple consiste à introduire, par aspiration ou par refoulement, une solution relativement diluée de phase stationnaire sur une petite partie de la longueur du tube capillaire. En progressant, un film de solution se dépose à la surface. Il suffit ensuite d’évaporer le solvant pour obtenir un film de phase stationnaire. Pour éviter des irrégularités de remplissage (apparition de vagues, voire des gouttelettes), d’autres variantes ont été développées telle que l’utilisation d’un index de mercure [4].
La méthode statique, quant à elle, consiste à remplir complètement la future colonne d’une solution assez diluée de phase stationnaire dans un solvant volatil, puis à obturer une extrémité de la colonne et à faire le vide à l’autre extrémité [5]. A la surface du minuscule
10 | P a g e ménisque de solution, le solvant s’évapore sans bouillir sous la pression réduite et laisse sur la paroi un film régulier de phase stationnaire.
2.2.3. Epaisseur du film et incidences sur les propriétés de séparation
a. Colonnes à films mince
L’utilisation d’une colonne à film mince permet à la fois la réduction de la durée de l’analyse et un faible ressuage. Une diminution de l’épaisseur du film réduit le facteur de rétention mais permet une augmentation de l’efficacité. Ces colonnes sont adaptées à l’analyse des substances peu volatiles. Un exemple de séparation d’hydrocarbures est indiqué dans la Figure I.4.
Figure I.4: exemple de chromatogramme de séparation d’hydrocarbures (>C6) sur une colonne capillaire, 250 µm ID,
épaisseur du film 0,25 µm.
b. Colonnes à films épais
La rétention augmente en fonction de l’épaisseur du film de phase stationnaire (Figure I.5). Ainsi, les films épais sont très utiles pour retenir les composés très volatils. Une autre utilisation des films épais consiste en la réalisation de pièges capillaires pour les substances présentes à l’état de traces [6].
11 | P a g e Figure I.5 : colonne capillaire à film épais, ED=700 µm, ID=530 µm, épaisseur du film 145 µm adaptée de [7].
2.3. Colonnes PLOT
En chromatographie gaz-solide : les colonnes comprennent un film de particules d’un adsorbant de type charbon actif, gel de silice, alumine, polymère organique ou tamis moléculaire. Elles sont souvent réservées à l’analyse des gaz. Que ce soit du charbon actif ou ses dérivés (noir de carbone, noir de carbone graphitisé ou du graphite), ces supports ont des surface spécifiques considérables (jusqu’à 1000 m2/g) et ont montré d’excellentes séparations des composés non polaires (notamment la phase Carbopack de Supelco). On parle alors de colonnes PLOT et elles sont surtout utilisées pour l’analyse de gaz. Les phases stationnaires principales utilisées sont indiquées dans le Tableau I.3.
Phase stationnaire Tmax °C Applications
Alumine 200 Alcanes, alcènes, hydrocarbures aromatiques Gel de silice 250 Hydrocarbures (C1-C4), éthers volatils, esters
et cétones Tamis moléculaire
(5X et 13X)
350 Hydrogène, oxygène, azote, méthane. hydrocarbures C1-C3sur 5X et jusqu’à C12 sur 13X
Q 310 Hydrocarbures C1-C10
S 250 Solvants organiques volatils C1-C6
U 190 Composés nitrés, eau, gaz inorganiques
Tableau I.3 : Principaux adsorbants et leurs températures limite d’utilisation.
Les tubes ouverts ne nécessitent pas l’utilisation de frittés contrairement aux colonnes remplies. De plus, les phases stationnaires préparées, de chimie versatile, présentent souvent un faible ressuage aux températures élevées. L’utilisation de colonnes de faibles diamètres
12 | P a g e accélère l’échange phase stationnaire- soluté. Afin d’augmenter la surface d’échange entre les solutés et la phase stationnaire, des colonnes longues peuvent être utilisées, où il est possible d’avoir recours aux phases polymériques poreuses plutôt qu’à des monocouches moléculaires. L’utilisation de couches de phase stationnaire plus épaisses permettra alors l’utilisation de colonnes courtes ce qui accélère la séparation mais au détriment de l’efficacité. La Figure I.6 illustre la séparation d’hydrocarbures légers sur une colonne PLOT.
