Identification of differentially expressed genes from multipotent epithelia at the onset of an asexual development
11
0
0
Texte intégral
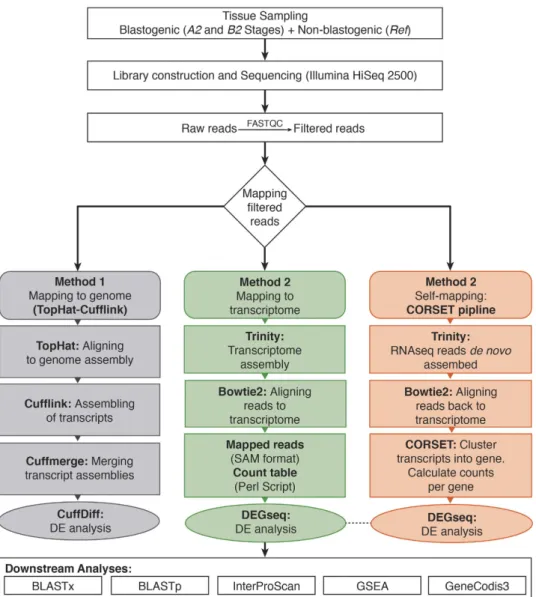
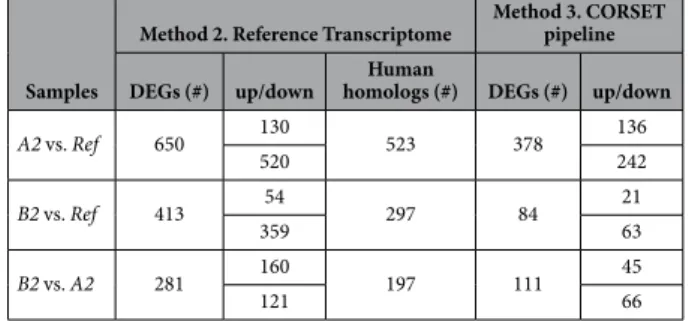
Figure

Documents relatifs
