1
Université de Picardie Jules Verne
Unité de Formation et de Recherche et de Médecine
Amiens
THESE D'ETAT DE DOCTEUR EN MEDECINE
Mention Spécialité
Oncologie-Radiothérapie
n° de thèse : 2015 – 153 Année 2014-2015
Par
Monsieur Farid BELKHIR
Né le 23/01/1982 à Ouaguenoun
(Algérie)
Présentée et soutenue publiquement le 07/10/2015
RADIOTHERAPIE ADJUVANTE DANS LES TUMEURS
EPITHELIALES THYMIQUES:
A PROPOS D’UNE ETUDE DE COHORTE RETROSPECTIVE DE 134
PATIENTS TRAITES A L’INSTITUT GUSTAVE ROUSSY DE VILLEJUIF
ENTRE 1990 ET 2011.
Le Président de Jury,
Monsieur le Professeur Claude KRZISCH
Les Juges,
Monsieur le Professeur Bruno CHAUFFERT
Monsieur le Professeur Pascal BERNA
Monsieur le Docteur Alexandre COUTTE
La directrice de thèse,
2
Table des matières
Remerciements
………...5Résumé
………..……….6Introduction
………..……….…..8L’objectif ………..………….……..8
1. Epidémiologie :………..………..8
2. Anatomie, embryologie et fonction du thymus :………..8
3. Classification histologique……….….10
4. Diagnostics différentiels :………..….16
5. La classification de Masaoka-Koga ……….….17
6. Manifestations cliniques et circonstances de découverte : ……….…….17
7. Le bilan pré-thérapeutique :………..……….……….20
8. Le traitement des TET………21
8.1. Chirurgie……….21
8.2. Radiothérapie……….22
9. La chimiothérapie……….26
10. Les thérapeutiques ciblées : ………..………..…….27
Matériels et méthodes
………271. Critères d’inclusion………..27
2. Critères d’exclusion ……….27
3
4. Statistiques ………..28
Résultats
……….………….281. Caractéristiques des patients………...28
1.1. Age et le sexe ……….……28
1.2. Performans Status selon l’OMS (PS) ………..….28
1.3. Circonstances de découverte………..28
2.
Les caractéristiques de la maladie……….………....292.1. La classification selon Masaoka-Koga………..……….29
2.2. Histologie des tumeurs et classification OMS 2004……….…………30
2.3. Syndromes paranéoplasiques……….……….30
3.
Traitements ………..…….32 3.1. Chimiothérapie d’induction ………..….32 3.2. Chirurgie tumorale ……….32 3.3. Radiothérapie………..……….33 3.4. Chimiothérapie adjuvante………343.5. Effets secondaires et complications de la radiothérapie………35
3.5.1. Toxicités aigues :……….35
3.5.2. Toxicités tardives :……….….35
4.
Evolution de la maladie………..……354.1. Rechutes locorégionales et métastatiques……….35
4.1.1. Rechutes locorégionales (tableau9) :……….36
4.2. Rechutes métastatiques………..41
4.3. Traitement des rechutes ………..………..43
4
4.4. Survie
……….………..……….434.3.1.Survie globale………..….43
4.3.2. Survie spécifique……….……….45
4.3.3. Survie sans progression………46
Discussion
………..471.
Discussion des résultats
……….………….471.1. Survie et facteurs pronostiques……….48
1.1.1. Survie globale et facteurs pronostiques………48
1.1.2. Survie sans progression et les facteurs pronostiques:……….…….49
2.
La réponse après chimiothérapie néo-adjuvante
:……….503.
Discussion de la méthodologie
………..……...….50Conclusion
………..……..515
Remerciements et dédicaces
Je tiens à remercier les membres du jury qui ont eu l’amabilité de juger et de lire mon travail :
- Monsieur le Professeur Claude KRZISCH
Professeur des Universités-Praticien Hospitalier (Cancérologie, radiothérapie). Oncopôle - Monsieur le Professeur Bruno CHAUFFERT
Professeur des Universités-Praticien Hospitalier (Oncologie médicale) Responsable du service d'Oncologie médicale. Oncopôle
- Monsieur le Professeur Pascal BERNA
Professeur des Universités – Praticien Hospitalier Responsable du service de chirurgie thoracique Pôle "Cœur Thorax - Vaisseaux"
- Monsieur le Docteur Alexandre COUTTE Praticien Hospitalier Contractuel
Service d'Oncologie-Radiothérapie CHU Sud AMIENS
- Madame le Docteur Cécile LEPECHOUX Praticien Hospitalier
Service de radiothérapie
Institut Gustave Roussy de Villejuif
Je remercie particulièrement le Dr Antonin Lévy qui m’a beaucoup aidé et orienté durant
toute la période qu’a duré ce travail. Il a été une aide précieuse pour moi avec ses remarques et ses conseils et surtout pour la réalisation des statistiques !
Je dédie ce travail et ma thèse à Ma fille Meriem Tiziri (13 mois) qui illumine mes journées et nuits avec sa lumière !
A ma femme Sara qui m’a aidé et soutenu durant toutes mes années passées en médecine !!!!!!
A tous mes amis (du collège, lycée et université). A mouloud khouas (cousin) et sa femme Kenza !! A mon ami Salem ouabas ( notre grand artiste) A mohamed YOUNSI, CHEHIMI, ALKAZEMI A amer OMEIRI ( un futur grand médecin)
A Ilan DARMON à qui je dis félicitation pour sa thèse soutenue le 21/09/2015
Aux différentes équipes de physique, dosimétrie et manipulatrices des différents hôpitaux que j’ai fréquenté notamment Amiens et St Quentin
A ma grande famille mes parents, mes grands-parents, mes frères et sœurs ainsi que mes neveux et nièces qui ont cru en moi malgré les difficultés
6
Résumé
RADIOTHERAPIE ADJUVANTE DANS LES TUMEURS EPITHELIALES THYMIQUES: A PROPOS D’UNE COHORTE RETROSPECTIVE DE 134 PATIENTS TRAITES A L’INSTITUT GUSTAVE ROUSSY DE VILLEJUIF ENTRE 1990 ET 2011.
Introduction.- Les tumeurs épithéliales thymiques (TET) sont des tumeurs rares avec une incidence de 250 nouveaux cas par an en France. Elles se développent à partir des cellules épithéliales du thymus et jamais à partir des cellules lymphocytaires. L’âge moyen de la survenue de ces tumeurs est de 50 ans mais elles peuvent se voir à tous les âges de la vie. Il n’existe pas de facteur de risque identifié à nos jours. Sur le plan histologique, elles sont classées actuellement selon la classification de l’OMS et leur extension se définit selon les stades de Masaoka-koga.
Le traitement principal des TET est la chirurgie. L’intérêt de la radiothérapie est discuté en fonction des stades Masaoka-Koga de la maladie.
Objectifs de l’étude.- L’objectif principal de notre étude est de comparer les survies globale et spécifique et le contrôle local, régional et métastatique des patients ayant une TET et traités à L’institut Gustave Roussy (IGR) avec les données de la littérature.
Patients et méthodes.- Il s’agit d’une étude rétrospective, monocentrique de type cohorte. Tous les patients inclus avaient une TET et ont été traités par radiothérapie adjuvante à l’IGR entre 1990 et 2011.
Résultats.- Cent-trente-quatre patients dont 74 hommes et 60 femmes (ratio H/F=1,23) ont été inclus. L’âge médian au diagnostic était de 53 ans [16-83]. Quatre-vingt-huit pour cent des patients avaient un Performans Status (PS) selon l’OMS à 0 et 12% à 1. La maladie a été révélée par un syndrome paranéoplasique chez 38% des patients dont 27,6% des patients avaient une myasthénie, des signes cardiorespiratoires chez 41% et découverte fortuite chez 10,4%. Parmi ces 134 patients, 52 (38,8%) patients avaient une TET de stade I/II de Msaoka-Koga, 37 (27,6%) patients avaient une maladie de stade III et 45 (33,6%) autres avaient une maladie de stade IV. Cent un (75,4%) patients avaient une TET de type thymome malin selon la classification OMS, 28 (20,9%) avaient une TET de type carcinome thymique et 5 (3,7%) avaient des TET non classées. Soixante-deux (46,3%) patients avaient développé un syndrome paranéoplasique dont 46 (34,3%) myasthénies.
Le suivi médian dans notre cohorte était de 8,8 ans (1,1-24,9 ans).
Concernant le traitement, 100% des patients avaient eu un traitement chirurgical de leur tumeur ainsi qu’une radiothérapie adjuvante dont 71 (53%) patients avaient eu une irradiation ganglionnaire prophylactique. Vingt et un (15,7%) patients avaient eu une chimiothérapie première et 13 (9,7%) patients avaient eu une chimiothérapie adjuvante. Cent (74,6%) patients avaient une chirurgie complète (R0), 29 (21,6%) patients avaient une chirurgie R1 et 5 (3,8%) autres patients avaient eu une chirurgie macroscopiquement incomplète (R2).