Figure I.6: exemple de chromatogramme de séparation d’hydrocarbures légers linéaires et ramifiés sur une colonne PLOT
0,32 mm ID, rampe de température 35°C à 240°C à 10°C/min puis 240°C pendant 10 min, He P=18 PSI.
3. Miniaturisation en CPG :
3.1. Intérêt de la miniaturisation
Une des tendances actuelles dans le domaine de l’analyse est d’intégrer les différentes étapes de l’analyse dans un seul dispositif automatisé. Ces systèmes sont souvent connus sous l’appellation TAS (de l’anglais « Total Analysis Systems »). La miniaturisation de ce type de systèmes (µTAS) présente un grand intérêt car les faibles dimensions facilitent l’intégration des différentes étapes ce qui augmente la vitesse des analyses (temps de cycle court). De plus, les dimensions réduites des µTAS présentent un intérêt particulier pour l’analyse de terrain de par leur facilité de transport. Un autre avantage de la miniaturisation est la faible consommation à la fois de l’échantillon et de réactifs éventuels ainsi que la consommation énergétique moindre. Enfin, la théorie prédit que la réduction des dimensions de la colonne de séparation devrait se traduire par l’amélioration des performances analytiques. C’est dans
13 | P a g e cette dynamique que différents projets de chromatographes en phase gazeuse sur puce ont été développés.
3.2. Méthode de fabrication des dispositifs miniaturisés
Le développement de microdispositifs est conjoint à l’utilisation de nouvelles méthodes de fabrication des systèmes à l’échelle du micromètre. Historiquement les techniques « Micro Electro Mechanichals systems », plus connues sous le sigle MEMS, dérivant directement des techniques de microfabrication ont été développées pour la microélectronique au cours des années 1970 dans l’industrie. Habituellement dans la fabrication des microsystèmes, on trouve des technologies utilisant le silicium [8-9], le verre [10], ou le plastique[11]. Principalement à base de silicium, les MEMS ont vu leur domaine d’application s’étendre aux méthodes analytiques grâce à la mise au point des méthodes de gravures profondes et de collages permettant de creuser des canaux dans un substrat puis de les refermer à l’aide d’un couvercle, l’étanchéité étant assurée par un processus de collage [12]. Plus récemment, des nouvelles méthodes de fabrication ont été développées pour satisfaire très rapidement à des besoins de réalisation de dispositifs à bas coût. Dans ce qui suit, nous allons décrire un ensemble de techniques de fabrication pour la réalisation de microcolonnes.
3.2.1. Méthodes conventionnelles
Rappelons que les méthodes conventionnelles de la microfabrication sont issues de l’industrie de la microélectronique. Pour réaliser un dispositif du type MEMS, les trois technique, i.e. la photolithographie (Figure I.7), la gravure (Deep Reactive Ion Etching, DRIE), et la soudure anodique sont essentielles [13-14]. On dépose d’abord une fine couche de résine photosensible. On utilise ensuite un masque optique et une lampe UV pour insoler localement la couche de résine. Cette étape est suivie par une révélation dans une solution chimique, résultant en une duplication des motifs du masque en négatif ou en positif selon le type de résine utilisée. Cette couche de résine est alors utilisée comme masque pour le micro usinage via une attaque chimique ou physique en phase liquide (gravure humide) ou gazeuse (gravure sèche). Après retrait de la résine, la plaquette contenant des microstructures gravées est finalement fermée par soudure anodique avec une plaque de pyrex. Ces techniques sont applicables à la production en masse de dispositifs microfluidiques de première génération composés de structures simples à réaliser (Figure I.8).
14 | P a g e Figure I.7.Gauche : Etapes de fabrication d’une puce chromatographique : (a) Dépôt d’une résine à l’aide d’une tournette
puis insolation par un faisceau parallèle d’U.V. (b) Photolithographie et gravure par le procédé DRIE (c) lift-off (d) collage anodique du wafer de silicium à une plaque en pyrex. Droite : Tournette (spin coater) pour étaler une couche de résine
photosensible sur un wafer en silicium.
