Modification of 15q11 — q13 DNA methylation imprints in unique Angelman and Prader — Willi patients
6
0
0
Texte intégral
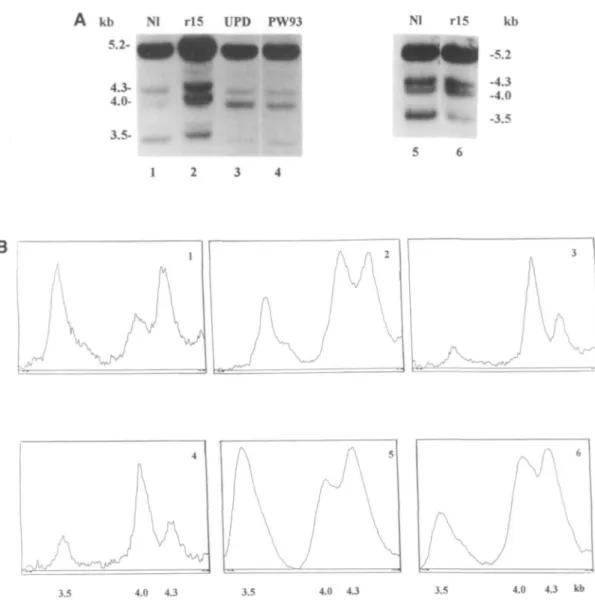
Figure


Documents relatifs