HAL Id: tel-01902746
https://tel.archives-ouvertes.fr/tel-01902746
Submitted on 23 Oct 2018HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
thaliana in a spatially heterogeneous environment
Lea Frachon
To cite this version:
Lea Frachon. Ecological genomics of adaptation of arabidopsis thaliana in a spatially heterogeneous environment. Cell Behavior [q-bio.CB]. Université Paul Sabatier - Toulouse III, 2017. English. �NNT : 2017TOU30098�. �tel-01902746�
Parce que je n’étais pas faite pour les études d’après ma prof de Terminale… Je tiens à remercier toutes les personnes qui ont cru en moi et qui m’ont soutenu lors de ces trois années de thèse.
Je tiens d’abord à remercier mon jury de thèse Christophe Thébaud, Irène Till-Bottraud, Joëlle Ronfort, Juliette De Meaux et Maxime Bonhomme, de prendre le temps d’évaluer mon travail. Je tiens également à remercier tous les membres de mes deux comités de thèse qui ont été plein de bons conseils ; Nina Hautekèete, Dominique Roby, Valérie Le Corre et Mathieu Gautier. Enfin je remercie les financeurs de ma thèse : la région Midi-Pyrénées et le LabEx TULIP.
Je continue ces remerciements par toi Fabrice, sans qui cette incroyable aventure aurait été complètement différente ! J’ai toujours une phrase en tête, entendu lors d’une pause au GEPV : « les étudiants en thèse avec Fabrice ont énormément de chance ». A l’époque, j’étais à mille lieux de penser qu’un jour c’est moi qui aurai ce privilège. En effet, tu m’as fait confiance dès le début alors que je n’y connaissais pas grand-chose en génomique ! Merci de m’avoir soutenue tout au long de ces trois ans dans mes projets scientifiques (ma thèse, l’organisation du colloque SPE, la création de l’AJS, la vulgarisation scientifique) mais également dans mes projets personnels (notamment avec la rénovation de la « pénichette »). Tu as toujours été très présent, notamment sur le terrain et en cette fin de thèse. Je ne pense pas qu’il y ait beaucoup de directeurs de thèse qui prennent autant de temps pour leurs étudiants. Pour tout cela je te remercie infiniment. Et « oui » tes doctorants sont privilégiés !
Affronter cette thèse a était rendu possible grâce à toute une équipe que je remercie grandement, par ordre de rencontre:
- Etienne ; merci d’avoir égaillé mes premiers mois de thèse avec ta bonne humeur et ton humour, tes « conneries » m’ont quand même bien manquées après !
- Claudia ; Ah Claudia !!! Qu’aurai été cette thèse sans toi ! Déjà scientifiquement, tu m’as beaucoup apporté et tu as toujours été là quand j’en avais besoin. Et puis viens notre amitié qui a commencé lors de nos pauses sportives assez intensives. Merci pour ta présence, tes nombreux conseils et tous ces bons moments passés ensemble.
- Cyril ; merci pour ta gentillesse et ta bonne humeur au quotidien. Tu as toujours le petit mot qui fait plaisir et valorise. Et puis parce que la vie à part le « poney-piscine », c’est aussi « tapply », merci !
- Baptiste ; merci pour ta patience pour m’expliquer la bio mol… Et merci pour ton aide sur le terrain.
- Jaishree ; Thanks for your happiness those last few months ! Have a good luck for your thesis!
- Je tiens aussi à remercier tous les stagiaires qui sont passés dans l’équipe ; Taeken, Arnaud, Mylène, Eve, Kevin et Thomas.
Parce que si j’en suis ici, c’est un peu votre faute… un immense merci à vous Nina et Yves. Mille mercis de m’avoir mis sur les voies de la recherche, sans vous je n’aurais surement jamais osé pousser les portes d’un labo ! Un remerciement tout particulier à Nina que je considère un peu comme ma mère scientifique. Tu as été là dans des moments difficiles et tu m’as toujours
«reboosté». Que ce soit avec les huiles essentielles à ma soutenance de M2, pour ma recherche de thèse et maintenant de postdoc ou dans ma vie perso. Merci !
Je tiens également à remercier toutes les personnes avec qui j’ai pu collaborer durant cette thèse ;
- Merci Mathieu Gautier de m’avoir formé à BayPass et surtout d’avoir toujours été ultra réactif à toutes mes questions.
- Merci beaucoup Dominique Roby pour ton aide sur toute la partie gènes candidats.
- I greatly thank Annie Schmitt for your hospitality in Davis laboratory during two months, your kindness, the time you took for me and the very constructive scientific exchanges. Thank you also to Miki and the Schmitt team. Thank you too Sharon Strauss, Jennifer Gremer and Daniel Runcie for your helpful scientific advices.
- Merci également à Jérôme Gouzy et Sébastien Carrère pour la partie traitement bioinfo, et merci à Ludo pour ta patience !
Je remercie les directeurs successifs du laboratoire LIPM Dominique Roby puis Claude Bruand. Je tiens également à remercier de manière plus générale tout le personnel du laboratoire, notamment ;
- Fabrice, Claudette, Jean Luc, Marine pour toute leur aide sur les différentes manips. - Christophe et Philippe pour la fabrication du quadrat pour le terrain.
- L’ensemble du service gestion, notamment Christophe que j’ai très souvent sollicité pour mes ordres de missions en pagaille …
- Soon et Isabelle qui ont toujours été là pour mes petits problèmes informatiques ! - Dominique, Christian et Florent pour votre gentillesse.
- Ariane, Aude, Thomas, Vincent, Pierre D et Lucas merci de votre enthousiasme pour l’organisation des journées SPE ! Ce n’était pas toujours facile, mais on a géré !!
- Merci également à Alissounette, Camille, Popo, Pierre B, Gaelle, Corine, Eliane, Mireille, Marielle, Carine, Medhi, Sylvie, Céline, Richard, Marta, Patrice, Fabienne, Sandra, Alice… - Merci à tous les membres fondateurs de l’AJS, cette aventure a été formidable. Merci Harold
(pour ta motivation à toutes épreuves !), Aude, Manon, Hélène, Mathilde, Rémi, Franck, Ariane….
- Florence et Shérifa, les femmes de ménages qui, tous les jours, passent avec un sourire et toujours une petite attention.
Finir cette thèse a également été possible grâce à de nombreuses personnes extérieures à la science ;
Merci donc à tous mes amis pour vos soutiens pendant ces trois années.
- Les « plus anciens » ; Poupoule, Delphine, Yohan, Réjou, Clémentine & Seb, Laure & PY. - Les « Lillois » : Zaza, Julie, Florence, Arnaud, Nico, Justine, Sophie, Cynthia & Pierre,
Benjamin, Franciane.
- Les « dérivés du labo » : Alissounette (merci pour ta folie ! Du premier aux derniers jours de ma thèse tu auras été d’un grand soutien), Camille, Popo (toujours pétillante et pleine d’attentions), Carlos, Pipou…
- Tous les grimpeurs, merci de m’avoir permis de m’évader dans la bonne humeur ; Ivanna (tout simplement mille mercis d’avoir été présente pour moi, tant de chose partagées ces deux dernières années), Anne, Clarisse, Quentin (Merci pour ces ptites pauses en fin de thèse indispensable à ma survie !), Jérôme, Sylvain T, Loïc, Adrien, Marie, Maud, Sylvain F (merci pour les tableaux renversés), David R, David P, Thomas, Yohan, Alice, Maria, Lisa et tous les autres. Merci à vous tous, vous êtes supers !!!
- Les « runneurs » du midi, indispensable pour se vider la tête : Claudia, Romain, Popo, Adrien, Marina, Yolaine, Mathew…
- Les musiciens ; Monsieur Massot, Catherine, Harold, Julien (Merci de m’avoir soutenu la dernière année ; toujours les bons mots pour me redonner confiance dans des moments clés de ma thèse !)
- Les Américains ; Jared, Monica, Hayley & Masha, Creg & Molly, Corey, Logan, Marla, Dana & Kent…. Thank you for all, you’re amazing !