Les dispositifs MEMS dits de deuxième génération, intégrant des actionneurs et des éléments de détection sur une même puce, sont en général beaucoup plus difficiles à réaliser. En principe, l’intégration de plusieurs types d’éléments sur la même puce ne doit pas poser de problème, mais la mise en œuvre beaucoup plus difficile ne peut être envisagée que pour une application à forte valeur ajoutée. La technique basée sur une technologie de micro usinage du silicium est un choix stratégique à long terme, puisqu’elle permet d’intégrer tous types de composants à base de silicium, microélectronique, micro-optique, micromécanique et microfluidique sur une même puce.
Figure I.8: Puce chromatographique de première génération.
3.2.2. Méthode non conventionnelle : La lithographie molle
La lithographie molle, plus connue sous sa désignation anglaise de « soft lithography », est une technique de microfabrication destinée à fabriquer ou reproduire des structures à partir de moules en élastomère, c'est à dire faits à partir de polymères présentant des propriétés élastiques [14-16]. Cette technique consiste à mouler un élastomère sur une surface micro-structurée. L'assemblage du bloc d'élastomère est ensuite collé sur un substrat choisi. Les
15 | P a g e interstices créés entre le substrat et l'empreinte des structures dans l'élastomère correspondent alors aux canaux microfluidiques (Figure I.9).
Figure I.9: Fabrication d'un dispositif microfluidique en PDMS. (a) Fabrication d'un moule par les techniques de
photolithographie à partir d'un masque transparent ; (b) Moulage de l'élastomère sur le moule. (c) Après démoulage de l'élastomère réticulé, les entrées et les sorties des fluides sont créées, il ne reste alors plus qu’à assembler le dispositif sur son substrat.
Ces dispositifs développés par lithographie molle sont de plus en plus utilisés dans les divers laboratoires scientifiques ou exploités par les industriels. Cependant, leur utilisation reste limitée à la microfluidique en phase liquide et demeure inadaptée aux gaz, notamment à cause de l’élasticité du polymère.
3.3. Phases stationnaires utilisées pour les MEMS en CPG
Cette section est présentée sous forme d’une revue. Elle décrit les phases stationnaires adaptées aux puces de chromatographie en phase gazeuse allant des films de polydimethylsiloxane aux dépôts moins conventionnels (telle la pulvérisation cathodique par exemple). Les différents types de phases stationnaires, leurs performances, leurs utilisations, leurs avantages et limitations sont largement discutés.
30 | P a g e
3.4. Programmation de température en chromatographie en phase gazeuse
En général, que ce soit en analyse de routine utilisant des chromatographes conventionnels ou en chromatographie sur puces, il est indispensable d’avoir recours à des rampes de températures rapides pour réduire le temps d’analyse. Dans les premières années de la chromatographie en phase gazeuse [17], un certain nombre de méthodes de chauffage de colonnes a été étudié. Beaucoup d’entre elles étaient basées sur la convection d’un fluide : bain de vapeur [17], bain d’huile [18] ou bain d’air [19]. La chaleur est ainsi transférée par convection à partir de la source de chauffe (par exemple une résistance électrique) vers la colonne, immergée dans un fluide. Plusieurs de ces méthodes ont disparu pour des raisons de sécurité ou d’ordre technique. Le four à bain d’air pour la CPG a été introduit au milieu des années 1950. Par rapport à l’huile et à la vapeur, l’air a une capacité thermique plus faible, mais cela permet des solutions technologiques propres, faciles à manipuler et n’ayant pas de limite supérieure de température de fonctionnement. Grâce à ces avantages, le four à bain d’air est devenu la méthode de chauffage standard pour la CPG dans les années 1960 et le reste encore aujourd’hui (Figure I.10).
Figure I.10 : schéma du four d’un chromatographe en phase gazeuse.
Des méthodes de chauffage par rayonnement ont été proposées. Gaisford et al. [20] ont conçu un four à micro-ondes destiné à une utilisation en chromatographie en phase gazeuse. Un système CPG basé sur ce principe a été commercialisé [21]. Walte et al. [22], quant à eux, ont développé un four à rayonnement infrarouge pour la CPG. La source de chauffe est une lampe à infrarouge pouvant atteindre des rampes de température de l’ordre de 1000°C/min.