Concernant le contrôle de la maladie, cinquante (37,3%) patients avaient eu une rechute locorégionale dont 26 patients avaient eu au moins une rechute locale et 42 patients avaient eu au moins une rechute régionale. Parmi les 26 rechutes locales, 12 (46,2%) étaient des rechutes pleurales, 7 (26,9%) étaient médiastinales, 4 (15,4%) étaient pleuro-médiastinales et 3 (12%) étaient pleuropéricardiques. parmi les 42 rechutes régionales, il y avait 36 (85,7%) récidives pleuro-diaphragmatiques, 2 (4,8%) récidives ganglionnaires, 3 (7,1%) récidives
7 pulmonaires et une (2,4%) récidive au niveau latéro-vertébral; Le taux de contrôle
locorégional à 5 était de: 67% (IC 95%: 59-76%), à 10 ans: 49% (IC 95%: 49-68%).
Douze (9%) patients avaient eu une progression métastatique de leur maladie ; Le taux de contrôle métastatique était de: 91% (IC 95%: 86-96%) à 5 ans, de 91% (IC 95%: 86-96%) à 10 ans. La survie spécifique à 5 ans était de 86% (IC 95%: 80-92%) et à 10 ans était de 72% (IC 95%: 63-82%). La survie globale à 5 ans était de 82% (IC 95%: 75-88%) et à 10 ans de 63% (IC 95%: 54-73%). Parmi les facteurs analysés pouvant influencer la survie globale seuls le Performans Status > 1 avec HR : 2,6 IC95% (1,2-5,5) avec p=0,01 et un stade IV de Masaoka-Koga avec un HR : 2,1 IC 95% (1,1-4) avec p=0,02 étaient sortis comme facteurs de mauvais pronostique en analyse multivariée. La survie sans progression à 5 ans était de 61% (IC 95%: 53-70%) et à 10 ans de 53% (IC 95%: 44-62%). La survie moyenne sans progression était de 13,2 ans (IC 95%: 11,2-15,2 ans) et la survie médiane sans progression était de 11,8 ans (IC 95%: 5,2-non atteint).
Conclusion.- notre étude conclut comme rapporté dans la littérature que la classification de Masaoka-Koga reste le seul outil pronostique des tumeurs épithéliales thymiques après un traitement chirurgical bien conduit. La radiothérapie adjuvante reste un traitement
indiscutable pour les stades III et IV par ailleurs pour les stades II avec résection complète (R0) son indication doit tenir compte de la taille tumorale, de son type histologique et sa validation doit être faite par un comité d’experts selon les recommandations du réseau RYTHIC.
Par ailleurs, des études prospectives doit être menées afin de mieux poser les indications des différentes modalités thérapeutiques, compte tenu de la complexité de ces tumeurs.
8
Introduction
Les tumeurs épithéliales thymiques (TET) sont des tumeurs rares pouvant se voir à tous les âges de la vie (6 mois -90 ans) avec un âge moyen au diagnostic de 53 ans (1)(2). Elles regroupent les thymomes malins (TM) et les carcinomes thymiques (CT) selon la définition de l’OMS 2004(3). Compte tenu de leur rareté, leur prise en charge doit se faire dans des centres spécialisés afin de garantir une prise en charge optimale au patient. La prise en charge doit être multidisciplinaire (chirurgien, radiothérapeute, oncologue médical et anatomopathologiste) et se baser sur les recommandations du réseau national (RYTHMIC) ou internationales (ITMIG).
L’institut Gustave Roussy de Villejuif (IGR) est l’un des centres experts de France dans la prise en charge et le traitement des TET.
L’objectif principal était d’étudier la survie globale et la survie spécifique et les contrôles
local, régional et métastatique des patients ayant une TET et traités à L’institut Gustave Roussy (IGR) comparés aux données de la littérature et cela en fonction des différents facteurs ( Stade de Masaoka-Koga, classification histologique de l’OMS, la qualité de la résection (R0, R1 ou R2), les doses de radiothérapie).
1. Epidémiologie :
Les TET sont des tumeurs rares, représentant environ 0.07% des 350 000 nouveaux cancers par an en France(4). L’incidence est d’environ 250 nouveaux cas/an en France soit un taux d’incidence de 0.4 pour 100 000 habitants(5). Aux USA, le taux d’incidence des TET est de 0.13 à 0.15 pour 100 000 habitants(6). Par ailleurs la prévalence réelle de ces tumeurs dans la population générale reste sous-estimée compte tenu de l’évolution longtemps
asymptomatique de ces tumeurs (découverte fortuite le plus souvent).
Les TET font partie des tumeurs médiastinales. Elles représentent 20% de toutes les tumeurs médiastinales et 50% des tumeurs du médiastin antérieur(5)(7). La localisation des TET est médiastinale dans 90% des cas, avec un développement à partir de la loge thymique. Il existe toutefois des formes ectopiques dues à la présence d’ilots thymiques ectopiques,
notamment au niveau cervical(8)(9).
2. Anatomie, embryologie et fonction du thymus
Le thymus est une glande située dans le médiastin antéro-supérieure (figure 1). Il est constitué de deux lobes, subdivisés en plusieurs lobules par une capsule conjonctive. Il est formé de deux contingents cellulaires qui sont les cellules épithéliales thymiques et les lymphocytes. On y distingue deux structures différentes : la corticale et la médullaire qui contient les corpuscules de Hassal (figure 2). Le thymus fait partie des organes lymphoïdes secondaires et joue un rôle important dans l’immunité et aussi dans la tolérance du soi. Il est le centre de maturation et d’éducation des lymphocytes T immatures ou
prothymocytes(10)(11). En effet, le thymus participe à la prolifération, la différenciation et la sélection des lymphocytes T immatures qui proviennent de la moelle osseuse. Les
9 lymphocytes T dirigés contre les antigènes du soi sont éliminés ou réprimés dans le thymus et n’arriveront pas à maturation afin d’éviter les différentes maladies auto-immunes(10)(11). Sur le plan embryologique, le thymus dérive à la fois de l’ectoderme et l’endoderme. La partie provenant de l’ectoderme forme la corticale et la partie provenant de l’endoderme la médullaire(10).
Figue 1 : situation de la loge thymique et ses rapports anatomiques
10
3.
Classification histologique de l’OMS
(3)(5)(8) :Les TET se développent à partir des cellules épithéliales du thymus. Le terme « tumeur épithéliale thymique » inclut les thymomes malins et les carcinomes thymiques. Les carcinomes thymiques regroupent les carcinomes épidermoïdes et les tumeurs
neuroendocrines thymiques. Les thymomes malins représentent environ 80% des TET, les carcinomes épidermoïdes 15% et les carcinomes neuroendocrines 5%(5)(12).
Les TET affectent préférentiellement l’adulte, avec un âge moyen au diagnostic de 53 ans. Cependant, ces tumeurs peuvent survenir à tous les âges de la vie [6 mois-90 ans])(1)(13). Les TET atteignent de façon équivalente l’homme et la femme avec un ratio H/F voisin de 1(5).
Historiquement de nombreuses classifications histo-pathologiques des TET ont été utilisées. Les plus utilisées sont celles de Marino et Muller-Hermelink(14)(15)(16)(17), Verley et Hollman(18), Lewis et al(19), Bernartz et al(20), Levine et Rosai(21).
Actuellement, la classification histologique des TET proposée par l’OMS est la plus utilisée et remplace ainsi les classifications sus-citées. Cette classification a été proposée pour la première fois en 1999(22) et a été actualisée en 2004(3) afin d’optimiser et d’harmoniser les pratiques. C’est une classification qui se base sur la morphologie des cellules épithéliales et le ratio lymphocytes/cellules épithéliales. Cette classification a défini 3 types tumoraux et utilise un système de lettres (A, B et C) et de chiffres (1, 2 et 3)(22)(3).
Les lettres font référence à la morphologie des cellules épithéliales. La lettre A correspond à « Atrophic » et la lettre B à « Bio-active ». Les chiffres 1, 2 et 3 correspondent au degré des atypies retrouvées et le ratio cellules tumorales / cellules lymphocytaires(23).
Cette classification peut être un outil pronostique comme l’a démontré Kondo dans son étude portant sur 100 patients traités pour une TET entre 1973 et 2001. La survie sans progression (PFS) et la survie globales à 10 ans étaient différentes selon les types
histologiques. La PFS à 10 ans était de 100% pour les types A et AB, de 83% pour les types B1 et B2, de 36% pour le type B3 et de 26% pour les carcinomes thymiques. La différence est significative pour la PFS entre le groupe A/AB et le groupe B1/B2 (P=0.046) et entre le groupe B3 et carcinomes thymiques avec un p=0,042.
La survie globale à 10 ans était de 100% pour les types A et AB, de 94% pour les types B1 et B2, de 92 % pour les type B3 et 58% pour les carcinomes thymiques(24).
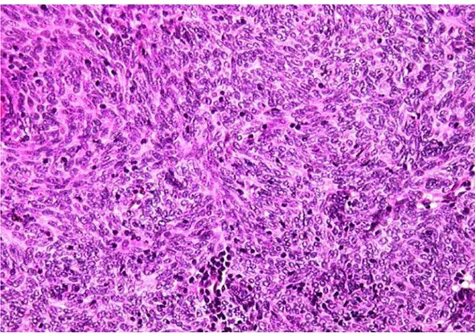
Les tumeurs thymiques de type A (figure3) : Elles représentent environ 4% à7% de toutes les
TET(12) et elles sont composées de cellules épithéliales fusiformes ou polygonales avec absence d’atypie cellulaire et correspondraient aux « thymomes bénins » bien encapsulés. La présence de lymphocytes est rare, ils sont souvent matures et co-expriment les
cytokératines et focalement le CD-20. Les diagnostics différentiels avec le type A sont les thymomes de type B3 et les tumeurs neuroendocrines.