- Enfin un immense merci à tous mes voisins portuaires qui ont été d’un formidable soutien les dernières semaines, vos sourires et vos attentions m’ont vraiment aidé à tenir le coup jusqu’au bout… Merci : Mélo, Alvaro, (Pascal et Valérie)x2, Abde, Greg, Quentin, Manu, Baptiste, Victor, Xavier, Mumu, Patrice, Michel, Anique, Christophe, Thibaut, Phiphi, Guy, Rosalia, Jean-François, Sisi, Carine, Agnès, Céline, Luc, Eric, René… Un merci ++ à Céline pour les massages shiatsu qui m’ont bien détendu ;) et enfin un grand merci à Noé qui m’a soutenu ces derniers mois, tu as été d’une gentillesse et d’une patience extrême avec moi !
Je finirai ces remerciements par ma famille. Un immense merci à « mes parents Toulousains » Thibault et Solenn ! Sans vous et vos princesses ma vie ici aurait été tout autre ! Merci pour tout ce que vous avez fais pour moi pendant ces trois années, c’est juste inestimable ! Merci à Cléanne et Elia, j’en suis complètement gaga, elles sont formidables !
Enfin, je remercie ma famille de leurs soutiens à toutes épreuves depuis de longues années ! Merci au G4 + bop’ power indispensable à ma survie ! Merci à Noé(mie!) et Margot d’être toujours là pour moi, c’est tellement important de savoir que je peux compter sur mes sœurs n’importe quand ! Merci Sylvain d’être là pour toutes les trois et de m’avoir un peu poussé à aller à la fac après la prépa ! Merci papa pour avoir accepté ce défi un peu fou de retaper le Lenoma qui m’a permis de vivre dans un cadre exceptionnel. Merci à toi et Lucile pour tous ces petits moments d’évasion à la Source. Enfin, un grand merci à toi maman d’avoir toujours cru en moi, de toujours nous avoir poussé vers le haut et d’avoir voulu pour moi et mes sœurs ce qu’il y avait de meilleur.
« C’est un beau fruit, mais il n’est pas mûr, et nous serons morts avant que le soleil de la pratique et
de l’expérience ne l’ait mûri »
I.
Introduction générale
A.
Adaptation aux changements globaux
……… 1Importance de l’effet des changements globaux sur la biodiversité ……….. 1
Se rapprocher d’un nouvel optimum phénotypique via la plasticité phénotypique ……… 4
Migrer pour fuir un milieu devenu défavorable ……….. 5
Adaptation locale via la sélection génétique ………. 6
B.
Identification des bases génétiques de l’adaptation
…….……….. 81. Méthodes d’analyses génome-environnement …………..………..…… 10
Approche individu-centré ……….. 11
Approche populationnelle ………... 12
Nécessité de tenir compte des pressions de sélection non seulement abiotiques, mais aussi biotiques ……… 15
2. Méthodes de QTL mapping sur des traits supposés adaptatifs……….……. 16
Replacer les études de QTL mapping dans un contexte écologiquement réaliste ……….… 22
C. Importance de l’échelle géographique considérée
……….. 24D. Objectifs de la thèse et modèle d’étude
………..……… 26Arabidopsis thaliana : une espèce modèle en génomique environnemental et en génomique écologique ………. 28
E. Plan de thèse
……….… 30II. Chapitre 1: Identification des pressions de sélection
potentielles agissant sur A. thaliana à une échelle
régionale
A. Introduction
………...…… 32Identification de 168 populations naturelles d’A. thaliana ………. 33
Caractérisation phénotypique ………. 34 Caractérisation écologique ……… 35 1. Climat ……….. 35 2. Sol ……….. 36 3. Communautés végétales ……… 37 4. Communautés microbiennes ……….. 39
B. Manuscrit en préparation: The putative selective agents acting on
Arabidopsis thaliana depend on the type of habitat
………..…... 41III. Chapitre 2: Identification des bases génétiques associées
aux agents sélectifs potentiels et étude de leurs signatures
de sélection
A.
Introduction
……….……….………. 74B.
Manuscrit: A genomic map of adaptation to local climate in
Arabidopsis thaliana
……….. 76Supporting information
………... 109C.
Manuscrit: Adaptation to plant communities across the genome of
Arabidopsis thaliana
……… 123Supporting information
………... 146D. Conclusion
……….…... 154IV. Chapitre 3: Evolution phénotypique et génomique d’une
population naturelle d’A. thaliana dans un habitat
spatialement hétérogène
A. Introduction
………. 156B. Manuscrit: Intermediate degrees of synergistic pleiotropy drive
adaptive evolution in ecological time
………..… 158Supporting information
………... 197C. Conclusion
………. 247V. Conclusion générale
………... 248Importance de considérer les stratégies reproductives dans la définition de la fitness ………. 249
Prendre en compte le réalisme écologique des populations passe par considérer le maillage complexe des agents sélectifs ……… 250
Architecture génétique de l’adaptation chez A. thaliana ……….. 252
Perspectives ……….. 254
VI. Bibliographie
……….... 260Publications scientifiques
NB: Les articles marqués d’une étoile sont directement liés à ma thèse
* 1. Frachon L.*, Libourel C.*, Villoutreix R., Carrère S., Glorieux C., Huard-Chauveau C.,
Navascues M., Gay L., Vitalis R., Baron E., Amsellem L., Bouchez D., Vidal M., Le Corre V., Roby D., Bergelson J. & Roux F. Intermediate degrees of synergistic pleiotropy drive adaptive
evolution in ecological time (Re-soumis à Nature Ecology and Evolution). *Authors
contributed equally to this work.
* 2. Bartoli C.*, Frachon L.*, Barret M., Rigal M., Zanchetta C., Bouchez O., Carrère S. & Roux
F. In situ relationships between microbiota and potential pathobiota in Arabidopsis thaliana
(Invitation à resoumettre dans eLife). *Authors contributed equally to this work.
* 3. Frachon L.*, Bartoli C.*, Carrère S., Bouchez O., Chaubet A., Gautier M., Roby D., Roux F.
A genomic map of adaptation to local climate in Arabidopsis thaliana (En révision dans New
Phytologist). *Authors contributed equally to this work.
* 4. Frachon L.*, Mayjonade B.*, Bartoli C.*, Hautekeete N.C. & Roux F. Adaptation to plant
communities across the genome of Arabidopsis thaliana. (La soumission sera effectuée une fois que le manuscrit n°3 sera accepté pour publication). *Authors contributed equally to this work.
5. Hautekèete N.C., Frachon L., Luczak C., Toussaint B., Van Landuyt W., Van Rossum F. & Piquot Y. (2015) Habitat type shapes long-term plant biodiversity budgets in two densely populated regions in north-western Europe. Diversity and Distributions 21: 631-642.
6. Tayeh A., Hufbauer R.A., Estoup A., Ravigne V., Frachon L. & Facon B. (2015) Biological invasion and biological control select for different life histories. Nature Communications
6:7268.
Communications scientifiques
- Organisation
Membre du comité d’organisation des « 8èmes journées des doctorants SPE » à Toulouse (30 juin – 2 juillet 2016)
1. Frachon L., Libourel C., Villoutreix R., Carrère S., Glorieux C., Huard-Chauveau C., Navascués M., Gay L., Vitalis R., Baron E., Amsellem L., Bouchez O., Vidal M., Le Corre V., Roby D., Bergelson J. & Roux F. (October 2016) Tracking the genetic bases of contemporary evolution in a spatially heterogeneous environment. International conference sfécologie, Marseille (France). Talk
2. Frachon L. (August 2016) The adaptive genetics of Arabidopsis thaliana in heterogeneous environments. Lab meeting in the group of Johanna Schmitt, Davis University (California, USA). Séminaire
3. Frachon L. (June 2016) The adaptive genetics of Arabidopsis thaliana in heterogeneous environments. LIPM meeting Toulouse (France). Séminaire
4. Frachon L. (March 2015) Plant-plant interactions: identification of genetics bases and characterization of their associated signatures of selection in A. thaliana. LIPM meeting Toulouse (France). Séminaire
- Posters
1. Frachon L., C. Bartoli, B. Mayjonade, T. Wijmer & F. Roux (December 2016) The selective agents acting on Arabidopsis thaliana depend on the type of habitat. British Ecological Society, Liverpool (UK).