31 | P a g e
3.5. Techniques de chauffage pour la chromatographie gazeuse sur puce
Les premiers chromatographes sur puces ont été conçus pour des analyses sur terrain [23-24]. Cependant, ces appareils ne comprenaient pas de système de chauffage des colonnes et les études portaient alors sur l’amélioration du procédé de fabrication et la réponse des détecteurs. L’absence de contrôle de température a limité leurs applications. Bien que le four du chromatographe soit souvent utilisé pour l’évaluation des performances des puces chromatographiques [25-27], une réflexion sur un autre moyen de chauffe s’est imposée. L’utilisation du four chromatographique est un obstacle contraignant à la portabilité (taille et consommation d’énergie). Le chauffage par résistance est le meilleur choix pour la micro chromatographie sur puces. Ainsi, les éléments chauffants peuvent être plaqués sur la puce ou directement incorporés et microfabriqués sur la puce (Figure I.11).
Figure I.11 : Chauffage et refroidissement d’un µCPG. (A) la colonne est chauffée par une résistance externe. (B) la colonne
est chauffée par une résistance gravée directement lors du procédé de fabrication. Adaptée de [28]
Les systèmes à effet Peltier sont généralement utilisés pour le piégeage et le refroidissement de la colonne en CPG. Ils peuvent également fonctionner comme des éléments chauffant. Cependant, ce dispositif, non résistif, est limité par une différence de température de 100°C entre le côté chaud et le côté froid. Ainsi, Lewis et al. [29] ont utilisé un film mince à base de polyimide comme dispositif de chauffage primaire et un système à effet Peltier comme élément chauffant d’appoint lors de la température programmée. Couplant la faible épaisseur de la puce (<500µm) à la faible masse thermique du polyimide, le chauffage ainsi que le refroidissement de la puce n’a consommé que 25 W. La température maximum atteinte était de 200°C.
32 | P a g e Des couches métalliques, résistives, peuvent être déposées sur la surface des colonnes microfabriquées pour les chauffer, grâce à une bonne conductivité thermique, une large gamme de températures et une faible inertie thermique. Généralement en Titane (Ti), il adhère facilement sur une couche de silicium [30]. D’autres procédés à base de chrome/or (Cr/Au) ont été rapportés [31]. Ce contact « intime » entre la colonne microfabriquée et la résistance permet des vitesses de chauffe extrêmement rapides (>2000 °C/min), ce qui est largement au delà des possibilités des fours CPG conventionnels (≈ 100 °C/min). La consommation d’énergie varie selon la taille et l’épaisseur de la puce (≈4 W/m). Le film métallique peut également fonctionner comme sonde de température ce qui est avantageux pour l’intégration du système [32].
4. Les phases stationnaires monolithiques
Il est peu connu que les premières colonnes monolithiques ont d'abord été utilisées en chromatographie en phase gazeuse (CPG) il y a plus de 30 ans [33-34]. Cependant, ils ne sont pas, à ce moment, apparus comme dignes d'intérêt car à la même époque Raymon et Dandeneau ont développé les colonnes capillaires ouvertes [35]. Grace au travail de pionniers de plusieurs groupes de recherche [36-38], les phases stationnaires monolithiques ont ré-émergé au début des années 1990, notamment pour des applications en phase liquide. Ces monolithes ont été préparés à partir de matériaux divers et dans une grande variété de formes (bâtonnets, disques…) [39]. Ce matériau tridimensionnel continu remplit entièrement le volume de la colonne et ne laisse pas de vides interparticulaires typiques des colonnes remplies. Sa structure est composée d’un squelette ayant un réseau poreux constitué de macropores interconnectés entre eux et de micro/méso- pores (par convention IUPAC [40], Ømicro ≤ 2 nm, 2 nm≤ Øméso ≤ 50 nm, Ømacro ≥50 nm). Le réseau macroporeux assure l’écoulement de la phase mobile à travers le réseau tridimensionnel, tandis que la texture micro/méso- poreuse du squelette contribue à la surface spécifique des monolithes et donc à la rétention chromatographique des solutés (Figure I.12). L’ensemble donne lieu à un flux favorisant un transfert de masse rapide des solutés entre la phase stationnaire et la phase mobile.