11
Figure 3: Thymome type A (X100). D’après THOMAS et al(8)
Les tumeurs thymiques de type AB (figure 4) : Elles constituent entre 28% à 34% des
TET(12). Elles sont constituées de zones de type A et de zones de type B ou anciennement appelées tumeurs mixtes. Elles sont composées à 50% de cellules lymphocytaires. Les cellules épithéliales les composant sont de petites tailles ainsi que leurs noyaux qui sont pales.
Figure 4: Thymome type AB (X100) D’après THOMAS et al(8)
Les tumeurs thymiques de type B1 (figure 5) : leur fréquence est de 9% à 20% de toutes les
TET(12) et sont constituées de zones dites sombres (corticales) où on retrouve des cellules épithéliales qui sont rares, polygonales avec un noyau pale et un petit nucléole et des cellules lymphocytaires normales les entourant. Des zones claires (médullaires) où on peut trouver des corpuscules de Hassal. Les diagnostics différentiels à évoquer sont les thymomes de type B2, l’hyperplasie thymique et les lymphomes lymphoblastiques T.
12 Figure 5: Thymome type B1 OMS (x100). D’après THOMAS et al(8)
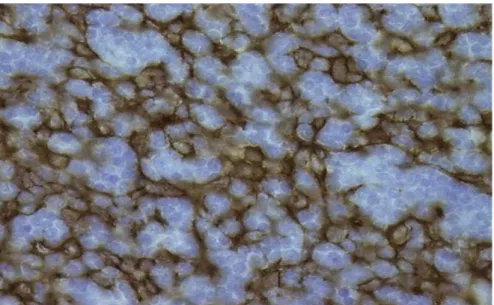
Les tumeurs thymiques de type B2 (figure 6 et 7) : Ce sont des tumeurs à prédominance
lymphocytaire et représentent environ 20% à36% de toutes les TET(12). Elles sont
constituées de grandes cellules épithéliales polygonales avec de gros noyaux à chromatine dense et des nucléoles proéminents. Ces cellules épithéliales sont clairsemées au sein de cellules lymphocytaires très abondantes formant ainsi des structures en réseau lâches mais peuvent également rarement former des formations organoïdes. Les espaces
péri-vasculaires sont fréquents. Un marquage par un marqueur épithélial à l’immunohistochimie montre cette structure en réseau (figure 7). Les diagnostics différentiels sont le thymome type B1 ou lymphome lymphoblastique T.
13
Figure 7 : Thymome type B2 immunohistochimie au marqueur épithélial Kl1 ( X200). D’après THOMAS et al(8)
Les tumeurs thymiques de type B3 (figure 8) : Elles représentent entre 10% à 14% des
TET(12) et sont formées de lobules à prédominance de cellules épithéliales polygonales ou rondes avec des noyaux irréguliers et nucléolés. La présence de lymphocytes est rare, souvent immatures. Ces tumeurs renferment assez souvent des espaces péri-vasculaires. Les diagnostics différentiels avec ce type de thymome et le type A ou le carcinome
thymique.
Figure 8: Thymome type B3 OMS (X200). D’après THOMAS et al(8)
Il existe deux autres types de tumeurs épithéliales thymiques non inclus dans cette
classification qui sont les thymomes micronodulaires à stroma lymphoïde B hyperplasique et les thymomes à stroma pseudo sarcomateux ou thymomes sarcomatoïdes.
Les tumeurs de type C (figure 9, 11) : Ces tumeurs sont regroupées sous le terme de
carcinomes thymiques dans la classification OMS 2004(3) , elles incluent les carcinomes épidermoïdes et les tumeurs neuroendocrines. Elles représentent environ 20% de toutes les TET(12).
14 - Les carcinomes épidermoïdes :
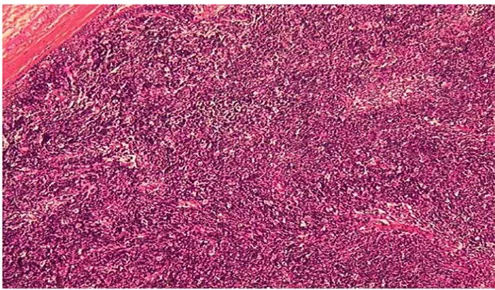
Ils sont caractérisés par l’absence de lobule et la présence d’importantes atypies cellulaires. Macroscopiquement, ils sont très infiltrants contrairement aux thymomes (figure 9 et 10). Microscopiquement, ils sont constitués de prolifération de cellules épithéliales malignes comportant de très importantes atypies renfermées dans un stroma fibreux, siège de réactions inflammatoires où on peut constater l’absence de lobule (figure11). Le diagnostic différentiel peut se poser avec une métastase thymique d’un carcinome bronchique ou d’autres organes.
Les carcinomes épidermoïdes sont subdivisés en tumeurs de bas grade de malignité (incluant les carcinomes épidermoïdes bien différenciés, les carcinomes basaloïdes et les carcinomes mucoépidermoïdes bien différenciés) et en tumeurs de moyen à haut grade de malignité (regroupant les tumeurs à cellules claires et les carcinomes indifférenciés).
Figure 9 : Image macroscopique d’un Figure 10 : Image d’un thymome.
Carcinome thymique. Daprès THOMAS et al(8) D’après THOMAS et al(8)
15
Figure 11 : Carcinome thymique (aspect histologique d’une tumeur malpighienne bien
différenciée) (X400). D’après THOMAS et al.(8)
Les tumeurs thymiques neuroendocrines (NE) figure 12 : Elles représentent environ 5% de
toutes les TET(12) et sont séparées en deux groupes dans la classification de l’OMS de 2004 : les carcinomes neuroendocrines bien différenciés, représentés par les carcinoïdes typiques et atypiques, et les carcinomes neuroendocrines peu différenciés, représentés par les carcinomes neuroendocrines à petites et à grandes cellules. Macroscopiquement, ces
tumeurs sont d’aspect homogène couleur beige-chamois et leur taille peut atteindre jusqu’à 20 cm. Microscopiquement, les carcinoïdes sont caractérisés par leur aspect endocrinoïde constitués de travées, cordons, nids et rosettes séparés par des tractus conjonctivo-vasculaires. L’index mitotique (inférieur ou supérieurs ou égal à 2 pour dix champs) et la présence ou non de nécrose classent ces tumeurs soit en typiques ou atypiques (figure 12). Sur le plan immuno-histochimique, ces tumeurs sont positives aux marqueurs des tumeurs neuroendocrines (chromogranine, synaptophysine et CD56) et elles sont également positives à la cytokératine.
Comme diagnostic différentiel de ces tumeurs, on peut évoquer une métastase thymique d’une tumeur neuroendocrine d’un autre site (poumon), le paragangliome médiastinal et le thymome de type A.
Les tumeurs endocriniennes thymiques peuvent se voir dans le cadre des néoplasies endocriniennes multiple de type 1 (NEM1).
16
Figure 12 :Tumeur carcinoïde (×400). D’après Thomas et al(8)
Cependant, il existe des formes frontières entre les différents types ce qui représente environ 15% des TET. Vingt-cinq pour cent des TET ont une histologie hétérogène
comportant plusieurs types à la fois(3)(5).
Parmi les autres classifications, on peut citer celle de Marino et Muller-Hermelink qui est une classification beaucoup utilisée avant la généralisation de la classification OMS. C’est une classification dite histo-fonctionnelle(14). Elle classe les TET en 6 groupes en fonction de la ressemblance entre la différentiation des cellules épithéliales tumorales et celle des cellules corticales ou médullaires normales. On a donc pu définir :
- les tumeurs médullaires,
- les tumeurs corticales,
- les tumeurs à prédominance corticale,
- les tumeurs mixtes,
- les carcinomes bien différenciés et
- les carcinomes de haut grade
4. Diagnostics différentiels :
Les deux principaux diagnostics différentiels des TET par ordre de fréquence sont les
lymphomes et les tumeurs germinales.
- Les lymphomes : Ils posent un problème diagnostique avec certaines TET compte
tenu de leur localisation cervico-médiastinale et de la richesse de certaines TET en cellules lymphocytaires. C’est le cas des formes indifférenciées ou à population Lymphocytaire presque exclusive (les thymomes B1 et B2).
- Les tumeurs germinales : Elles font partie des diagnostics à évoquer devant toute
tumeur médiastinale. Leur élimination est rendue plus facile avec les nouvelles techniques d’immunohistochimie et les marqueurs tumoraux spécifiques (B-HCG et AFP).
17
5. La classification de Masaoka-Koga
La classification de Masaoka-Koga est une classification anatomo-clinique, se basant sur les résultats de la pièce opératoire et non sur la biopsie (classification post opératoire). Elle a été proposée pour la première fois en 1981 par Masaoka et al(1) puis révisée en 1994par Koga et al(25). Cette classification a un intérêt pronostique, car plusieurs études avaient démontré la relation entre les différents stades et la survie(26). Elle classe les tumeurs thymiques en quatre stades de I à IV en fonction de l’atteinte des différentes structures médiastinales (Tableau 1).
Tableau 1 : Proposition ITMIG de 2011 des critères principaux pour la classification Masaoka
Koga(5)(26).