2. Frachon L., R. Villoutreix, C. Libourel, E. Baron, S. Carrère, C. Glorieux, L. Amsellem, V. Le Corre, J. Gouzy, J. Bergelson & F. Roux (May 2016) Adaptive genomics to fine-grained spatial heterogeneity in a natural population of Arabidopsis thaliana. Journée des doctorants de l’école doctorale SEVAB, Toulouse (France).
3. Frachon L., R. Villoutreix, C. Libourel, E. Baron, S. Carrère, C. Glorieux, L. Amsellem, V. Le Corre, J. Gouzy, J. Bergelson & F. Roux (July 2015) Adaptive genomics to fine-grained spatial heterogeneity in a natural population of Arabidopsis thaliana. 7ème journée des doctorants du département INRA- SPE, Rennes (France).
Introduction
générale
1
A. Adaptation aux changements globaux
Importance de l’effet des changements globaux sur la biodiversité
Année 2016 : les géologues ont validé le changement d’ère géologique dû à l’impact de l’Homme sur la planète (Waters et al. 2016). L’Anthropocène place ainsi l’Homme comme la source majeure des changements sur l’écosystème terrestre.
Ce n’est plus à prouver : l’Homme a un réel impact sur la planète. Dès le Pléistocène, les activités humaines auraient conduit à l’extinction de la mégafaune via une chasse excessive (Martin & Steadman 1999, Wolverton 2010, Johnson 2002). Les pratiques agricoles ont modifié la diversité des communautés biotiques dès le Néolithique (Lopez-Garcia et al. 2013). Le développement des moyens de transport ont par la suite augmenté les échanges entre les continents, entraînant une modification importante de la répartition géographique des principales espèces cultivées et de nombreuses espèces sauvages (Beinart & Middleton
2004), certaines d’entre elles étant décrites comme envahissantes dès le 19ème siècle
(Richardson & Pysek 2007) comme l’Ajonc d’Europe introduit dès 1825 sur l’île de la Réunion (Udo et al. 2017) ou encore le lapin introduit en Australie en 1859 (Ratcliffe 1959).
Depuis quelques décennies, les activités humaines se sont multipliées et intensifiées à un rythme sans précédent (Figure 1), modifiant ainsi en profondeur (i) la diversité, la structure et la dynamique des communautés biotiques, et par conséquent (ii) le fonctionnement des écosystèmes, notamment via une altération des services écosystémiques (Chapin III et al. 2000, Sala et al. 2000, Millennium Ecocystem Assessment 2005). Avec un taux d’extinction du nombre d’espèces depuis 1900 de 100 à 1 000 fois plus important que les 1 à 10 millions d’années précédentes (Pimm et al. 1995, Pimm et al. 2014), le déclin observé de la biodiversité est sans précédent, affectant plus de trois quarts de la surface des biomes terrestres (Ellis et al. 2012).
2
Figure 1: Composantes des changements globaux menant à un déclin de la biodiversité. L’utilisation des terres comprend la perte et la fragmentation des habitats, l’intensification des pratiques agricoles, l’étalement urbain et l’érosion des sols. Figure modifiée d’après Vitousek et al. (1997).
Parmi les modifications environnementales majeures liées aux activités humaines récentes, nous pouvons citer le changement climatique observé à une échelle mondiale. Le climat étant un des principaux facteurs abiotiques déterminant les aires de distribution géographique des espèces, une augmentation de température de seulement 1°C entraîne à la fois un déplacement des niches climatiques et une remontée importante des espèces vers de plus hautes latitudes (Thuiller 2007, Kelly & Goulden 2008, Felde et al. 2012). A une échelle régionale ou à une échelle des paysages, une forte progression du tourisme et des échanges commerciaux a entraîné une augmentation du nombre d’introductions d’espèces exotiques dans les pays (Beinart & Middleton 2004, Carruthers et al. 2011), perturbant potentiellement les interactions au sein des communautés biotiques (Elton 1958, Gilman et
al. 2010). Ainsi, depuis le début du siècle, de nombreuses épidémies sur les plantes cultivées
résultent de sauts géographiques d’espèces pathogènes, comme le chancre bactérien du kiwi causé par la bactérie Pseudmonas syringae pv. actinidiae qui s’est propagée à travers le monde via des plantules importées de kiwi (Bartoli & Roux 2017). Aux mêmes échelles géographiques, des modifications de l’environnement comme la perte des habitats et leur fragmentation (Aguilar et al. 2006), l’intensification des pratiques agricoles ou l’urbanisation,
Population humaine
(taille et utilisation des ressources)
Agriculture
(pâturage, activité forestière…)
Industrie Augmentation de CO2 Altération du cycle biochimique Composés organiques persistants Utilisation des terres et changement du couvert végétale Récoltes des populations naturelles Invasions biologiques Changement climatique global Perte de biodiversité
3
vont entraîner l’apparition de nouvelles barrières géographiques auxquelles les espèces n’ont jamais été confrontées. Par exemple, de par la construction de routes et de bâtiments qui divisent et isolent les habitats naturels, les zones urbaines deviennent de véritables barrières pour les espèces animales et végétales (Van Rossum & Triest 2010). Par ailleurs, selon les régions géographiques du globe, le dépôt d’azote atmosphérique, l’augmentation de CO2, la production et la prolifération de composés organiques persistants (e.g. fluorochrome) ou la surexploitation des stocks de ressources biologiques ont été signalés comme des changements globaux ayant des effets non négligeables sur la biodiversité (Vitousek et al. 1997, Sala et al. 2000).
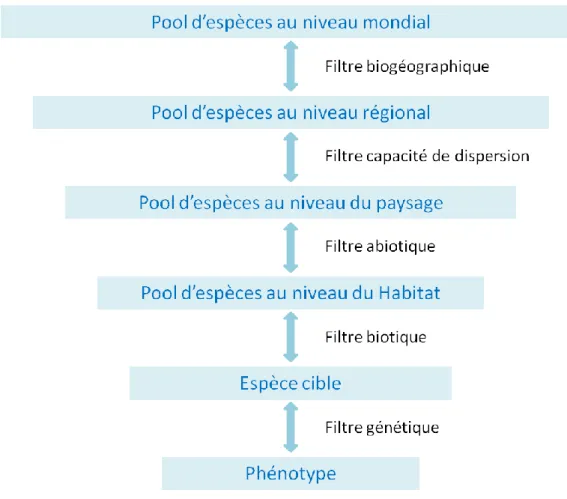
L’hétérogénéité des changements globaux en cours et les différences d’échelles spatiales auxquelles ils sont observés vont se superposer aux filtres environnementaux existants (Figure 2), augmentant ainsi la multiplicité des pressions de sélection et leurs interactions auxquelles devront faire face les espèces.
Figure 2: Différents filtres environnementaux agissant à différentes échelles spatiales. Ce sont la combinaison de différents agents sélectifs qui créeront le maillage de ces filtres environnementaux. Figure modifiée d’après Gugerli et al. (2013).
4
En présence de ce nouveau réseau de pressions de sélection, trois types de réponse non-exclusifs peuvent être adoptés par les espèces (Hansen et al. 2012). Sur le court terme, les individus peuvent s’acclimater aux changements de conditions environnementales via la plasticité phénotypique, en exprimant des phénotypes particuliers en réponse aux conditions environnementales locales. Sur le long terme, les organismes peuvent migrer vers des sites plus favorables, potentiellement sur de longues distances. Le troisième type de réponse correspond à la sélection génétique amenant à l’adaptation locale. Ci-dessous, je décris plus précisément ces trois types de réponse.