Les monolithes offrent de nombreux avantages par rapport aux colonnes particulaires comme l'ont démontré un grand nombre d'études publiées dans la littérature scientifique [41]. Cette activité a conduit à l'acceptation des colonnes monolithiques comme phase stationnaire chromatographique. Leurs applications en chromatographie liquide à haute performance
33 | P a g e (HPLC) et électrochromatographie capillaire (CEC) ont été récemment décrites dans plusieurs études [42-45].
(A) (B) (C) (D)
Figure I.12 : Structure poreuse de monolithes obtenue par images MEB. (A) mésopores, (B) macropores, (C) monolithe
inorganique (silice vierge), (D) monolithe organique (poly divinylbenzène) [46-47]
Les monolithes sont classés selon la composition chimique de base du squelette. On définit ainsi deux grandes catégories : les monolithes organiques et les monolithes inorganiques.
La Figure I.12 montre que les monolithes organiques (cf. contour bleu, D) présentent un squelette dense avec une géométrie globulaire ayant peu ou pas de texture micro-poreuse. Du fait de leur configuration plutôt macroporeuse (faible porosité et faible surface spécifique), ces monolithes sont plutôt adaptés aux séparations de macromolécules (bio- polymères, polymères synthétiques…). En revanche, les monolithes inorganiques sont moins compacts et leur squelette est plus homogène (cf. contour vert Fig, 12 C). Le système micro/méso- poreux (cf. contours rouge et jaune, Fig, 12 B et A) leur confère une surface spécifique plus importante que celle des monolithes organiques ce qui permet la diffusion de petites molécules dans la structure.
Du fait de leurs structures purement organiques, les monolithes polymériques présentent l’inconvénient de subir un effet de gonflement à haute teneur en solvants organiques contrairement aux supports inorganiques. En effet, les déformations élastiques irrégulières qui en résultent conduisent souvent à des problèmes de répétabilité de la rétention chromatographique.
4.1. Les monolithes inorganiques
Les monolithes inorganiques regroupent l’ensemble des réseaux dont le squelette est à base de silice (SiO2) [48] ou d’oxydes de métaux de transition, issus du groupe IV du tableau
périodique, tels que la zircone (ZrO2) [49], l’oxyde de titane ou l’oxyde de hafnium (HfO2)
34 | P a g e Seuls les réseaux monolithiques à base de silice ont été largement exploités en chromatographie liquide et en électrochromatographie et quasiment jamais en chromatographie en phase gazeuse [50-51]. Introduits en 1996 pour la chromatographie liquide [52-53], les monolithes de silice semblaient présenter un intérêt majeur pour les techniques séparatives mais leurs premières utilisations faisaient apparaître de nombreux problèmes de reproductibilité de synthèse. Les monolithes inorganiques sont obtenus par voie sol-gel, alors que les monolithes organiques sont obtenus par simple polymérisation [50]. A partir de 2001, et suite à l’amélioration du procédé sol-gel, les premières colonnes de silice monolithique commerciales étaient disponibles grâce à la technologie exclusive « Chromolith® » de la société Merck (KGaA, Darmstadt, Allemagne), puis « Onyx® » suite au rachat de la licence par Phenomenex (Torrance, CA, Etats Unis). La morphologie structurale de ces monolithes est constituée d’un squelette de 1,5 µm de taille, de macropores ayant 2 µm de diamètre et des mésopores ayant 13 nm de diamètre.
Comparés aux supports particulaires, les monolithes de silice offrent des performances hydrodynamiques (perméabilité) et cinétiques (efficacité) prometteuses pour leur emploi dans les techniques séparatives. Afin de mieux appréhender leurs atouts chromatographiques, les grandeurs indispensables à la caractérisation de leur perméabilité et efficacité sont rappelées brièvement dans le chapitre 2.
4.2. Synthèse et caractérisation des monolithes de silice
La première polymérisation sol-gel a été réalisée par Ebelmen, qui décrivit dès 1845 la conversion en verre solide de l’acide silicique exposé à l’air humide [54]. Cependant, le véritable commencement de la polymérisation sol-gel date des années 1930 avec l’utilisation pour la première fois, par la firme allemande Schott, d’un procédé sol-gel pour fabriquer des récipients en verre.
