The interaction of iron and the genome: For better and for worse
Texte intégral
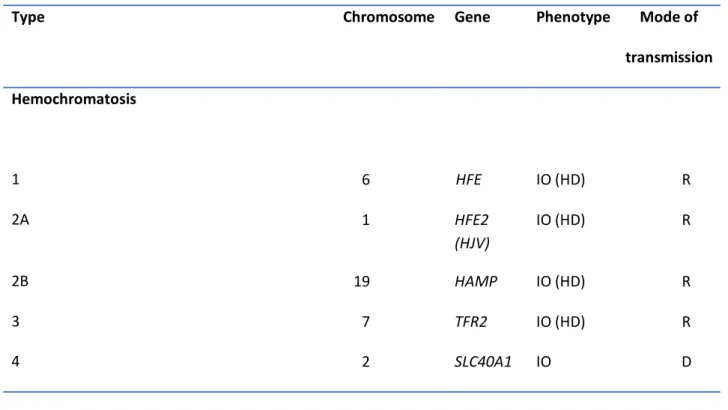
Figure
Documents relatifs
Afin de représenter explicitement une auréole autour des granulats recyclés et en même temps économiser le "coût numérique", à l’échelle mésoscopique une épaisseur de
- Le vingt quatrième Président des USA a été Président deux fois d’une façon discontinue (le 22ème et le 24ème) ; Il a précédé et succédé au petit-fils du neuvième
4. Disponibilité de la version PDA de Micromedex avec prochaine mise à jour 2. Bonne idée d’acheter le bouquin sur JCAHO quand l’édition de 1998 sera renouvelée pour avoir une
On s’intéresse au nombre de clients au bureau de poste de La Rivière des pluies à Sainte- Marie. Une étude statistique montre qu’en moyenne, pendant les heures d’ouverture, il
Abstract.- The binding state of tin atoms segregated at the grain boundary of fine grained iron and iron alloys is investigated by Mossbauer source experiments.. It is found that
We were able to highlight the influence of some milling parameters on the morphology, size and distribution of various powders constituting our alloys based on
The total model combined the calculations of the heat-mass transfer model pre-.. Meat is a multi-composite structure and juice expulsion during cooking is the result of
The identification of the anisotropy energy of nickel with the energy states near the point X turns out to be successful : it results in a value for the first