Figure 3: Réponses potentielles d’une espèce face à différents agents sélectifs, lui permettant de survivre à ce nouvel environnement. D’après Matesanz & Valladares (2014).
Se rapprocher d’un nouvel optimum phénotypique via la plasticité phénotypique
La plasticité phénotypique correspond à la capacité d’un génotype à produire plusieurs phénotypes en fonction de l’environnement biotique ou abiotique auquel il est
5
exposé (Sultan 2000, Agrawal 2001). Ainsi, la plasticité phénotypique permettrait à court terme une réponse rapide d’une espèce en modifiant son phénotype sans modification génétique (Matesanz & Valladares 2014). L’hétérogénéité environnementale favorise la plasticité phénotypique (Moran 1992, Sultan & Spencer 2002). Ceci est d’autant plus vrai dans le cas d’une hétérogénéité temporelle où tous les individus font face à une modification de l’environnement. A l’inverse, dans le cas d’une hétérogénéité spatiale, des refuges peuvent toujours persister permettant ainsi aux génotypes fixés adaptés localement de se maintenir.
Les modèles théoriques prédisent que la plasticité phénotypique adaptative peut aider les populations naturelles à se rapprocher d’un nouvel optimum phénotypique (Lande 2009, Chevin et al. 2010). Face aux changements globaux en cours, il est donc attendu que la plasticité phénotypique soit une réponse adaptative répandue entre les espèces. Cependant, malgré ses bénéfices théoriques, la plasticité phénotypique adaptative n’est pas aussi fréquente qu’on pourrait l’espérer (Charmantier et al. 2008). Cette contradiction entre théorie et observations peut résulter de coûts et de limites qui entravent l’évolution de la plasticité phénotypique adaptative. Parmi les nombreux coûts et limites répertoriés (DeWitt
et al. 1998), nous pouvons citer dans le cadre du changement climatique le manque de
fiabilité des signaux environnementaux, amenant les individus à des réponses plastiques non-adaptatives ou mal-adaptatives (van Kleunen & Fischer 2005, Chevin et al. 2010, Price et
al. 2013, Murren et al. 2015). De tels signaux peuvent correspondre à des évènements
climatiques extrêmes en dehors de la gamme historique des conditions climatiques rencontrées par les populations. Par ailleurs, la plasticité phénotypique est théoriquement favorisée dans des environnements où les variations environnementales sont régulières et prédictibles (Moran 1992, Sultan & Spencer 2002). Or, le changement climatique est non seulement associé à une augmentation moyenne des températures mais aussi à une augmentation du niveau de fluctuation des conditions climatiques entre les années.
Migrer pour fuir un milieu devenu défavorable
Face aux changements globaux, un deuxième type de réponse correspond à la migration des espèces depuis leurs milieux d’origine devenus défavorables vers de nouveaux milieux correspondant à leur niche écologique (Hansen et al. 2012). Ce type de réponse est
6
notamment attendu dans le cadre du changement climatique où un déplacement des enveloppes climatiques vers de plus hautes latitudes est d’ores et déjà observé.
Durant ces dernières décennies, il a été observé une migration des espèces avec un taux moyen de déplacement de 17.6 km/décennie et une remontée moyenne des espèces en altitude de 11 m/décennie (méta-analyse réalisée par Chen et al. 2011). Cependant, la dynamique de migration est très variable entre les espèces (Chen et al. 2011), entraînant par conséquent une modification de la diversité et de la composition des communautés et des interactions interspécifiques inhérentes (Gilman et al. 2010, Singer et al. 2013). De manière intéressante, il peut même être observé une augmentation provisoire de la biodiversité à l’échelle du paysage, avec l’arrivée rapide de nouvelles espèces qui est concomitante à une extinction plus lente d’autres espèces déjà présentes dans les communautés (Jackson & Sax 2010, Hautekèete et al. 2014). Ces étapes transitoires suggèrent l’importance d’étudier la dynamique de la biodiversité à différentes échelles géographiques : déclin de la biodiversité à une échelle mondiale vs augmentation provisoire de la biodiversité à l’échelle du paysage.
Pour suivre le déplacement géographique des enveloppes climatiques, les espèces végétales dispersant leur pollen et leurs graines sur de longues distances (i.e. espèces anémochores, espèces zoochores…) seront favorisées par rapport aux espèces végétales ayant des distances de dispersion limitées (i.e. espèces barochores). Cependant, même pour les espèces végétales avec de longues distances de dispersion, la probabilité qu’une propagule arrive dans un milieu favorable peut être fortement réduite à cause de la présence de barrières naturelles (court d’eau, montagne, forêt…) mais surtout à cause de l’augmentation de la présence de barrières anthropiques (route, urbanisation, champs…).
Adaptation locale via la sélection génétique
Le troisième type de réponse pour répondre aux changements globaux concerne l’adaptation locale via la sélection génétique (Hansen et al. 2012). Ce type de réponse repose sur la disponibilité dans les populations de variants génétiques qui seront sélectionnés lors de la marche adaptative vers un nouvel optimum phénotypique. Plusieurs sources peuvent être à l’origine de ces variants génétiques (Bay et al. 2017) :
- Immigration d’allèles à partir de populations proches (i.e. ‘genetic rescue’) (Hoffman & Sgrò 2011).
7
- Apparition de nouvelles mutations dans les populations naturelles (i.e. de novo mutations).
- Variation génétique préexistante dans les populations naturelles (i.e. ‘standing genetic variation’).
Toutefois, ces trois sources de variants génétiques n’apparaissent pas équivalentes en termes de vitesse de réponse à des changements globaux rapides. En effet, les contraintes imposées par l’attente de nouvelles mutations peut entraîner une limite adaptative rapidement atteinte par les populations (Stapley et al. 2010, Hancock et al. 2011). Les flux de gènes peuvent effectivement permettre l’arrivée dans une population d’allèles pré-adaptés à des modifications environnementales. Cependant, les flux de gènes entre populations proches restent limités sur une courte période de temps ; et ce phénomène est d’autant plus accentué par l’augmentation des barrières anthropiques comme décrit précédemment. Par ailleurs, une contrepartie associée aux flux de gènes est l’immigration simultanée d’allèles maladaptés augmentant potentiellement le fardeau génétique des populations.
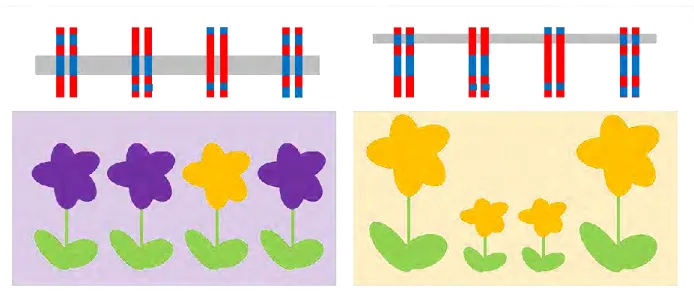
Il apparaît donc indispensable que les populations possèdent une variation génétique préexistante suffisante pour répondre rapidement aux changements globaux. Une population génétiquement diversifiée permet d’augmenter la probabilité que certains individus aient une combinaison génétique favorable pour répondre à de nouveaux stress environnementaux, et ainsi permettre d’atteindre un nouvel optimum phénotypique plus rapidement (Chevin et al. 2010, Figure 4).
8
Figure 4: Illustration de l’importance de la diversité génétique préexistante dans les populations naturelles pour répondre rapidement aux changements globaux. Trois populations de quatre individus homozygotes sont représentées ici. La population 1 est génétiquement monomorphe alors que les populations 2 et 3 présentent une diversité génétique. Lors d’un changement environnemental symbolisé par la flèche noire, une combinaison d’allèles devient alors optimale. Cette combinaison « idéale » pour le nouvel environnement est représentée sur le côté gauche du schéma. Les allèles adaptatifs vis-à-vis du nouvel environnement sont colorés, alors que les allèles neutres sont en gris. La combinaison « idéale » n’étant pas présente dans la population 1, elle a de fortes chances de s’éteindre. En revanche, la combinaison « idéale » est déjà présente dans les deux autres populations, leur permettant de s’adapter rapidement à ce nouvel environnement et ainsi de se maintenir.
