Approche clinique et génétique des syndromes autoinflammatoires
Texte intégral
Figure
![Figure 1 : présélection des articles [4]](https://thumb-eu.123doks.com/thumbv2/123doknet/1963072.210/21.892.156.766.85.446/figure-preselection-des-articles.webp)
![Tableau I : Niveau de preuve scientifique de la littérature et force des recommandations (Score de Sackett modifié) [4]](https://thumb-eu.123doks.com/thumbv2/123doknet/1963072.210/22.892.102.819.263.797/tableau-niveau-preuve-scientifique-litterature-recommandations-sackett-modifie.webp)
![Tableau III : Age de début de la maladie périodique dans 7 séries de la littérature [1] (Parmi 989 patients provenant de 7 publications)](https://thumb-eu.123doks.com/thumbv2/123doknet/1963072.210/35.892.99.794.634.853/tableau-maladie-periodique-series-litterature-patients-provenant-publications.webp)
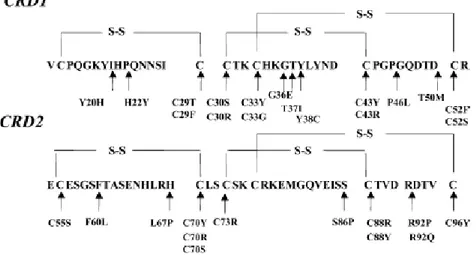
![Figure 3 : Schéma de la protéine Pyrine/ Marénostrine, codée par le gène MEFV [63]](https://thumb-eu.123doks.com/thumbv2/123doknet/1963072.210/40.892.236.643.152.262/figure-schema-proteine-pyrine-marenostrine-codee-gene-mefv.webp)
![Figure 4: Pedigree illustrant le modèle de transmission autosomique récessive [76]](https://thumb-eu.123doks.com/thumbv2/123doknet/1963072.210/41.892.198.618.84.363/figure-pedigree-illustrant-le-modele-transmission-autosomique-recessive.webp)
![Figure 5 : Schéma montrant la structure des deux formes membranaire et soluble des récepteurs 1A du TNF [106]](https://thumb-eu.123doks.com/thumbv2/123doknet/1963072.210/46.892.243.615.86.348/figure-schema-montrant-structure-formes-membranaire-soluble-recepteurs.webp)
![Figure 6: Les pedigrees de deux familles atteintes de TRAPS A- Famille Australienne d’origine Ecossaise [ 108]](https://thumb-eu.123doks.com/thumbv2/123doknet/1963072.210/47.892.209.626.79.573/figure-pedigrees-familles-atteintes-famille-australienne-origine-ecossaise.webp)
Outline
Documents relatifs
Reasonable correla- ultimate bearing capacity from a soil anics laboratory of the University of tion, however, is gained using the study are obvious. of such
recognised as effectively as samples A and C in the assay of 5B in comparison with other assays indicating that antigenic determinants in the TNF - molecule are
Je suis ravie de t’accueillir en ce 1 er jour dans ma classe!. Je t’invite à aller t’assoir à
First, the affinity of F5 for the feline epitope may be lower or the actual number of the relevant epitopes may be less; second, the polyclonal rabbit anti- hrTNFα may recognize a
Modification du territoire d’expression de gènes du développement chez le serpent... Modification du territoire d’expression de gènes du développement chez
Plusieurs stratégies peuvent être utilisées pour réduire le risque de réactions immédiates et retardées à la perfusion : traitement d’induction à 3 doses (0, 2 et 6
We evaluated the effect of the TNF antagonists infliximab (Ifx), adalimumab (Ada) and etanercept (Eta) on anti-mycobacterial immune responses in two conditions: with ex vivo
Absence de tumor necrosis factor (TNF) circulant dans une pneumonie expérimentale à Pasteurella haemolytica biotype A1 (Pha1) chez le veau... with several other members of the