Les monolithes de silice qui nous intéressent sont synthétisés par voie sol-gel. Cependant les procédés classiques présentés dans la littérature et développés pour des applications en phase liquide devront subir de substantielles adaptations afin de prendre en considération les spécificités requises pour la chromatographie en phase gazeuse (porosité et perméabilité). Dans un premier temps seront présentés les principaux aspects liés à la synthèse de monolithes de silice par voie sol-gel en mettant l’accent sur les principales adaptations de ce procédé nécessaires pour disposer d’un matériau dont les caractéristiques morphologiques sont optimales pour les méthodes séparatives. Il s’en suivra une étude des différentes
35 | P a g e techniques de caractérisation (directes ou indirectes), permettant de définir au mieux ces matériaux.
4.3. Généralités sur le procédé sol-gel
Le principe du procédé sol-gel, autrefois appelé « chimie douce » repose sur l’utilisation d’une succession de réactions d’hydrolyse-condensation de précurseurs d’alcoxydes de silicium tels que le tétraméthoxysilane (TMOS) ou le tétraéthoxysilane (TEOS) en solution. L’adjonction d’eau permet l’hydrolyse des précurseurs de silice, souvent catalysée en milieu acide ou basique. Elle est suivie de polycondensation de produits d’hydrolyse en oligomères. Les réactions mises en jeu dans ce procédé sont présentées dans la Figure I.13.
OR Si OR OR OR OR Si OR OR OR H O H OR Si OH OR OR OR Si O H OR OR OR Si OR OR O OR Si OR H OR O H OR Si OH OR OR OR Si OR OR OR OR Si OR OR O Si OR OR OR H O R H O H OR Si OR OR O R H O H OR Si OH OR OR Si O Si OR OR OR OR OR OR Si O Si OR OR OR OR OR OR + + + + H2O + + ROH (1) (2) (3) -+ - + ROH + -+
-Figure I.13 : La réaction du procédé sol-gel.
Au cours de cette croissance des oligomères, on assiste à une fluctuation progressive de composition du milieu conduisant d’un côté à la formation de zones riches en silice qui constitueront le squelette du monolithe et de l’autre côté à la formation de zones riches en solvant qui formeront les pores du monolithe. Ce phénomène constitue la séparation de phases due à la décomposition spinodale. Cette dernière est précédée, accompagnée ou suivie d’une étape de gélification (transition sol-gel) qui figera le matériau à un moment donné de l’évolution de cette séparation de phases.
La structure ainsi obtenue sera conditionnée par les cinétiques relatives de ces deux processus ainsi que par la taille des oligomères au début de la séparation de phases. Nakanishi et al. [55] ont montré que pour une même cinétique de séparation de phases et de transition sol-gel, la taille des oligomères (liée à la composition initiale du mélange réactionnel) lors du début de la
36 | P a g e séparation de phases influence fortement la morphologie finale du matériau. Le maillage du réseau devient de plus en plus dense dans l’étape de vieillissement du gel (Figure I.14).
Figure I.14 : Evolution de la morphologie du matériau au cours de la séparation des phases en fonction de la taille des
oligomères [55].
4.4. Procédé sol-gel dédié à la chromatographie
En fonction des conditions de réaction (vieillissement, séchage…) ou de traitement post-synthèse (traitement thermique…), le procédé sol-gel permet d’aboutir à des matériaux bien distincts en termes de propriétés morphologiques et/ou physico-chimiques.
La chromatographie implique l’emploi de matériaux à la fois macroporeux (assurant la diffusion des solutés) et mésoporeux (surface spécifique). Le procédé sol-gel doit donc subir quelques adaptations dont l’introduction d’un porogène pour générer la macroporosité et l’introduction d’une étape permettant de générer des mésopores de taille adaptée à la chromatographie quelle que soit sa nature (Figure I.15).