B. Identification des bases génétiques de l’adaptation
Comme nous l’avons vu précédemment, une réponse des espèces à des changements globaux rapides passera en partie par la sélection génétique, notamment à partir de la variation génétique préexistante. Afin de déterminer le potentiel adaptatif des populations naturelles face aux changements environnementaux d’origine abiotique et/ou biotique globaux (Bergelson & Roux 2010, Bay et al. 2017), un des enjeux majeurs en écologie évolutive est donc d’étudier l’architecture génétique de l’adaptation et implique de s’intéresser aux questions suivantes (liste non exhaustive) :
- Quel est le nombre de gènes sous-jacents à l’adaptation locale ?
Changement de pressions de sélection Combinaisons d’allèles favorables à ce nouvel environnement
Population 1
Population 2
Population 3
9
- Quelle est la distribution des effets alléliques ?
- Quelle est l’identité des gènes adaptatifs et des fonctions biologiques associés? - La pléiotropie et l’épistasie contribuent-elles à la marche adaptative vers un
nouvel optimum phénotypique ?
En tirant bénéfice du développement récent de technologies de séquençage haut débit (Next Generation Sequencing technologies, NGS) permettant d’obtenir un nombre sans précédent de marqueurs génétiques (notamment de type Single Nucleotide Polymorphism, SNP), quatre approches complémentaires peuvent être utilisées pour étudier l’architecture génétique de l’adaptation, et plus particulièrement pour cartographier finement les gènes sous-jacents à l’adaptation (Table 1).
Table 1: Différentes approches permettant d’identifier les bases génétiques de l’adaptation locale (d’après Hoban et al. 2016). SNP: single nucleotide polymorphism, QTL: quantitative trait loci, BC: backcross, RIL: recombinant inbred lines, GWAS: genome-wide association studies.
Les méthodes ‘genetic differentiation outlier tests’ et ‘population-specific selective sweeps’ ne s’appuient que sur des données génomiques et cherchent à identifier des régions génomiques dont les patrons de diversité et de sélection s’écartent des attendus neutres.
10
Bien que largement utilisées, ces deux méthodes fournissent une liste de gènes candidats qu’il est souvent difficile de relier à des agents sélectifs potentiels ou à des traits phénotypiques adaptatifs. Pour palier ce manque, des méthodes statistiques permettant d’identifier le long du génome des polymorphismes génétiques associés soit à des variables écologiques (‘Genome-Environment Association’, GEA), soit à des traits phénotypiques (‘QTL mapping traditionnel’ et ‘GWAS’) ont été développées. Ci-après, je me focalise plus particulièrement sur ces méthodes d’analyses d’association génome-environnement et de QTL mapping sur des traits supposés adaptatifs.
1. Méthodes d’analyses génome-environnement
Les analyses de type association génome-environnement (Genome-Environment Association, GEA) reposent sur l’effet de gradients écologiques sélectifs spatiaux sur la variation génomique d’une espèce donnée (Lasky et al. 2012). En effet, un environnement hétérogène au niveau abiotique ou biotique conduit à différents optima phénotypiques locaux, qui se traduiront par une différenciation spatiale des variants génétiques sous-jacents aux phénotypes impliqués dans la réponse aux agents sélectifs. Ce type d’analyses permet donc non seulement d’identifier des gènes potentiellement impliqués dans l’adaptation mais aussi de décrire les facteurs écologiques responsables de leur divergence génétique entre les populations (Pluess et al. 2016, Figure 5).
Réalisées dans un premier temps par des approches individu-centré où un seul individu par population était caractérisé au niveau génomique, les analyses de type GEA ont été réalisées par la suite en adoptant des approches populationnelles afin de tirer bénéfice des informations apportées par la variation génétique intra-population.
11
Figure 5: Principe de l’analyse d’Association Génome-Environnement (GEA). Les génomes sont représentés pour sept populations. A droite, la caractérisation d’une variable édaphique et d’un descripteur des communautés végétales est indiquée pour chacune des populations. En bas du schéma, les coefficients de corrélation entre la variation génétique et la variation écologique (violet : variable édaphique, vert : richesse spécifique) sont tracés en fonction de la position des marqueurs génétiques polymorphes le long du génome. Approche individu-centré
Une des premières études de type GEA le long du génome chez une espèce sauvage a été réalisée par une approche individu-centré (Hancock et al. 2011). Pour réaliser cette étude, les auteurs se sont basés sur 948 accessions européennes de l’arabette des dames (Arabidopsis thaliana) génotypées pour 215k SNPs et caractérisées pour 13 variables climatiques (extrêmes et saisonnalité des températures et des précipitations). Pour identifier les régions génomiques associées aux variations climatiques, les auteurs ont utilisé un test partiel de Mantel basé sur le calcul du coefficient de corrélation de Spearman entre un SNP donné et une variable climatique, tout en contrôlant pour l’effet de l’histoire démographique d’A. thaliana en utilisant une matrice d’apparentement entre les 948 accessions calculée à partir des 215k SNPs. Ces analyses ont permis d’établir une carte
12
génomique de l’adaptation au climat chez A. thaliana. En effet, les SNPs les plus fortement corrélés au climat étaient significativement enrichis en variants génétiques correspondant à des changements d’acide aminé, suggérant que l’approche individu-centré de type GEA a bien permis d’identifier des loci adaptatifs.
A partir de 202 accessions de la luzerne tronquée (Medicago truncatula) génotypées pour environ 2 millions de SNPs, une approche individu-centré a aussi été adoptée pour identifier les loci candidats sous-jacents à l’adaptation à trois gradients climatiques, à savoir la température annuelle moyenne, les précipitations du mois le plus pluvieux et l’isothermalité (Yoder et al. 2014). Pour identifier les SNPs les plus associés à ces trois variables climatiques, les auteurs ont utilisé un modèle linéaire mixte incluant une matrice d’apparentement pour contrôler les effets confondants associés à l’histoire démographique de Medicago truncatula. En se basant sur l’étude des signatures de sélection dans les régions génomiques encadrant les SNPs les plus fortement corrélés au climat, les auteurs ont conclu que les loci sous-jacents à l’adaptation au climat étaient soumis à un balayage sélectif basé sur de la variation génétique préexistante (i.e. ‘soft selective sweeps’).
A partir de 1943 cultivars africains et indiens génotypés pour 404 627 SNPs et caractérisés pour des variables climatiques et édaphiques, une approche individu-centré a été récemment adoptée chez une plante d’intérêt agronomique, i.e. le sorgo commun (Sorghum bicolor) (Lasky et al. 2015). Un modèle mixte linéaire incluant une matrice d’apparentement a aussi été utilisée pour identifier les SNPs les plus associés au climat et au sol. Comme précédemment observé chez A. thaliana, les SNPs les plus fortement corrélés aux variables écologiques étaient significativement enrichis en variants génétiques correspondant à des changements d’acide aminé. Ces résultats ont ainsi permis aux auteurs de proposer des gènes candidats de réponse à la sécheresse et de tolérance à la toxicité à l’aluminium qui pourront être intégrés dans des programmes d’amélioration variétale.
Approche populationnelle
Bien que très puissantes, les approches individuelles négligent la variation génétique au sein des populations. Cependant, comme cela a été énoncé précédemment, il est
13
important de considérer la variation génétique intra-populationnelle pour obtenir une meilleure estimation du potentiel adaptatif des populations naturelles. Ainsi, une étude effectuée à partir de simulations a démontré qu’une approche populationnelle permettrait d’augmenter la puissance statistique des associations entre variation des fréquences alléliques des populations et variation des facteurs écologiques (Lotterhos & Whitlock 2015). Ces approches populationnelles ont été peu utilisées jusqu’à présent car le séquençage de plusieurs individus par population peut se révéler très coûteux. Pour palier ce problème, une alternative a été proposée par l’utilisation de l’approche Pool-Seq. Cette approche consiste pour une population donnée, à extraire l’ADN de chaque individu, puis de créer un bulk de manière équimolaire et de séquencer ce bulk (Schlöterrer et al. 2014). Plusieurs études ont démontré que les fréquences alléliques estimées à partir d’une approche Pool-Seq étaient fortement corrélées aux fréquences alléliques obtenues à partir d’un séquençage individuel (Schlöterrer et al. 2014, Fracasetti et al. 2015).