Stades de Masaoka Critères diagnostiques et consensus ITMIG
Stade I - Tumeur complètement encapsulée, macroscopiquement et microscopiquement
- Pas d’extension à la graisse médiastinale
Ce groupe inclut les tumeurs invasives ne franchissant pas la capsule et les tumeurs sans capsule mais sans invasion des tissus périphériques
Stade IIA - Invasion microscopique trans-capsulaire (≤ 3mm, confirmation anatomopathologique)
Stade IIB - Infiltration macroscopique dans la graisse péri-thymique - Invasion macroscopique du thymus normal ou de la graisse
péri-thymique confirmée à l’anatomopathologie
- Adhérences macroscopiques -mais sans invasion- à la plèvre médiastinale ou au péricarde
Stade III - Extension macroscopique aux organes adjacents (péricarde, gros vaisseaux et les poumons)
Sont inclues dans ce stade les tumeurs avec invasion microscopique de la plèvre médiastinale ou viscérale, du
péricarde, invasion directe du parenchyme pulmonaire, invasion du nerf phrénique ou du nerf vague ou des gros vaisseaux et cela à l’examen anatomo-pathologique
Stade IVA - Tumeurs avec greffes pleurales et ou péricardiques différentes de la tumeur principale
Stade IVB - Métastases ganglionnaires médiastinales, cervicales ou extra-thoraciques
- Métastases hématogènes (parenchyme pulmonaire) et extra-thoraciques
6. Manifestations cliniques et circonstances de découverte
L’évolution des TET est lente et souvent asymptomatique. Leur diagnostic se fait chez un tiers des patients fortuitement lors de réalisation d’examens d’imagerie tels qu’une radiographie thoracique ou un scanner thoracique pour d’autres indications(27)(28). Seulement 35% des cas sont révélés par des signes cliniques locaux notamment douleur thoracique, dyspnée, toux et syndrome cave supérieur(28). Les manifestations
paranéoplasiques révèlent environs 30% des TET avec pour les tymomes la myasthénie, erythroblastopénie, hypo-gammaglobulinémie ou syndrome de good et hypercalcémie pour les carcinomes thymiques ou les NEM1 pour les tumeurs NE(27)(28).
18
a. Les syndromes paranéoplasiques et TET
Les TET sont connues pour être associées à des syndromes paranéoplasiques. Les
manifestations paranéoplasiques sont différentes en fonction du type tumoral. Concernant les thymomes, les syndromes paranéoplasiques souvent rencontrés sont la myasthénie, l’erythroblastopénie (EBP), hypo-gammaglobulinémie (syndrome de good) et d’autres syndromes plus rares (tableau2)(28)(5)(29).
Pour les carcinomes épidermoïdes thymiques , le syndrome paranéoplasique le plus fréquent est l’hypercalcémie par hypersécrétion de PTH-rp(28).
Pour les carcinomes neuroendocrines, ils sont souvent associés à des troubles endocriniens dans environs 30% des cas(28)(30) tels que le syndrome de cushing , les néoplasies
endocriniennes multiples de type 1 (NEM1) qui se caractérisent par l’association chez un même patient d’au moins 2 des 5 atteintes suivantes : hyperparathyroïdie, tumeur endocrine du duodénum ou du pancréas, tumeur de l’hypophyse, atteinte des glandes surrénales, tumeur endocrine du thymus ou des bronches(30).
- La myasthénie :
Environ 30% à 50% des thymomes sont associés à une myasthénie(29)(28). La myasthénie est associée dans 50% des cas à une hyperplasie thymique et dans 15% à 20% des cas à une TET. Les TET souvent associées à la myasthénie sont de type B1 ou B2, survenant chez des patients âgés de plus de 40 ans(31)(32).
La myasthénie peut révéler une TET ou elle peut survenir dans un certain nombre de cas après le traitement de la TET. Dans 5% des cas de TET avec myasthénie on retrouve un ou plusieurs autres syndromes paranéoplasiques associés(28).
La myasthénie est une maladie auto-immune, neuromusculaire qui engendre une fatigabilité musculaire. Elle est due à la synthèse d’auto-anticorps dirigés contre les récepteurs de l’acétylcholine (anti RACh) bloquant ainsi la transmission du flux nerveux au niveau de la jonction neuromusculaire(33).
Le diagnostic de myasthénie est basé sur un faisceau d’arguments clinico-biologiques : Sur le plan clinique, l’atteinte des muscles oculomoteurs est la plus fréquente se traduisant par une diplopie et un ptosis.
D’autres atteintes musculaires sont à rechercher notamment une fatigabilité des membres et des muscles du cou, une atteinte bulbaire (voix nasonnée, fausses routes, trouble de la motricité linguale).
Devant toute suspicion de myasthénie, un dosage des anticorps anti-RACh est légitime. La positivité de ces anticorps et la présence de signes cliniques confirment le diagnostic. Si les anticorps reviennent négatifs en présence de signes cliniques évocateurs un
électromyogramme (EMG) peut affirmer ou infirmer le diagnostic. Le signe recherché sur l’EMG est la présence de décrément. (5)(33).
Si la myasthénie est confirmée, sa prise en charge doit être faite par un médecin référent (neurologue, interniste) pour stabiliser la myasthénie et anticiper ainsi une éventuelle décompensation (crise myasthénique sévère) lors du traitement chirurgical de la TET(5).
19 Tableau 2 : les différentes manifestations paranéoplasique rencontrées dans les TET(5)(28)(29)
Les différents syndromes paranéoplasiques Manifestations hématologiques :
- Erythroblastopénie - Thrombopénie - Pancytopénie
- Anémie hémolytique auto-immune - Myélome multiple Troubles neuro-musculaires : - Myasthénie - Syndrome de Lambert-Eaton - Syndrome de Morvan - Encéphalite limbique
- Atteinte neuronale périphérique - Syndrome de l’homme rigide Maladies de système auto-immunes
- Lupus érythémateux disséminé
- Syndrome des anticorps anti-phospholipides (SAPL) - Syndrome de Sjogrën
- Polyarthrite rhumatoïde (PR) - Polymyosite et myosite - Sarcoïdose
- Myocardite non infectieuse - Sclérodermie
- MICI (maladies inflammatoires intestinales) Troubles endocriniens : - Syndrome de cushing - Maladie d’Addison - Thyroïdite d’Hashimoto - Hyperparathyroïdie Déficit immunitaire :
- Hypogammaglobulinémie (Syndrome de Good) Pathologies rénales :
- Syndrome néphrotique Maladie dermatologiques :
- Pemphigus
- Candidose cutanéomuqueuse chronique Second cancer :
- Cancers solides (poumon, sein, colon, thyroide, glioblastome - Hémopathies malignes (lymphomes, myélomes multiples) - Cutanés (sarcome de kaposi)
- L’hypogammaglobulinémie ou le syndrome de Good :
C’est l’association d’une hypogammaglobulinémie (déficit immunitaire) et d’infections broncho-pulmonaires ou de la sphère ORL sévères et récurrentes dans le cadre d’une tumeur thymique. Il représente environ 7 à 13% des hypo-gammaglobulinémies acquises et touche souvent l’adulte entre 40 ans et 70 ans(34)(35).
20 Le diagnostic est souvent fait de façon concomitante à la tumeur thymique (80% des cas). Le syndrome peut également survenir jusqu’à 5 ans après le traitement d’une tumeur thymique d’où l’intérêt d’une surveillance attentive chez tout patient traité pour une TET(35).
Le diagnostic se base sur des éléments clinico-biologiques. Outre le contexte sus cité, l’électrophorèse des protéines sériques retrouve une hypo-gammaglobulinémie avec un myélogramme normal (éliminer un myélome).
Le traitement du syndrome de Good est basé sur le traitement de la tumeur thymique et d’une éventuelle infection par une antibiothérapie et des injections d’immunoglobulines tant que persiste l’hypo-gammaglobulinémie(34).
7. Le bilan pré-thérapeutique :
a- L’examen clinique :
L’examen clinique est un élément essentiel dans la prise en charge de tout patient. Il comporte l’interrogatoire et l’examen physique. A l’interrogatoire, le médecin doit rechercher une altération de l’état général, des signes cliniques pouvant évoquer un syndrome paranéoplasique (signe myasthéniques, épisode infectieux), les antécédents personnels et familiaux afin de mieux cerner le terrain.
L’examen physique recherchera des signes liés à la tumeur elle-même (dyspnée,
épanchement pleural, syndrome cave supérieur et des adénopathies sus-claviculaires et cervicales) et également des signes pouvant être en relation avec un syndrome
paranéoplasique associé.
b- Le bilan biologique :
Le bilan biologique doit comporter un bilan préopératoire (ionogramme sanguin, groupe sanguin, RAI, et TP, TCA), un bilan à la recherche d’un syndrome paranéoplasique: anticorps antiRAch (myasthénie), électrophorèse des protéines avec dosage pondéral des
immunoglobulines (syndrome de Good et myélome multiple), numération formule sanguine avec réticulocytes ( érythroblastopénie), anticorps antinucléaires, la TSH et enfin un dosage de l’alpha foetoprotéine, B-HCG afin d’éliminer une tumeur germinale.
Si le patient est en âge de procréer une cryoconservation du sperme ou d’ovules est indispensable, surtout si une chimiothérapie est envisageable(5).
c- Les examens d’imagerie :
Le scanner thoracique injecté avec des coupes abdominales hautes est l’examen de
référence dans le bilan pré-thérapeutique des TET, qui permet d’apprécier la taille tumorale, son extension aux structures voisines, la présence d’adénopathies médiastinales ou des lésions du parenchyme pulmonaire(5).