37 | P a g e Nakanishi et al. [57] ont introduit le polyéthylène glycol comme porogène afin de générer la macroporosité du matériau. Ce polymère est introduit pour favoriser la séparation de phases au cours du procédé sol-gel avec création d’une phase gel riche en complexe « silice-polymère » et une phase riche en solvant.
Dans le détail, les différentes étapes du procédé sol-gel sont :
4.4.1. Hydrolyse
Le mélange réactionnel classique réalisé en milieu aqueux en présence est constitué d’un acide (CH3COOH 0,01 M) pour catalyser l’étape d’hydrolyse, d’un précurseur de silice
(TMOS ou TEOS) et d’un polymère organique inerte tel que le PEG (polyéthylène glycol) utilisé comme porogène pour générer les macropores. Ce dernier doit être soluble en milieu aqueux et capable de former d’un côté, des liaisons hydrogènes avec les silanols, et de l’autre, d’établir des interactions répulsives avec la phase sol pour favoriser la séparation de phases (Figure I.16).
4.4.2. Condensation et gélification
Les réactions (2) et (3) de la Figure I.13 sont mises en jeu dans cette étape où la croissance du polymère s’accompagne de fortes interactions entre les groupements silanols et le PEG. Au cours du temps, on assiste à une diminution progressive de la solubilité du complexe silice-PEG dans la phase sol, conduisant à un enrichissement en silice de certaines zones du mélange. Ce phénomène est à l’origine d’un mécanisme de séparation de phases qui conduit, d’un côté à une structure riche en complexe silice-PEG, et de l’autre côté à une phase riche en solvants. La Figure I.16 illustre l’évolution de la structure du milieu au cours de la séparation de phases en fonction du temps dans le cas de l’obtention d’une structure macroporeuse. Cette structure évolue par accroissement de la taille du squelette et par augmentation de l’écart de composition entre les deux phases. Le milieu passe progressivement d’une structure nanoporeuse à une structure co-continue et homogène. La polymérisation se poursuit même après l’induction de la séparation de phases, et le squelette s’épaissit davantage. Dans le même temps, le solvant est exclu vers la phase sol qui s’accroit également. On peut aboutir au final, à une fragmentation des domaines (avec domaine = macropore + squelette) et à la formation de particules agrégées (t > 75 min).
38 | P a g e Figure I.16 Evolution de la structure du monolithe en fonction du moment d’intervention de la gélification lors de la
séparation de phases. t = 10 min à t = 75 min : images réalisées au microscope confocal a balayage laser (MCBL), échelle = 50 μm. Adaptée de [58]
Au cours de la polycondensation, lorsque le réseau tridimensionnel devient fortement réticulé, une gélification se produit dans le milieu (transition sol-gel), figeant ainsi la séparation de phases à un certain stade de son évolution. Si la transition sol-gel intervient au tout début de la séparation de phases, on obtiendra un réseau plutôt « nanoporeux ». A contrario, une gélification tardive peut entrainer la formation de particules (agrégats) au lieu d’un réseau « co-continu ». Cela indique que la morphologie du monolithe (macroporosité et taille du squelette) est essentiellement fixée par les cinétiques relatives de la séparation de phases et de la gélification. Celles-ci dépendent largement des propriétés physico-chimiques de la phase sol ainsi que des conditions opératoires adoptées. Les performances chromatographiques (rétention, efficacité, résistance au transfert de masse) étant directement liées aux caractéristiques morphologiques du matériau, il est important de les contrôler de façon judicieuse [58-59] . Il s’en suit une étape de vieillissement (à 30 ou 40°C) qui intervient après le point de gélification. Cette étape implique des changements structurels, essentiellement au niveau du réseau mésoporeux si le gel est maintenu en solution. En effet, même si la taille de la macroporosité et du squelette est globalement fixée, des monomères et des oligomères peuvent continuer à réagir dans le liquide interstitiel.
La synérèse ou contraction du monolithe est due à la formation de nouvelles liaisons par condensation de groupements silanols de surface du squelette, ce qui entraine une contraction du réseau pouvant aller jusqu’à 30% en volume avec des gels de silice [60-61]. Afin de lutter contre cette contraction du réseau, Motokawa et al. [62] ont suggéré en 2002, dans le cadre de la synthèse in-situ de monolithes en format capillaire, l’introduction avec le TMOS d’un co-précurseur de silice : le methyltriméthoxysilane (MTMS) avec un ratio de 3/1 (v/v) Les groupements CH3 du MTMS permettent de diminuer la densité de silanols à la surface et
39 | P a g e entrainent une réduction de la condensation, se traduisant par une diminution du phénomène de contraction du monolithe.