Les analyses de type GEA basées sur des données de fréquences alléliques peuvent être réalisées suivant différentes méthodes (De Villemereuil et al. 2014, Lotterhos et Whitlock 2015), dont en voici un aperçu (liste non exhaustive):
- Méthodes basées sur la différentiation génétique entre populations naturelles vivant dans des habitats écologiques contrastés.
(i) BayeScan (Foll & Gaggiotti 2008). Basée sur l’estimation d’un indice de
différenciation génétique entre populations (FST), cette méthode Bayésienne permet d’identifier des marqueurs génétiques présentant des valeurs extrêmes de FST entre des populations écologiquement différentes.
(ii) BayeScEnv (De Villemereuil & Gaggiotti 2015). Cette méthode (elle-aussi Bayésienne) est une extension de BayeScan incorporant une information environnementale sous la forme d’une différenciation environnementale continue entre populations. Cette méthode permet ainsi d’associer une variable environnementale spécifique à une forte différenciation génétique.
14
- Méthodes permettant d’estimer l’intensité d’une relation entre les fréquences alléliques et un gradient écologique.
(iii) Modèle mixte incluant des facteurs latents (Latent factor Mixed Model,
LFMM) (Frichot et al. 2013). Dans un premier temps, cette méthode génère
des variables latentes modélisant l’histoire démographique de l’espèce (i.e. structure génétique des populations) par une approche comparable à une Analyse en Composantes Principales. Afin de limiter le taux de faux positifs (fausses association génome-environnement), ces variables latentes sont par la suite incorporées comme co-variables dans les modèles de régression entre les fréquences alléliques et les variables écologiques.
(iv) BayEnv (Coop et al. 2010). Pour tenir compte de l’histoire démographique de l’espèce, cette méthode Bayésienne estime dans un premier temps une matrice de covariance des fréquences alléliques entre populations à partir d’un sous-jeu de marqueurs génétiques, permettant ainsi d’estimer un modèle nul auquel les corrélations entre fréquences alléliques à un marqueur génétique donné et une variable écologique sont comparées. La significativité de cette comparaison est approximée par l’estimation d’un facteur bayésien (Bayes Factor, BF). Récemment, une nouvelle version de BayEnv (BayEnv2) a été mise à disposition afin d’intégrer les données obtenues à partir d’une approche Pool-Seq (Günther et Coop, 2013).
(v) BayPass (Gautier 2015). Elaborée à partir de la méthode BayEnv2, cette méthode Bayésienne permet une meilleure estimation de la matrice de covariance populationnelle via une modification du paramétrage des priors concernant la fréquence moyenne des allèles de référence. Par ailleurs, BayPass propose des stratégies alternatives de modélisation de la relation entre fréquences alléliques et variables écologiques. Par exemple, au-delà du modèle STD (‘Standard covariate model’) qui correspond à une extension du modèle développé dans BayEnv, BayPass propose le modèle AUX (‘Auxiliary variable covariate model’) qui introduit dans le modèle STD une variable auxiliaire binaire qui est attachée au coefficient de régression ‘fréquences alléliques – variation écologique’ de chaque marqueur génétique testé, permettant ainsi de classifier chaque marqueur génétique testé comme
15
associé ou non (‘binary decision’) à une variable écologique donnée. Comme pour BayEnv2, la méthode BayPass est aussi adaptée aux données obtenues à partir d’une approche Pool-Seq.
Les études de GEA basées sur une approche populationnelle restent peu nombreuses chez les espèces sauvages mais deviennent de plus en plus populaires. Par exemple, à partir de cinq populations d’arabette de Haller (Arabidopsis halleri) caractérisées pour cinq variables topo-climatiques et pour lesquelles les fréquences alléliques le long du génome ont été obtenues par une approche Pool-Seq, 175 gènes ont été identifiés (à partir de test partiels de Mantel) comme significativement associés à une ou plusieurs des cinq variables topo-climatiques (Fischer et al. 2013).
Une étude comparative menée sur trois espèces de chênes (Quercus petraea, Quercus
pubescens, Quercus robur) en Suisse a été réalisée à partir de 71 populations (~20 individus
par population) caractérisées (i) par une approche Pool-Seq d’amplicons de gènes candidats (Rellstab et al. 2016) et (ii) pour 31 variables abiotiques (topographie, climat et sol). En utilisant la méthode LFMM, les auteurs ont identifié sept gènes communs entre les 3 espèces, comme significativement associés aux mêmes facteurs abiotiques (précipitations et teneur en argile dans les sols).
A partir de 10 populations de l’épinoche à trois épines (Gasterosteus aculeatus) localisées dans la Mer Baltique et caractérisées pour plus de 30 000 SNPs obtenus par une approche Pool-Seq, de nombreuses régions génomiques ont été identifiées via la méthode BayEnv comme associées à des gradients de température et de salinité (Guo et al. 2015).
Nécessité de tenir compte des pressions de sélection non seulement abiotiques, mais aussi
biotiques
L’approche de type GEA s’avère très puissante pour identifier les bases génétiques associées à des variables écologiques, permettant ainsi d’obtenir une meilleure compréhension des processus sous-jacents à l’adaptation locale de certaines espèces à leur environnement. Cependant, à notre connaissance, toutes les études GEA réalisées jusqu’à présent se sont intéressées uniquement à des variables abiotiques. Notamment, du fait de la
16
disponibilité d’un nombre important de bases de données climatiques, la majorité des études de type GEA ont porté sur l’identification de régions génomiques associées au climat (Bay et al. 2017). Dans une moindre mesure, des études de type GEA ont porté sur des variables édaphiques, soit par identification de régions génomiques spatialement différenciées entre des populations échantillonnées sur deux habitats édaphiques très contrastés (Arabidopsis lyrata sur des sols serpentiniques et des sols non-serpentiniques, Turner et al. 2010), soit par identification de régions génomiques associées à un gradient édaphique (Lasky et al. 2015, Pluess et al. 2016, Rellstab et al. 2016).
Or, au cours de son cycle de vie, un individu ne répond pas seulement à des conditions abiotiques. En effet, un individu interagit simultanément et séquentiellement, de manière directe ou indirecte, avec une large gamme de partenaires biotiques, dont les relations peuvent varier du mutualisme à la pathogénicité en passant par la compétition avec d’autres espèces ou avec ses congénères (Roux & Bergelson 2016). Par ailleurs, il est prédit que les interactions interspécifiques peuvent fortement affecter la réponse des espèces et des communautés biotiques aux changements climatiques (Gilman et al. 2010, Singer et al. 2013). Ainsi, de nombreux agents sélectifs potentiels y compris les facteurs biotiques doivent être considérés afin d’obtenir une vision plus complète du paysage génomique de l’adaptation locale.
2. Méthodes de QTL mapping sur des traits supposés adaptatifs
Comprendre l’architecture génétique de l’adaptation d’une espèce peut également passer par l’étude des relations existantes entre variation génétique et variation naturelle de traits phénotypiques supposés adaptatifs. Ci-dessous, je présente les deux grandes catégories de méthodes qui peuvent être utilisées pour cartographier les QTLs (Quantitative Trait Loci) associés à la variation naturelle de traits phénotypiques (Bazakos et al. 2017).