L’IRM thoracique est rarement utile sauf lors d’une suspicion d’atteinte cardiaque ou des gros vaisseaux(36) ou doute sur une hyperplasie thymique(5).
Le TEP scan est de plus en plus utilisé surtout lorsque la tumeur est invasive ou en cas de récidive(5). Il permet de mieux apprécier l’atteinte ganglionnaire quand persiste un doute au scanner et également il constitue une aide appréciable au radiothérapeute pour mieux guider le traitement de radiothérapie (fusion des imageries).
21
d- Les biopsies tumorales :
La pratique de biopsies pour avoir une preuve histologique devant la suspicion d’une tumeur thymique est abandonnée. Les progrès en imagerie permettent en effet le plus souvent de poser le diagnostic de tumeur thymique. La biopsie reste cependant indispensable quand subsiste un doute sur la nature de la tumeur médiastinale (doute sur un lymphome) ou lorsque la tumeur est inopérable d’emblée(27).
e- Autres examens :
En cas de suspicion d’atteinte bronchique la fibroscopie bronchique est justifiée.
Les explorations respiratoires fonctionnelles sont réalisées avant le traitement afin d’estimer la fonction respiratoire(5)
8. Le traitement des TET
Le réseau RYTHMIC a mis en place des réunions de concertation pluri-disciplinaires
régionales et nationales où les modalités thérapeutiques de chaque patient présentant une tumeur épithéliale thymiques sont discutées.
8.1. Chirurgie
La chirurgie est le traitement principal des TET, elle doit être réalisée chez tous les patients présentant une TET en absence de contre-indication chirurgicale. L’abord de la cavité médiastinale par voie trans-stérnale médiane est la voie d’abord de première
intention(37)(38). D’autres voies d’abord peuvent être privilégiées quand la tumeur présente une extension très importante. Par exemple, réalisation d’une thoracotomie antérieure bilatérale avec stérnotomie transverse ou longitudinale partielle avec extension
antérolatérale quand la tumeur présente une atteinte pleurale ou lorsqu’une exérèse pulmonaire est prévue(5). La chirurgie mini-invasive vidéo assistée peut être réalisée dans le cas où la tumeur est de petite taille et bien encapsulée. Dans ce cas, la pièce opératoire est extraite via un sac protecteur à travers un orifice bien adapté à la taille du sac afin de ne pas fragmenter la pièce opératoire(38).
La première étape dans la chirurgie des TET est l’inspection de tout le médiastin et les cavités pleurales par le chirurgien. Le chirurgien évalue ensuite l’aspect macroscopique de la loge thymique, la présence ou non d’une invasion de la capsule thymique, de la graisse médiastinale et la présence ou non d’adhérences avec les différentes structures
médiastinales (la plèvre, le péricarde, les poumons, le cœur, les nerfs phréniques, les gros vaisseaux et les ganglions). Tout ceci doit être noté dans le compte rendu opératoire afin de classer la tumeur selon les stades de Masaoka-Koga et guider la radiothérapie adjuvante. La chirurgie doit consister en une thymectomie et thymomectomie en un seul bloc sans
effraction de l’interface tumorale(39). La chirurgie doit être la plus complète possible et cela en faisant des résections élargie aux organes atteints notamment à la plèvre, au péricarde, au parenchyme pulmonaire et sacrifice d’un nerf récurrent(40). Si la tumeur présente un envahissement des structures voisines, une chirurgie en bloc est de mise selon les
recommandations ITMIG(38). Si le chirurgien n’est pas sûr d’obtenir une résection complète à un endroit donné, un marquage par des clips est nécessaire afin d’optimiser le traitement de cette zone par la radiothérapie(28)(38).
22 Pour les thymomes malins, le curage ganglionnaire n’est recommandé que pour les stades III et IV de Masaoka-Koga et ne doit concerner que le médiastin antérieur. Le taux d’atteinte ganglionnaire de ces tumeurs reste faible : environ 2%, avec moins de 1% pour les Stades I à 6% pour les stades III(41). Par ailleurs, un curage ganglionnaire étendu (médiastin antérieur, les autres sites intra-thoraciques, l’aire sus-claviculaire et la base du cou) est recommandé pour les carcinomes thymiques qui donnent assez souvent des atteintes ganglionnaires diffuses(37).
La résection complète est obtenue dans 100% des stades I, environ 85% des stades II, 47% pour les stades III et 26% pour les stades IV(27).
La mortalité post opératoire est d’environ 2,5% avec un intervalle allant de 0,7% à 4,9%(27). Les autres complications post opératoires sont estimées à 20%(42).
La chirurgie peut être réalisée dans un but de réduction tumorale ou « débulking » afin d’optimiser un traitement par radiothérapie.
Figure 13 et 14 : Orientation d’une pièce opératoire par le chirurgien. D’après THOMAS et
al(8)
8.2. Radiothérapie
- Indications
La radiothérapie est l’un des principaux traitements des TET. Elle est soit réalisée en
adjuvant après un traitement chirurgical bien conduit comme elle peut aussi, plus rarement, être réalisée en première intention (néo-adjuvant ou exclusive) ou à titre palliatif. La
radiothérapie préopératoire a en général pour objectif principal une réduction tumorale afin d’obtention un traitement chirurgical optimal.
23
a- Radiothérapie adjuvante ou radiothérapie post opératoire :
Après un traitement chirurgical bien conduit, la radiothérapie a pour objectif d’améliorer le contrôle local, de la survie sans progression et donc de la survie globale en réduisant le risque de récidive locorégionale. L’indication de la radiothérapie adjuvante dépend du stade Masaoka-Koga, des limites de résection (complète ou non) et du type histologique selon l’OMS (thymomes vs carcinomes thymique). Dans tous les cas, la radiothérapie doit être réalisée dans les 3 mois suivants le traitement chirurgical(5)(37)(28).
Jusque dans les années quatre-vingt, la radiothérapie adjuvante était réalisée chez tous les patients traités pour une tumeur thymique, indépendamment de son stade ou des limites de sa résection (complète ou non complète). Cette attitude a été remise en question par des travaux récents.
Pour les stades I de Masaoka-Koga, La radiothérapie adjuvante n’est plus indiquée. En effet Zhang et al avaient démontré dans une étude prospective randomisée de 29 patients traités pour une TET de stade I de Masaoka-Koga l’absence de bénéfice de la radiothérapie
adjuvante. La survie globale à 10 ans était de 88% pour le bras radiothérapie adjuvante contre 92% pour le bras chirurgie seule avec P>0.05(43). Le taux de récidive locorégionale de ces tumeurs de stade I était faible : entre 2 à 3% à 10 ans(44)(45)(2). Forquet et al n’avaient pas retrouvé de bénéfice à la radiothérapie adjuvante pour les stades I. La survie globale à 5 ans était de 91% pour les patients traités par radiothérapie adjuvante contre 98% pour les patients traités par chirurgie seule(45).
L’intérêt de la radiothérapie post opératoire chez les patients traités pour des tumeurs de stade II et III de Masaoka-Koga en résection complète après un traitement chirurgical a été remis en cause.
Le taux de récidive tumorale pour les tumeurs de stade II est estimé entre 5 à 15%, celui des tumeurs de stade III entre 20% à 40% et celui des stades IV entre 25% et 50%(5).
Les indications de la radiothérapie adjuvante selon le dernier référentiel RYTHMIC ont été définies en fonction de la qualité de la résection (R0 ou R1/R2), du stade Masaoka-Koga, et du type histologique selon la classification de l’OMS(5).
Les indications de la radiothérapie sont les suivantes :
- si la résection est complète, la radiothérapie n’est pas retenue chez les patients ayant
une maladie de stade I de Masaoka-Koga, de stade IIA de type A à B2 et de stade IIB de type A à B1. En revanche, la radiothérapie est indiquée chez les patients avec une tumeur thymique de stade IIA de type histologique B3, de stade IIB de type
histologique B2 et B3, de stade III et IV quel que soit le type histologique et les carcinomes thymiques quel que soit leur stade de Masaoka-Koga(5)(46).
- Si la résection est incomplète (R1), la radiothérapie est indiquée chez tous les
patients. Si la résection est R2 ou dite de débulking, la radiothérapie dans ce cas est souvent associée à une chimiothérapie(5)(46). Ces indications ont été justifiées après les résultats de plusieurs études de larges effectifs (bases de données américaines et japonaises)(45)(47)(48).