4.4.3. Restructuration de la microporosité
Les monolithes obtenus par le procédé sol-gel de Nakanishi et al.[55] ne possèdent pas de mésopores mais uniquement des micropores et des macropores. Afin d’obtenir des mésopores (≈ une dizaine de nanomètres), une étape de restructuration des micropores en mésopores peut être effectuée.
Il s’agit d’équilibres de dissolutions-reprécipitations sélectives de la silice mis en jeu en milieu basique (mûrissement d’Ostwald). Ce milieu basique est généralement obtenu en remplaçant le milieu réactionnel par l’hydroxyde d’ammonium (1 M à 120°C pendant 9h) [59]. En 2000, Tanaka et al. [63] simplifient leur mode de synthèse en introduisant l’urée dans le mélange initial à la place de l’hydroxyde d’ammonium pendant l’étape de lavage afin de générer cette mésoporosité lors du traitement hydrothermique. L’avantage engendré par l’utilisation de l’urée se situe dans la suppression de l’étape d’échange de solvant avec l’hydroxyde d’ammonium. Le pH, initialement à une valeur avoisinant 3-4, augmente au fur et à mesure de l’hydrolyse de l’urée en ammoniaque sous l’effet de la température, pour atteindre un pH avoisinant 11-12 permettant ainsi la restructuration du réseau poreux.
La Figure I.17 montre l’accroissement du diamètre des mésopores ainsi que l’obtention d’une distribution plus large de ces mésopores avec l’augmentation du temps de traitement à l’urée à 120°C.
Figure I.17 : Distribution relative de la taille des pores d’un monolithe de silice en fonction de la durée de traitement
40 | P a g e
4.4.4. Elimination du porogène
Après vieillissement du monolithe, comprenant ou non l’étape de restructuration de la microporosité, il est nécessaire d’éliminer le porogène lié aux agrégats de silice par des liaisons hydrogènes. Cette dernière étape du procédé sol-gel peut se faire de deux manières différentes :
4.4.4.1. Par calcination
Cette étape est réalisée à haute température (~600°C) et permet ainsi la décomposition du porogène. Cependant, elle doit être précédée d’un séchage souvent critique. En effet, une pression capillaire du liquide sur le monolithe peut engendrer un « stress » lors de l’évaporation du solvant et ainsi faire apparaître des craquelures au sein du réseau monolithique [64-65]. Ce stress permettra à des groupements silanols de se rapprocher et ainsi se condenser pour former un pont siloxane entraînant la contraction du réseau de silice. De plus, ces forces de pressions capillaires peuvent entraîner la destruction des pores eux-mêmes dans le cas des pores de très petits diamètres.
L’étape de calcination est réalisée (à l’issu du séchage) durant 24 h à une température de l’ordre de 600°C dans le cas des macro-monolithes de silice et inférieure à 330°C dans le cas de la synthèse in-situ dans des capillaires (température relativement basse pour éviter la décomposition de la gaine polyimide du tube capillaire)[60].
4.4.4.2. Par lavage
Puy et al. [66] ont remplacé l’étape de séchage-calcination par une étape de lavage avec un solvant polaire (eau [67], méthanol [68]) susceptible de générer des liaisons hydrogènes et ainsi favoriser l’élimination efficace du porogène (>70%) tout en maintenant une bonne stabilité du monolithe de silice.
5. Influence des différents paramètres sur la morphologie du monolithe de
silice :
La structure globale du monolithe (squelette, macro et méso- pores) est dictée par la nature et les proportions relatives des réactifs utilisés dans le mélange réactionnel initial ainsi que par les conditions opératoires mises en œuvre dans le procédé sol-gel.
La Figure I.18 illustre l’évolution de la morphologie du monolithe en fonction de la composition initiale d’un mélange ternaire composé de silice (précurseur), de solvant et de