Détection innée des cellules infectées au VIH-1 par les cellules dendritiques plasmacytoïdes : étude du rôle régulateur de la protéine Vpu de souches pandémiques et non pandémiques
Texte intégral
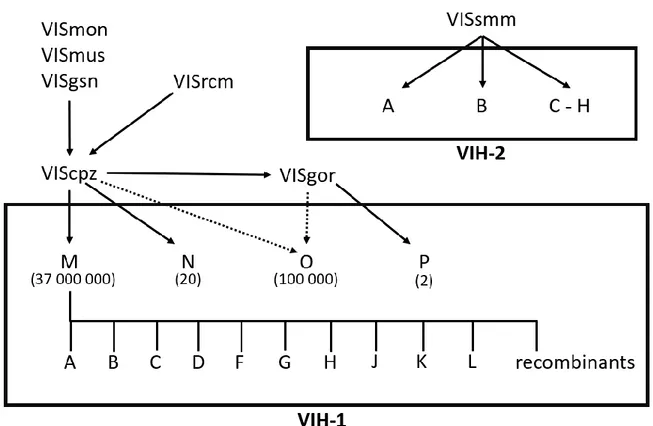
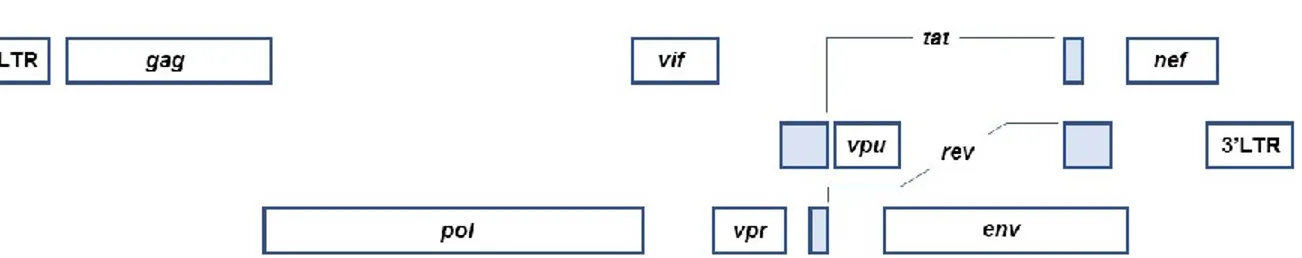
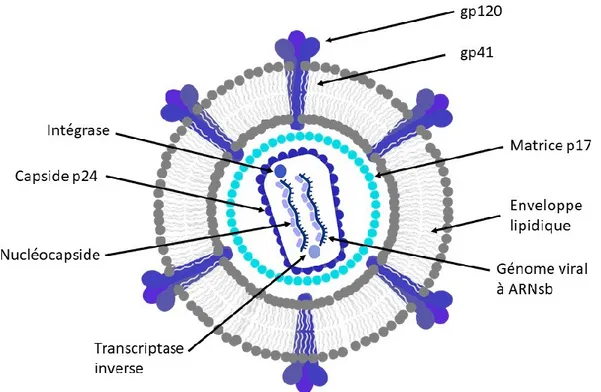
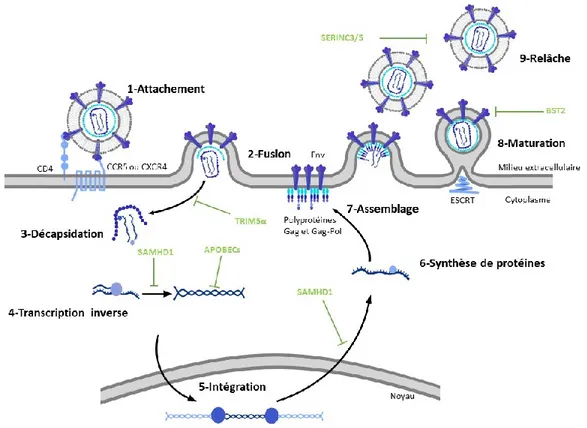
Figure
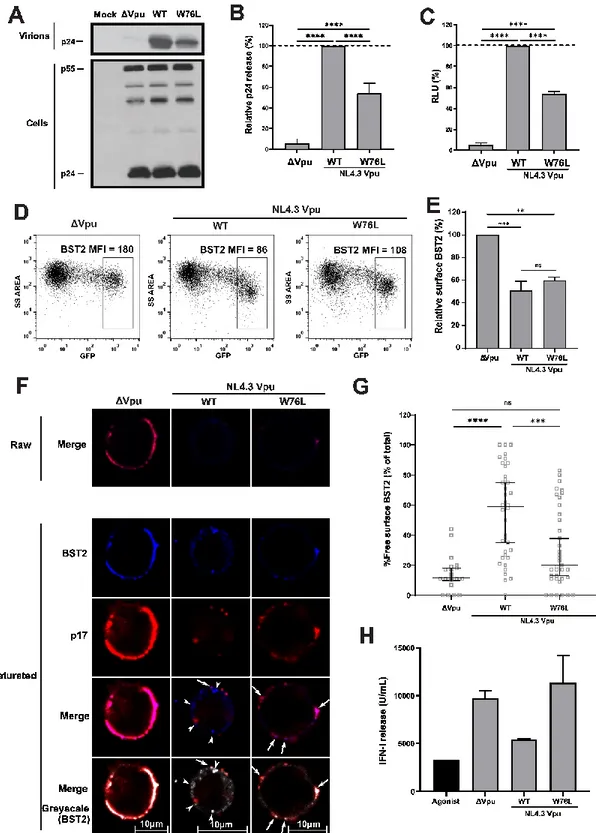
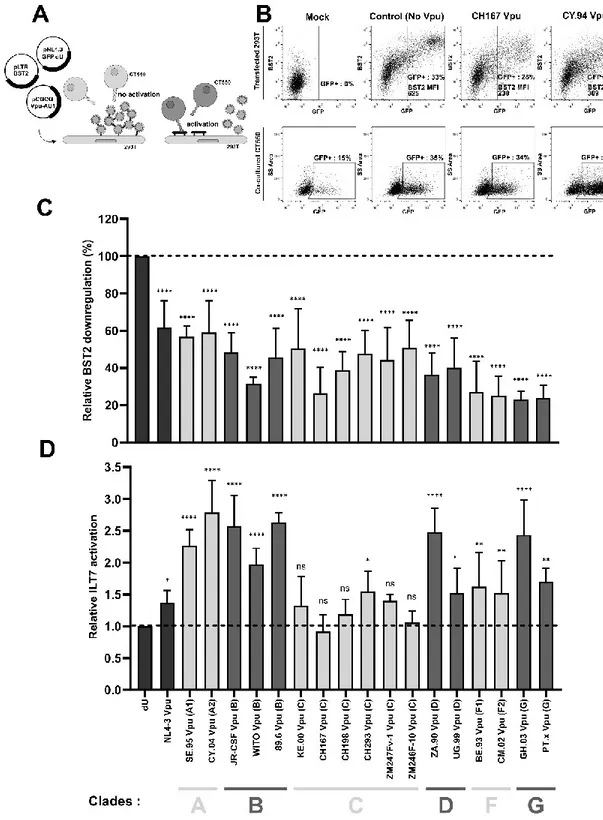
Documents relatifs
سيدقتّ سكش ىأ لاإ ،َلضفّ للها يه قيفْتب عضاْتولا لوعلا ارُ ماوتإ دعب يٌعسيلا ىلع سابع ةدٌسْب زْتكدلا ذاتسلأا ىلإ ىاٌتهلااّ سكشلا ليزجب َجْتا
3 ( فﯾﺿ ﻲﻗوﺷ ﻲﺑدﻷا بﯾﻘﺣﺗﻟاو : ﻷا ﺔﺳارد ﻲﻓ فﯾﺿ ﻲﻗوﺷ ﺔﻘﯾرط لﺛﻣﺗ نﯾﺳﺣ ﮫط ﮫﻛﻠﺳ يذﻟا هﺎﺟﺗﻼﻟ ﺎﻣﻋد بد رﻌﺷﻠﻟ ﻲﺧﯾرﺎﺗﻟا روطﺗﻟا فﯾﺿ ﻲﻗوﺷ دﺻر دﻘﻓ ،ﻲﻠھﺎﺟﻟا بدﻸﻟ ﮫﺗﺳارد
Nous avons choisi, pour pouvoir estimer les indices de similarite´, de faire la mise en correspondance de chaque point 3D du premier ensemble dit de re´fe´rence avec le point le
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Figure 3: Comparison of microstructure evolution: top: grain size distributions (equivalent diameter in number) from MC (blue) and LS (red) simulations at different time.. The
RÉSUMÉ : Cet article traite du développement des études comparatistes sur les langues indo-européennes et sémitiques en France au XIXe siècle en prenant pour départ leur
Auff wel- ches nach gethaner umbfrag die Herren gesandten in namen ihren Rhä- ten und gmeinden sich eines gutten willens, dasselbig zu verrichten, angedeüttett; iedoch ist
Les émotions ressenties par les enseignants dans le cadre professionnel et la façon dont ils les gèrent sont des facteurs déterminants pour maintenir une éthique