En effet, Forquet et al avaient analysé 626 patients de la base de données américaine (SEER), traités pour une tumeur épithéliale thymique de stade II ou III de Masaoka-Koga par
24 chirurgie et radiothérapie adjuvante. Ils avaient démontré l’existence d’un bénéfice en faveur de la radiothérapie adjuvante pour les stades II et III avec une survie globale à 5 ans de 76% pour les patients traités par radiothérapie adjuvante contre 66% pour les patients traités par chirurgie seule avec un p =0,01. En analyse de sous-groupes (résection complète versus résection incomplète), le bénéfice de la radiothérapie adjuvante n’était retrouvé que chez les patients ayant eu une résection incomplète (R1 ou R2) de leur tumeur. En d’autres termes, la radiothérapie adjuvante pour les stades II et III en résection complète dans cette analyse n’avait pas de bénéfice en terme de survie sans progression ni en survie globale(45). Omasa et al dans leur étude comprenant 1265 patients de la base de données japonaise des tumeurs thymiques (JART) et traités pour une TET entre 1991 et 2010 au japon, avaient retrouvé un bénéfice de la radiothérapie adjuvante dans les carcinomes thymiques et pas pour les thymomes de stade II et III(47). Dans cette étude, il y avait 155 carcinomes
thymiques de stade II et III et 1110 thymomes de stade II et III dont 895 de stade II et 370 de stade III. Quatre cent trois (31,9%) des 1265 patients avaient eu une radiothérapie adjuvante dont 80 patients avaient un carcinome thymique, 200 patients avaient un thymome de stade II et 123 patients avaient un thymome de stade III. La survie sans progression à 5 ans était de 78% pour les patients ayant bénéficié d’une radiothérapie post opératoire contre 83,5% pour les patients ayant été traités par chirurgie seule. Ces résultats tendent à démontrer un effet délétère de la radiothérapie avec un p=0,056. En analyse ajustée sur les différents facteurs (le type histologique, le stade Masaoka de la maladie et la qualité de la résection), il n’y avait pas de différence significative entre les deux groupes de patients (radiothérapie post opératoire versus chirurgie seule) avec un HR : 0,76 IC 96% (0,58-1,01) et un p=0 ,116. Pour les carcinomes thymiques, il y avait 52 patients de stade II dont 25 avaient bénéficié d’une radiothérapie adjuvante et 27 autres patients traités par chirurgie seule. La survie sans progression à 5 ans chez ces 2 groupes de patients respectivement était de 91,3% contre 68,1%. Pour les 95 patients de stade III, 55 patients avaient eu une radiothérapie adjuvante contre 41 patients étaient traités par chirurgie seule. La survie sans récidive était de 50,5% chez les patients traités par radiothérapie adjuvante contre 26,1% chez les patients opérés sans radiothérapie. Le HR de la PFS en analyse ajustée sur le stade de Masaoka-Koga et la qualité de la résection était HR : 0,48 IC 95% (0,3-0,78) avec un p=0,003. Pour les patients ayant un thymome, la survie sans progression (PFS) à 5 ans pour les stades II et III était de 93,4% pour les patients traités par radiothérapie adjuvante contre 92,3% pour les patients traités par chirurgie seule. En analyse ajustée sur le stade de Masaoka-Koga et la qualité de la résection ( R0 ou R1) le HR de la PFS était à 0,98 IC 95% (0,7-1,37) avec un p =0,905(47). Berman et al avaient rapporté un taux de récidive de 0% chez les patients ayant eu de la radiothérapie adjuvante contre 8% chez les patients traités par chirurgie seule (p=0,15) et une mortalité globale de 3,2% avec un décès dans chaque bras. Cette étude avait été menée de 1990 à 2008 et portée sur 62 patients traités pour une tumeur thymique par chirurgie complète dont 37 patients avaient bénéficié d’une radiothérapie adjuvante et 25 autres avaient eu un traitement chirurgical seul(49).
25
b- Radiothérapie définitive en cas de tumeurs non opérées :
La radiothérapie peut être exclusive dans le cas où la tumeur est non opérable ou en cas contre-indications médicales à la chirurgie. Dans ce cas, la radiothérapie est souvent associée à une chimiothérapie soit concomitamment soit d’une façon séquentielle(5).
c- Dose de radiothérapie
Les doses de radiothérapie doivent être déterminées en fonction de la qualité de résection de la tumeur (R0, R1 ou R2). Le réseau RYTHMIC préconise en cas de résection complète (R0) une dose entre 45 Gy et 50 Gy sur la totalité du volume cible. Si la résection est incomplète (R1 ou R2) la dose doit être d’au moins 56 Gy avec éventuellement un complément au niveau des zones à haut risque préalablement repérées par des clips (pendant l’acte chirurgical). Le fractionnement doit être un fractionnement classique avec une dose par séance comprise entre 1,8 Gy et 2 Gy et un nombre de 5 séances par semaine(5)(46).
d- Quels champs d’irradiation
Afin de définir les volumes cibles de radiothérapie post opératoire, le radiothérapeute doit s’aider de l’imagerie initiale afin d’estimer la taille tumorale initiale (scanner, TEP scan), le compte rendu opératoire afin d’évaluer la vraie extension de la tumeur et ses rapports avec les structures médiastinales et le compte rendu anatomopathologique afin de savoir si la résection était complète ou non.
Les volumes cibles optimaux pour la radiothérapie adjuvante ne sont pas définitivement établis. La définition de ces volumes repose sur l’accord des experts. Il est donc
recommandé d’inclure la globalité de la loge thymique ainsi que les éventuelles extensions de la tumeur aux structures voisines (le péricarde, la plèvre, le parenchyme pulmonaire et les gros vaisseaux)(5).
Si le patient a bénéficié d’une chimiothérapie première, le volume cible doit être défini avec l’aide de l’imagerie post chimiothérapie(5).
Les limites supérieures des champs d’irradiation sont situées au niveau du défilé costo-diaphragmatique et les limites inférieures au niveau du médiastin moyen. L’irradiation des creux sus-claviculaire n’est pas recommandée par les experts car il n’y avait pas d’étude démontrant son efficacité(5).
Dans la littérature, les pratiques sont différentes en fonction des écoles et des centres. Certains centres pratiquent une irradiation médiastinales totale même si la résection est complète (R0), d’autres appliquent une irradiation du lit opératoire avec des marges de sécurité et d’autres encore suggèrent une irradiation hémi-thoracique afin de réduire les récidives pleurales (basales). Ogawa et al avaient démontré qu’une irradiation à hauteur de 40 Gy de la totalité du médiastin prévenait les récidives médiastinales chez les patients avec des tumeurs complètement réséquées. Par contre cette irradiation serait insuffisante quand une atteinte pleurale est documentée(50). Sugie et al suggèrent qu’une dose de 30 à 40 Gy au niveau médiastinal pour des tumeurs de Stade II complètement réséquées soit suffisante et une dose de 50 Gy à 55 Gy pour les stade III et une irradiation hémi-thoracique à hauteur soit de 11,2 Gy en 7 fractions ou de 15 à 16 Gy en 10 fractions quand une atteinte pleurale initiale est présente(51). L’élargissement des champs d’irradiation n’était pas sorti comme facteur améliorant la survie à 5 ans(52).
26
8.3. La chimiothérapie
La chimiothérapie est l’une des modalités thérapeutiques des TET parmi les autres
modalités. Elle peut être réalisée soit en néo-adjuvant (chimiothérapie première) quand la tumeur est localement avancée (TET de stade III ou IVA) et non accessible à un traitement chirurgical en première intention et cela dans le but de la rendre opérable. Soit elle peut être réalisée comme traitement définitif quand la chirurgie est définitivement écartée. Dans ce cas, la chimiothérapie est administrée de façon séquentielle avec la radiothérapie. Enfin elle peut être réalisée à visée palliative en cas de TET métastatique ou récidivante non opérable. Dans ce cas, la chimiothérapie exclusive est le standard(5). Le taux de réponse globale à la chimiothérapie néo-adjuvante est d’environ 70% à 80%(37)(53) et le taux de réponse en cas de chimiothérapie palliative ou de tumeur récidivante est variable allant de 20% à 60%(5).
La chimiothérapie post opératoire (adjuvante) lorsque la résection est complète (R0) ou microscopiquement incomplète (R1) n’est pas recommandée(37). Pour les tumeurs avec un résidu macroscopique (R2), ce cas est considéré comme une tumeur en place pour laquelle la combinaison de la radio-chimiothérapie est le traitement de choix(5)(46) et la chirurgie dans ce cas n’est pas considérée comme le traitement principal surtout lorsqu’il s’agit d’un carcinome thymique(46). La chimiothérapie peut être administrée soit de façon séquentielle avec la radiothérapie, soit de façon concomitante. La dose de la radiothérapie doit être d’au moins 60 Gy dans ce cas(5).
Le type de chimiothérapie de choix est une association de sels de platine (Cisplatine ou carboplatine) et d’anthracyclines (Adriamycine) et pour les carcinomes neuroendocrines les sels de platine (Cisplatine ou carboplatine) et Etoposide (VP16)(5)(54)(55).
Pour la chimiothérapie néo-adjuvante, dont le but est de rendre les tumeurs opérables, un bilan de réévaluation doit être fait après deux à quatre cures(37). Le taux de réponse post chimiothérapie première est compris entre 70% et 80% [40-100](37) et l’obtention d’une résection complète est de 50 %(37)(53). Si la tumeur demeure inopérable pour des raisons médicales ou chirurgicales, un traitement par radiothérapie ou radio-chimiothérapie
définitive est indiqué. Environ 10 à 15% des patients ayant eu une chimiothérapie première ne bénéficieront pas de traitement radical (chirurgie et/ou radiothérapie)(37).
La chimiothérapie est également utilisée lors des récidives ou lors des progressions
métastatiques (chimiothérapie palliative) et dans ce cas les molécules utilisées sont à base d’Anthracycline (Adiamycine), sels de platine (Cisplatine ou Carboplatine), Ifosfamide et Etoposide (VP16). Le nombre de cures recommandé avant une réévaluation est entre 4 à 6 cures. Le taux de réponse dans ces cas ne dépasse pas les 40% avec un intervalle de survie globale compris entre 37 et 72 mois(37). En deuxième ligne de chimiothérapie les molécules utilisées sont le paclitaxel et le pemetrexed.