La première catégorie de méthodes correspond aux analyses de liaison ou bien encore appelée QTL mapping traditionnel (Bergelson & Roux 2010). Ces méthodes ont vu le jour dès la fin des années 80 et sont basées sur une carte génétique correspondant à une représentation de la position de marqueurs génétiques les uns par rapport aux autres, avec les distances entre marqueurs exprimées en termes de fréquence de recombinaison. Le QTL
17
mapping traditionnel est réalisé à partir de populations ségrégées, i.e. des populations artificielles découlant de croisements entre deux ou plusieurs génotypes parentaux. On comprend alors aisément que ces méthodes de cartographie QTL traditionnelle aient été plus largement adoptées chez les espèces végétales que chez les espèces animales. En effet, chez les plantes, les croisements entre génotypes d’une même espèce génèrent en moyenne un plus grand nombre de descendants que les croisements au niveau intraspécifique chez les animaux. Par ailleurs, les croisements interspécifiques peuvent être plus facilement réalisés chez les espèces végétales que chez les espèces animales, permettant de s’intéresser à l’architecture génétique d’une adaptation propre à une espèce donnée.
Plusieurs types de populations de QTL mapping traditionnel peuvent être utilisés pour cartographier les marqueurs génétiques associés à la variation naturelle phénotypique (Figure 6). Les premières populations de QTL mapping traditionnel qui ont été développées correspondent à des populations F2, issues généralement de l’autofécondation d’un individu F1 hybride obtenu par croisement entre deux génotypes parentaux. L’avantage de ce type de populations est qu’elles permettent une estimation de la dominance des QTLs identifiés, ce qui peut être une information précieuse pour comprendre l’architecture d’un trait adaptatif. Cependant, un inconvénient majeur des populations F2 est que chaque individu F2 phénotypé doit aussi être génotypé (à l’exception des espèces végétales avec un mode de multiplication végétative).
Pour résoudre ce problème, les populations de lignées recombinantes consanguines (Recombinant Inbred Line RIL, Figure 6 et Figure 7) ont été développées et correspondent à des lignées issues de descendants F2 qui ont subi plusieurs générations d’autofécondation jusqu’à obtenir des lignées quasi-homozygotes représentant une mosaïque unique des deux génomes parentaux (Bazakos et al. 2017). Ces populations sont devenues très populaires car les lignées RILs peuvent être phénotypées presque indéfiniment mais génotypées qu’une seule fois puisqu’il s’agit de lignées quasi-homozygotes (Savolainen et al. 2013, Bazakos et al. 2017). Malgré ces avantages, la diversité génétique présente au sein d’une famille RIL est limitée à la diversité génétique des deux génotypes parentaux.
18
Figure 6: Illustration des différentes populations de QTL mapping traditionnel pouvant être utilisées pour cartographier les marqueurs génétiques associés à la variation naturelle phénotypique. RILs: Recombinant inbred line, AI-RILS: Advanced intercross-recombinant inbred lines, HIF: Heterogeneous inbred family, MAGIC: multiparent advanced generation inter-cross lines, NIL: near-isogenic line. D’après Bergelson & Roux 2010.
Ainsi, une troisième type de population a vu le jour il y a quelques années et correspond aux lignées MAGIC (multiparent advanced generation intercross, Figure 6) (Cavanagh et al. 2008) qui ont été développées à la fois chez des espèces sauvages comme
A. thaliana (Kover et al. 2009) ou des espèces cultivées comme le blé ou le riz (Huang et al.
2012, Bandillo et al. 2013). Contrairement aux familles RILs, des croisements multiples sont effectués entre plusieurs génotypes parentaux et ceci sur plusieurs générations. Les descendants à partir de ce schéma de croisement sont ensuite autofécondés sur plusieurs générations, permettant de créer des lignées quasi-homozygotes comme pour les familles RILs. Ces populations MAGIC ont donc à la fois l’avantage d’être très diversifiées génétiquement mais aussi de contenir des lignées quasi-homozygotes. Par ailleurs, les générations de croisement supplémentaires permettent d’augmenter le nombre d’événements de recombinaison et donc d’obtenir une meilleure précision que les familles RILs quant à la localisation des QTLs le long du génome.
19
Figure 7: Illustration de la méthode de QTL mapping avec des familles RILs. Le QTL mapping permet d’associer une variation phénotypique au polymorphisme de certains marqueurs génétiques.
Malgré tout, les populations de QTL mapping traditionnel restent très peu résolutives et difficiles à mettre en place chez la plupart des espèces animales. Ainsi, la méthode de Genome-Wide Association (GWA) mapping est apparue comme une alternative puissante pour cartographier finement les régions génomiques associées à la variation phénotypique naturelle (Figure 8) (Mitchell-Olds & Schmitt 2006).
Figure 8: Illustration de la méthode de GWA mapping bénéficiant des événements de recombinaison passés. La figure du haut représente une population d’origine avec différentes versions d’un chromosome. Le chromosome noir symbolise une version ancestrale où une mutation s’est produite (représentée par un triangle) et responsable d’un nouveau phénotype. Après des milliers de générations, des événements de recombinaisons se sont accumulés. Les chromosomes de la population actuelle représentée en bas sont ainsi caractérisés par une mosaïque de régions génomiques dérivées de la population d’origine. Dans la nouvelle population, les trois individus du bas auront un phénotype différent des trois autres individus, dû à la mutation arrivée dans la population d’origine (triangle). Le marqueur moléculaire ‘b’ étant proche de ce polymorphisme, il sera ainsi corrélé avec le phénotype. En revanche, les marqueurs ‘a’ et ‘c’ sont distants de ce polymorphisme causal et ne seront donc pas corrélés au phénotype. D’après Mitchell-Olds & Schmitt (2006).
20
En effet, en tirant bénéfice des évènements de recombinaison qui se sont accumulés sur plusieurs centaines de milliers d’années (voir millions d’années), le GWA mapping utilise le déséquilibre de liaison (DL) naturelle présent dans une collection de génotypes naturels. Les estimations du DL sont très variables entre les espèces et dépendent principalement du régime de reproduction : d’environ 1bp chez la mouche du vinaigre (Drosophila
melanogaster) (MacKay et al. 2012) jusqu’à 500kb chez la variété temperate japonica du riz
(Oryza sativa) qui a un régime de reproduction fortement autogame (Mather et al. 2007), par exemple. La contrepartie d’un DL court est la nécessité d’avoir un nombre de marqueurs génétiques suffisants afin de balayer l’ensemble du génome lors des analyses de GWA mapping. Cependant, avec le développement des NGS, cet inconvénient deviendra de plus en plus rare dans les années à venir.
Deux autres inconvénients majeurs de la méthode GWA mapping (qui ne pourront être résolus par l’utilisation de NGS) restent les faux positifs et l’hétérogénéité génétique et/ou allélique. Comme pour les analyses de GEA, les faux positifs correspondent à de fausses associations génotype-phénotype qui résultent de l’effet de l’histoire démographique de l’espèce (Figure 9). Comme pour les analyses de GEA, plusieurs méthodes statistiques peuvent être utilisées pour corriger ces faux positifs. L’une des plus populaires consiste à intégrer dans un modèle mixte une matrice d’apparentement entre les génotypes utilisés dans l’étude phénotypique (Kang et al. 2010). Bien que très performantes, ces méthodes statistiques entraînent aussi l’apparition de faux négatifs (Figure 9), c’est-à-dire des marqueurs génétiques réellement associés à la variation phénotypique naturelle (i.e. marqueurs causaux) mais qui sont perdus après correction pour l’effet de l’histoire démographique de l’espèce (Brachi et al. 2010).
21
Figure 9: Illustration des faux positifs et faux négatifs dans les études de GWA mapping. D’après Bergelson & Roux (2010).