27
8.4. Les thérapeutiques ciblées :
Environ 2 % des thymomes et 80% des carcinomes thymiques ont une surexpression du facteur KIT (facteur de croissance transmembranaire)(37)(56). Pour cela plusieurs molécules ont été testées et utilisées chez des patients comme Imatinib, Sunitinib ou Sorafénib. Les anti EGVR et VEGF tels que Bévacizumab n’ont pas démontré une efficacité suffisante justifiant leur utilisation(37)(53).
Matériels et méthodes
Notre étude est une étude rétrospective, monocentrique de type cohorte ayant inclus des patients traités à l’IGR par radiothérapie adjuvante entre 1990 et 2011 pour une tumeur épithéliale thymique (TET). Les patients ont été choisis à partir de la base de données du comité « pathologies thoraciques ».
1. Critères d’inclusion
- Patients traités à l’institut Gustave Roussy pour une TET ;
- Patients ayant été traités initialement par chirurgie radicale puis radiothérapie ou
radio-chimiothérapie adjuvante ;
- Les patients peuvent également recevoir un traitement par chimiothérapie soit en néo
adjuvant ou en adjuvant ;
- Aucune limite d’âge
2. Critères d’exclusion
- Patients traités pour une TET mais n’ayant pas eu de radiothérapie adjuvante, ou ayant
eu une radiothérapie ou radio-chimiothérapie seule sans chirurgie ;
- Patients avec des métastases viscérales d’emblée ;
- suivi inférieur à 3 ans.
3. Recueil des données
Le recueil des données a été effectué courant 2015 en remplissant une fiche d’informations contenant les items suivants :
- Caractéristiques des patients et circonstance de découverte de la maladie: âge au
diagnostic, le sexe, le Performans Status selon l’OMS (PS).
- Circonstance de découverte de la maladie : syndrome paranéoplasique (myasthénie,
lupus etc..), douleur thoracique, dyspnée ou découverte fortuite.
- Caractéristiques de la maladie : histologie et classification OMS, stade de la maladie
selon Masaoka-Koga, les structures envahies.
- Traitements : type de traitements (chirurgie, chimiothérapie et radiothérapie), qualité
de la chirurgie (R0, R1 ou R2), type de la radiothérapie (3D, IMRT), champs de la radiothérapie (lit opératoire ou lit opératoire avec irradiation ganglionnaire prophylactique) la dose délivrée, son fractionnement, type de la chimiothérapie et nombre de cures réalisées.
- Complications de la radiothérapie (aigues et tardives) : pulmonaires, cardiaques, œsophagiennes et fibrose.
- Evolution de la maladie : récidives (locales, régionales et métastatiques), temps
jusqu’à progression, traitements de la première récidive, état des patients au moment du recueil (vivants ou décédés), cause du décès si le patient est décédé.
28
4. Statistiques
Les résultats sont exprimés en médiane pour les variables continues et en nombre absolu et en pourcentages pour les variables qualitatives.
Le suivi a été estimé par la méthode de Kaplan-Meier inversée. Les analyses de survie globale (SG), survie sans progression (PFS), survie spécifique (SS), le contrôle locorégional (CLR) et le contrôle métastatique (CM) ont été estimés par la méthode de Kaplan-Meier. Les taux de survie ont été définis comme le temps entre le début du traitement jusqu’au premier événement. Les événements considérés étaient le décès de toute cause pour la SG, la mort ou la progression tumorale pour la PFS et la mort liée au cancer traité pour la SS. Pour les taux de CLR et de CM, la mort sans rechute et les rechutes ultérieures ont été censurées. Les courbes de survie ont été comparées en utilisant le test du log-rank pour l'analyse univariée et par le modèle de Cox à risque proportionnel pour l'analyse multivariée. Dans le modèle de Cox, les variables avec p < 0,2 ont été utilisées et les variables continues ont été dichotomisées. Les analyses statistiques ont été réalisées avec le logiciel SPSS, version 19 (SPSS Inc, États-Unis). Les valeurs de p inférieures à 0,05 ont été considérées comme significatives.
Résultats
Parmi les patients traités à l’institut Gustave Roussy entre 1990 et 2011 pour une TET, 134 patients qui correspondaient aux critères d’inclusion avaient été retenus et inclus dans notre cohorte pour analyse.
1. Caractéristiques des patients
Elles sont résumées dans le tableau 3.1.1. L’âge et le sexe
L’âge médian des patients était de 53 ans [16-83], et la moyenne d’âge était de 50 ans. Parmi les patients, il y avait 74 (55,2%) hommes et 60 (44,8%) femmes soit un sex ratio H/F de 1,23. L’âge moyen au diagnostic pour les hommes était de 48 ans contre 52 ans pour les femmes.
1.2. Le Performans Status selon l’OMS ( PS)
Au moment du diagnostic, 118 (88,1%) patients avaient un score OMS à 0 et 16 (11,9%) patients avaient un score OMS à 1. Aucun patient n’avait un score OMS supérieur à 1 au moment du diagnostic.
1.3. Les circonstances de découvertes
La circonstance de découverte principale était une symptomatologie cardiorespiratoire. Parmi les autres signes cliniques ayant conduit au diagnostic de la maladie dans notre cohorte il y avait une symptomatologie cardiorespiratoire telle que la dyspnée, la douleur thoracique, la toux ou la péricardite chez 55 (41%) patients (tableau1). La tumeur thymique a été révélée par un syndrome paranéoplasique chez 51 (38,1%) patients dont 37 (27,5%) des patients avaient eu une myasthénie révélatrice et 14 (10,5%) autres patients avaient d’autres syndromes paranéoplasiques (tableau 1). Chez quatorze (10,5%) patients, leur TET était de découverte fortuite et 14 (10,5%) autres patients il n’y avait pas d’information sur les circonstances de découverte de leur maladie.
29
2. Les caractéristiques de la maladie (
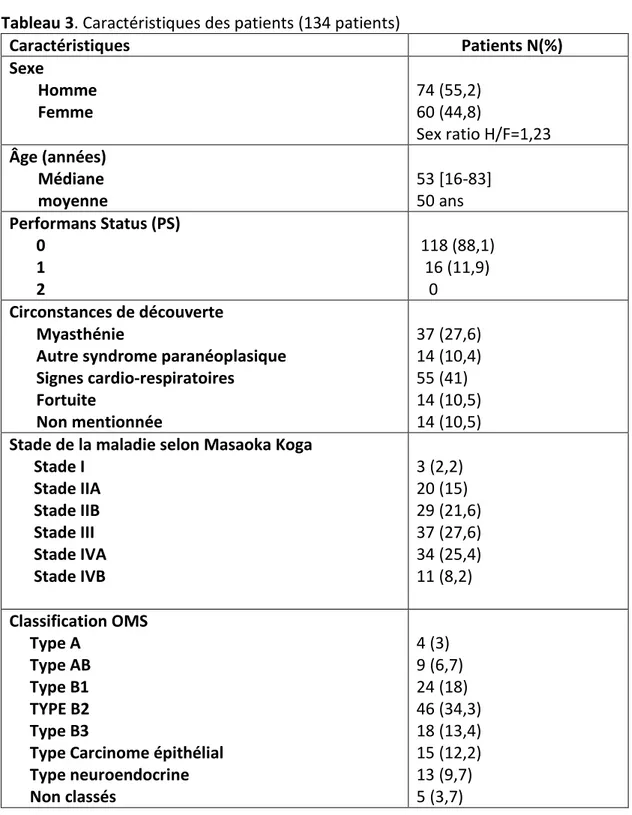
tableau 3 et 4)Tableau 3. Caractéristiques des patients (134 patients)
Caractéristiques Patients N(%) Sexe Homme Femme 74 (55,2) 60 (44,8) Sex ratio H/F=1,23 Âge (années) Médiane moyenne 53 [16-83] 50 ans Performans Status (PS) 0 1 2 118 (88,1) 16 (11,9) 0 Circonstances de découverte Myasthénie
Autre syndrome paranéoplasique Signes cardio-respiratoires Fortuite Non mentionnée 37 (27,6) 14 (10,4) 55 (41) 14 (10,5) 14 (10,5)
Stade de la maladie selon Masaoka Koga Stade I Stade IIA Stade IIB Stade III Stade IVA Stade IVB 3 (2,2) 20 (15) 29 (21,6) 37 (27,6) 34 (25,4) 11 (8,2) Classification OMS Type A Type AB Type B1 TYPE B2 Type B3
Type Carcinome épithélial Type neuroendocrine Non classés 4 (3) 9 (6,7) 24 (18) 46 (34,3) 18 (13,4) 15 (12,2) 13 (9,7) 5 (3,7)
2.1. La classification selon Masaoka-Koga
La classification de la maladie selon les stades de Masaoka-Koga a été faite à partir du compte rendu anatomopathologique définitif.
Trois (2,2%) patients avaient une maladie classée de stade I, 49 (36,6%) patients avaient une maladie classée stade II dont 20 (15%) patients de stade IIA et 29 (21,6%) de stade IIB, 37 (27,6%) avaient une maladie de stade III et 45 (33,6%) patients étaient classés en stade IV dont 34 (25,4%) patients de stade IVA et les 11 (8,2%) autres avaient une maladie de stade IVB.
30
2.2. Histologie des tumeurs et classification OMS 2004
La classification OMS a été mise en place à partir de 1999. Pour cela, les patients traités avant cette date, leur maladie a été classée initialement selon d’autres classifications. Pour pouvoir reclasser ces patients selon la classification internationale (OMS), le Professeur De Montpreville (médecin anatomopathologiste référent des tumeurs épithéliales thymique) du Centre Chirurgical Marie Lannelongue (CCML) nous a fourni une grille de lecture qui nous a permis d’obtenir une équivalence OMS de ces tumeurs.
Il y avait 47 (35%) patients traités avant 1999 qui sont concernés par la reclassification histologique.
Dans notre cohorte, il y avait 4 (3%) patients classés type OMS A, 88 (65,7%) patients avaient un type B dont 24 (18%) patients de type B1, 46 (34,3%) de type B2 et 18 (13,4%) de type B3, 9 (6,7%) AB, 28 (20,9) patients avaient un carcinome thymique dont 15 (11,2%) carcinomes épidermoïdes et 13 (9.7%) carcinomes neuroendocrines et 5 (3,7%) patients avaient une tumeur non classée.
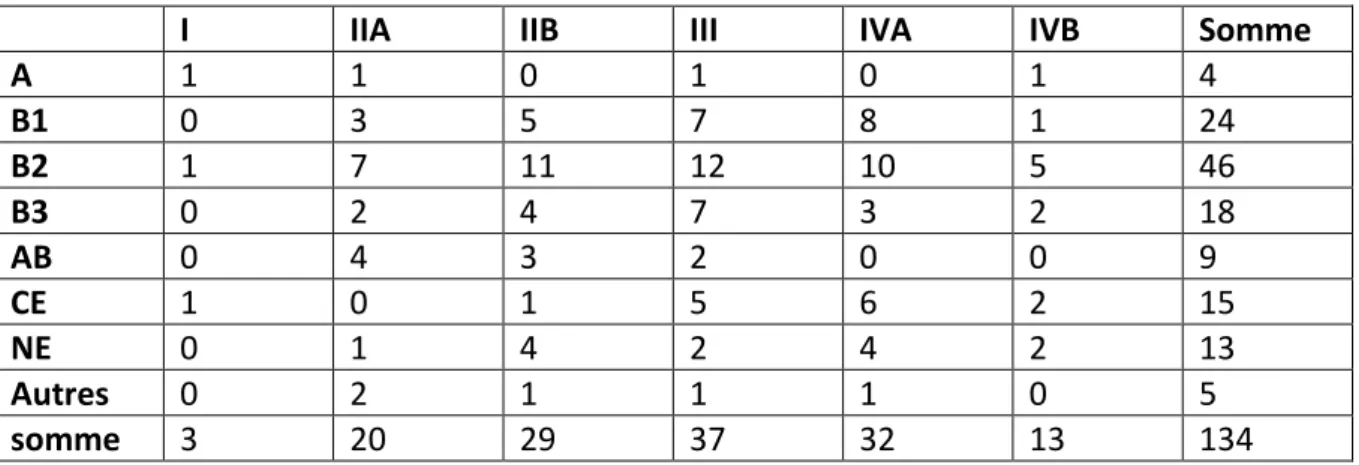
Tableau 4 : Stade de la maladie en fonction de la classification OMS
I IIA IIB III IVA IVB Somme
A 1 1 0 1 0 1 4 B1 0 3 5 7 8 1 24 B2 1 7 11 12 10 5 46 B3 0 2 4 7 3 2 18 AB 0 4 3 2 0 0 9 CE 1 0 1 5 6 2 15 NE 0 1 4 2 4 2 13 Autres 0 2 1 1 1 0 5 somme 3 20 29 37 32 13 134
2.3. Les syndromes paranéoplasiques(Tableau 5)
Soixante-deux (46,3%) patients avaient au moins un syndrome paranéoplasique. Parmi les syndromes paranéoplasiques, la myasthénie était présente chez 46 (34,3%) patients et représente 74,2% de tous les syndromes paranéoplasiques. Les autres syndromes
paranéoplasiques rencontrés sont : 3 (2,2) myosites, 2(1,5%) syndrome de Good, 2 (1,5%) érythroblastopénies, 3 (2,2%) lupus et SAPL, 5 (3,7%) syndromes de cushing, un patient avait une maladie de basedow, un autre avait une urticaire, une neuropathie périphérique chez un autre patient , une fièvre récurrente chez un autre patient, une diarrhée chronique chez un autre patient et un autre patient avait une hémophilie par la présence d’anticorps anti-facteur VIII.
Parmi les patients ayant développé des syndromes paranéoplasiques, 5 (8%) patients avaient deux syndromes paranéoplasiques associés. Les cinq patients avaient la myasthénie avec un autre syndrome paranéoplasique : une érythroblastopénie, un syndrome de Good, une diarrhée chronique, une urticaire et une maladie de basedow.
31
Tableau 5 : Les syndromes paranéoplasiques
Syndrome paranéoplasique Patients N(%)
Myasthénie Myosite et polymyosite Lupus et SAPL Syndrome de cushing Syndrome de Good Erythroblastopénie Maladie de basedow Neuropathie périphérique Anticorps anti facteur VIII Fièvre récurrente Urticaire récidivante Diarrhée chronique 46 (34 ,3) 3 (2,2) 3 (2,2) 5 (3,7) 2 (1,5) 2 (1,5) 1 1 1 1 1 1 SAPL : syndrome anticorps anti-phospholipides
- Les syndromes paranéoplasiques en fonction de l’âge et du sexe (tableau 6):
La répartition des syndromes paranéoplasiques était équitable selon le sexe avec 34 (54,8%) femmes et 28 (45,2%) d’hommes.
La répartition des syndromes paranéoplasiques selon les âges était la suivante : quatre (3%) patients étaient âgés de moins de 30 ans, 10 (7,5%) autres patients étaient âgés entre 30 et 39 ans, 19 (114,2%) patients étaient âgés entre 40 et 49 ans, 20 (15%) patients avaient entre 50 et 59 ans et 9 (6,7%) patients étaient âgés de plus de 60 ans. Trente-neuf (62,9%) patients ayant eu un syndrome paranéoplasique avaient un âge compris entre 40 et 60 ans au
moment du diagnostic de leur TET.
- La répartition des syndromes paranéoplasiques en fonction de la classification OMS (tableau 6) :
Cinquante et un (82,3%) patients avec un syndrome paranéoplasique avaient une tumeur de type B dont 12 (19,4%) de type B1, 32 (51,6%) de type B2 et 7 (11,3%) de type B3. Il y’avait 3 (4,8%) tumeurs de type AB, 5 (8,1%) carcinomes neuroendocrines et 3 (4,8%) patients avaient des tumeurs non classées histologiquement.
Il n’y avait pas de syndrome paranéoplasique pour les carcinomes épidermoïdes ni pour le type A. Les carcinomes neuroendocrines sont accompagnés de syndrome de cushing chez 5 (3,7%) patients.
- La répartition des syndromes paranéoplasiques en fonction des stades de Masaoka Koga (tableau 6) :
Vingt-huit (45,2%) patients des 62 patients ayant un syndrome paranéoplasique étaient de stade II de Masaoka-Koga dont 12 (19,4%) patients étaient de stade IIA et 16 (25,8%) patients de stade IIB. Trente-quatre (54,8%) patients avaient une maladie localement avancée dont 15 (24,2%) patients étaient de stade III, 18 (29%) patients de stade IVA et un patient était de stade IVB.
32
Tableau 6 : les syndromes paranéoplasiques en fonction de l’âge, sexe et types OMS
Caractéristique Patients avec syndrome paranéoplasique N(%)
Age < 30 ans 30 -39 ans 40 -49 ans 50 -59 ans > 60 ans 4(3) 10 (7,5) 19 (14,2) 20 (15) 9 (6,7) Sexe : Homme femme 28(45,2) 34 (54,4) Classification OMS Type A Type AB Type B1 Type B2 Type B3 Carcinome épidermoïde Carcinome neuroendocrine autres 0 3(4,8) 12 (19,4) 32 (51,6) 7 (11,3) 0 5 (8,1) 3 (4,8) Stade Masaoka Koga
Stade I Stade IIA Stade IIB Stade III Stade IVA Stade IVB 0 12 (19,4) 16 (25,8) 15 (24,2) 18 (29) 1 (1,6)
3. Traitements
Cent pour cent des patients ont été traités à visée curative. Les traitements sont résumés dans le tableau 7.
3.1
.
Chimiothérapie d’inductionVingt et un (15,7%) patients avaient eu une chimiothérapie première avant la chirurgie. Cent pour cent des chimiothérapies néo-adjuvantes avaient été à base de sel de platine. Treize patients avaient eu une chimiothérapie par VIP (Vincristine, Ifosfamide et Cisplatine), 6 autres patients avaient eu un protocole CAP (Endoxan, Adiamycine et Cisplatine), un patient avait eu un PAVI, un autre avait eu du Cisplatine et de l’Etoposide. Le nombre moyen des cures était de 4 [2-6].
Pour le taux de réponse de la chimiothérapie néo-adjuvante, se référer au chapitre chirurgie tumorale partie limites de résection.
3.2. Chirurgie tumorale
Cent pour cent des patients avaient eu une chirurgie de leur TET avant leur traitement de radiothérapie. Cent treize (84,3%) patients avaient eu une chirurgie première et 21 (15,7%) un traitement chirurgical après une chimiothérapie néo-adjuvante.