L’hétérogénéité génétique provient du fait qu’une même valeur phénotypique peut résulter de différentes combinaisons de QTLs (Figure 10). Par ailleurs, différents allèles à un même gène peuvent avoir le même effet et/ou des effets contrastés pour un phénotype donné, entraînant des phénomènes d’hétérogénéité allélique (Figure 10). Ces observations sont particulièrement bien documentées pour la date de floraison dont les bases génétiques ont été très étudiées chez différentes espèces végétales comme A. thaliana (Atwell et al. 2010) ou bien encore le maïs (Zea mays) (Buckler et al. 2009). Pour limiter les effets de l’hétérogénéité génétique et de l’hétérogénéité allélique, une solution proposée est de travailler à une échelle géographique régionale où la diversité génétique reste importante tout en étant restreinte par rapport à la diversité génétique observée sur l’ensemble de l’aire de répartition d’une espèce (Bergelson & Roux 2010). Travailler à une échelle géographique régionale présente aussi l’avantage de limiter les effets de l’histoire démographique de l’espèce considérée sur la détection des régions génomiques associées à la variation phénotypique naturelle (i.e. diminution des taux de faux positifs et de faux négatifs).
22
Figure 10: Illustration de l’effet de l’hétérogénéité génétique et de l’hétérogénéité allélique sur la détection de QTLs dans les études de GWA mapping. Cas de la date de floraison. D’après Bergelson & Roux (2010).
Bien que puissante et très prometteuse, (i) l’utilisation de l’approche GWA mapping reste encore l’apanage de quelques espèces sauvages modèles et de plus en plus des espèces cultivées (Bartoli & Roux 2017), et (ii) l’identité des pressions de sélection agissant sur les traits phénotypiques supposés adaptatifs est souvent suggérée mais rarement testée.
Replacer les études de QTL mapping dans un contexte écologiquement réaliste
La majorité des études de QTL mapping ont été réalisées soit dans des environnements contrôlés de laboratoire pour les espèces sauvages, soit dans des conditions agricoles (élevage ou champs cultivées) pour les espèces domestiquées. Cependant, la variation génétique d’une espèce est exposée à la sélection naturelle dans des habitats écologiquement contrastés. L’étude de l’architecture génétique de l’adaptation dans un contexte écologiquement réaliste fait ainsi appel aux approches développées en génomique écologique (Figure 11) (Ungerer et al. 2008).
23
Figure 11: Figure illustrant le concept de génomique écologique. D’après Ungerer et al. 2008.
Il est donc important de considérer le contexte écologique dans lequel sont mesurés les phénotypes. En effet, au cours de leur cycle de vie, les individus perçoivent des signaux environnementaux complexes et variés. Ainsi, quelques études de QTL mapping réalisées chez A. thaliana ont pu mettre en évidence que l’architecture génétique de la date de floraison était très différente entre des conditions de phénotypage de serre et des conditions de phénotypage sur un terrain expérimental (Weinig et al. 2002, Brachi et al. 2010). Plus précisément, une étude de GWA mapping réalisée à partir de 184 accessions mondiales d’A. thaliana a montré qu’une majorité des gènes associés à la date de floraison mesurée sur un terrain expérimental dans le Nord de la France étaient impliqués dans la régulation de l’horloge circadienne, voie de régulation peu citée dans les études de QTL mapping de la date de floraison mesurée dans des conditions contrôlées de laboratoire (Brachi et al. 2010).
Cependant, les conditions écologiques rencontrées par les plantes sur un terrain expérimental peuvent être encore très éloignées des conditions rencontrées par les populations dans leurs habitats naturels. Ainsi, une étude de QTL mapping traditionnel basée sur une famille RIL (i) issue d’un croisement entre deux populations d’A. thaliana localement adaptées (une population localisée en Suède et une population localisée en Italie) et (ii) phénotypée dans les habitats d’origine des populations, a permis de mettre en évidence que
24
deux tiers des QTLs associés au nombre de fruits étaient spécifiques aux habitats d’origine (Ågren et al. 2013).
Replacer les études de QTL mapping dans un contexte écologiquement réaliste permettrait donc à termes de retracer les trajectoires évolutives des traits adaptatifs dans les populations naturelles.
C. Importance de l’échelle géographique considérée
A quelle échelle géographique étudier l’adaptation locale et l’architecture génétique sous-jacente ? Cette question est loin d’être anodine. En effet, bien que l’adaptation locale ait pu être observée à des échelles spatiales très variées (de l’ordre de quelques mètres à l’échelle d’un continent) (Richardson et al. 2014), les études de type GEA ou de GWA mapping ont la plupart du temps été réalisées à partir d’une collection de génotypes échantillonnées à une large échelle spatiale (i.e. monde, continent). Ce constat est particulièrement bien illustré par les études de type GEA portant sur l’adaptation au climat (Hancock et al. 2011, Lasky et al. 2012, Yoder et al. 2014, Lasky et al. 2015) ou les études de GWA mapping peu importe le trait phénotypique considéré (Atwell et al. 2010, Huang et al. 2012, Bartoli & Roux 2017).
Le choix de travailler à une large échelle géographique peut être expliqué par plusieurs raisons :
- Il est communément admis que travailler sur une collection de génotypes échantillonnés sur l’ensemble de l’aire de distribution d’une espèce permettra d’avoir accès à toute la variation naturelle phénotypique présente au sein de cette espèce. Bien que cela puisse effectivement être le cas pour certains traits, d’autres traits phénotypiques peuvent présenter presque autant de diversité au sein d’une population locale que sur l’ensemble de l’aire de distribution. Chez A.
thaliana, cela a été observé pour la résistance qualitative et quantitative à des
bactéries phytopathogènes (Huard-Chauveau et al. 2013, Karasov et al. 2014) ainsi que pour des traits phénologiques comme la date de floraison (Brachi et al. 2013).
- Dans le cadre des changements globaux, un des objectifs est d’estimer le potentiel adaptatif des espèces afin de prédire l’évolution de la biodiversité au
25
cours des prochaines décennies. Ainsi, identifier les bases génétiques de l’adaptation à l’échelle de l’aire de distribution d’une espèce devrait permettre de prédire le devenir des populations locales. Or, comme nous l’avons évoqué précédemment, les bases génétiques de l’adaptation à une pression environnementale peuvent être très différentes d’une région géographique à l’autre (i.e. hétérogénéité génétique) et même entre des populations locales proches géographiquement (Hoekstra et al. 2006, Brachi et al. 2013).
- Une raison beaucoup moins scientifique concerne le coût de la caractérisation génomique des génotypes utilisés dans les études de type GEA ou de GWA mapping, nécessitant la mise en place de consortiums internationaux où chaque laboratoire veut caractériser les génotypes collectés dans son propre pays.
Toutefois, certaines études ont indiqué la complémentarité de travailler à différentes échelles spatiales. Ainsi, l’importance de travailler à différentes échelles géographiques a été mise en évidence dans une étude de type GEA réalisée sur l’arabette des Alpes (Arabis
alpina) au sein des Alpes (Alpes européennes, Alpes françaises et trois massifs montagneux
dans les Alpes françaises) (Manel et al. 2010). En effet, le pourcentage de loci ALFP corrélés à 8 variables climatiques était dépendant de l’échelle géographique considérée (Européenne : 12% des loci AFLP, régionale : 11% locale : variable entre 3 et 17% suivant le massif montagneux). De même, une étude de GWA mapping réalisée sur A. thaliana à cinq échelles géographiques différentes (mondiale, européenne, française, régionale et locale) a révélé que les bases génétiques associées à la phénologie étaient très dépendantes de l’échelle géographique considérée (Brachi et al. 2013). Ces études suggèrent que deux types de réponse adaptative peuvent être considérées : (i) une adaptation locale site-spécifique dû à des pressions de sélection variant à une échelle spatiale fine, et (ii) une adaptation plus générale en réponse aux pressions de sélection agissant à une plus grande échelle spatiale.
Comme préconisé dans certaines études (Bergelson & Roux 2010), l’échelle géographique à laquelle étudier l’adaptation locale et l’architecture génétique sous-jacente doit donc être déterminée en fonction des échelles de variation spatiale des pressions de sélection auxquelles est confrontée l’espèce étudiée. Déterminer l’échelle géographique à laquelle travailler nécessite donc dans un premier temps d’identifier les