INFORMATION TO USERS
This manuscript has been reproduced from the microfilm master. UMI films the
text
directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer.The quallty of this reproduction is dependent upon the quality of the
copy submitted. Broken or indistind print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction.
ln the unlikely event that the author did not send UMI
a
complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material hadto
be removed, a note will indicate the deletion.Oversize materials (e.g.,
maps,
drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps.Photographs included in the original manuscript have been reproduced xerographically in this
copy.
Higher qualitys·
x g• black and white photographie prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contad UMI directly to order.ProQuest Information and Leaming
300 North Zeeb Road, Ann Arbor, Ml 48106-1346 USA 800-521-0600
Université de Sherbrooke
Épissage alternatif dans la région carboxy-terminale du gène suppresseur de tumeurs p53
par
Mélanie Laverdière Département de biochimie
Mémoire présenté à la Faculté de médecine en vue de lobtention du grade de maitre ès sciences (M.Sc.) en biochimie
l+I
National Library of CanadaAcquisitions and Acquisitions et
Bibliographie Services services bibliographiques
395 Welinglon StrMt 395, rue Welington
011awa ON K1A ON4 Ollawa ON K1A ON4
c.'8da c...da
The author bas granted a
non-exclusive licence allowing the
National Library of Canada
to
reproduce, loan, distn'bute or sell
copies of this thesis
in
microform,
paper or electronic formats.
The author retains ownership of the
copyright
in
this thesis. Neither the
thesis nor substantial extracts from it
may be printed or otherwise
reproduced without the author's
permission.
L'auteur a accordé une licence non
exclusive permettant
à
la
Bibliothèque nationale du Canada de
reproduire, prêter, distribuer ou
vendre des copies de cette thèse sous
la
forme de microfiche/film, de
reproduction sur papier ou sur format
électronique.
L'auteur conserve la propriété du
droit d'auteur qui protège cette thèse.
Ni la thèse ni des extraits substantiels
de celle-ci ne doivent être imprimés
ou autrement reproduits sans son
autorisation.
0-612-67299-9
TABLE DES MATIÈRES
Table des matières ... .1
Liste des illustrations ... III Liste des abréviations ... V Résumé Introduction ... 1
Épissage constitutif ... 1
Épissage alternatif ... 7
Mécanisme de sélection des sites d'épissage ... 9
Séquences stimulatrices d'épissage ... 9
S ' equences inhi"b" . d' ' . 1tnces ep1ssage ... . 11
Gène suppresseur de tumeurs p53 ... 14
Épissage alternatif du gène suppresseur de tumeurs p53 ... l S Résultats ... 19
Chapitre I ... 20
Abstract ... 21
Introduction ... 22
Materials and methods ... 25
Results ... 31
Ack.nowlegements ... 53 References ... 54 Chapitre II ... 59 Abstract ... 60 Introduction ... 61 Experimental procedures ... 64 Results ... 70 Discussion ... 85 Ack.nowlegements ... 89 References ... 90 Discussion ... 96 Conclusions et perspectives ... l 08 Remerciements ... l 12 Références ... 114 Annexe ... 139
LISTE DES ILLUSTRATIONS
Introduction :
Figure l : Séquences du pré-ARNm nécessaires au processus d'épissage ... 3
Figure 2 : Étapes de la fonnation du complexe d'épissage ... 5
Tableau 1: Séquences des éléments exoniques inhibiteurs d'épissage identifiés
jusqu'à maintenant ... 12
Figure 3 : Représentation de l'événement d'épissage alternatif dans l'extrémité
carboxy-terminale du gène suppresseur de tumeurs p53 murin ... 16
Chapitre 1:
Figure l : Alternative splicing of the p53 tumor suppressor gene ... 32
Table l : Cell lines tested for alternative splicing in the C-terminal region of
p53 between exons IO and 11 ... 33
Figure 2 : Analysis of p53 mRNA levels in cell lines ... 34
Figure 3: Diagrammatic representation of the PCR amplification products used for DNA sequencing of the mouse, bu~ rat and hamster p53 exon
l 0-11 region ... 36
Figure 4: Sequence alignment of the mouse, rat, h~ gorilla and hamster p53
exon l 0-11 region ... 38
Table 3 : Percentage ofhomology in intron 10 from the mouse, rat and human
p53 gene ... 39
Figure 5 : RNase protection analysis of the alternative splicing pattern of the mouse, rat and human p53 gene in cell lines from ditîerent species
after transient transfection ... 44
Chapitre II :
Figure 1 : Alternative splicing of the p53 tumor suppressor gene ... 71
Figure 2: ln vitro splicing of pre-mRNAs containing or lacking the AH element
(ESS) ... 72
Figure 3 : Analysis of splicing complex formation by native gel electrophoresis ... 74
Figure 4: ln vitro splicing ofMp53 pre-mRNA containing ditîerent portions of the
AH element ... 75
Figure 5: ln vitro splicing of analysis of adenovirus (Ad~ILI) pre-mRNA containing
mutations in the AH element ... 77
Figure 6: Stimulation of in vitro splicing of Mp53R pre-mRNA by competitor RNA containing the Ahmin sequence ... 79
Figure 7: Electrophoretic mobility gel shift assay with AHmin and mutants AH
RN A transcripts ... 80
Figure 8: Specific binding of a 64kDa factor to the p53 AH element and Western
LISTE DES ABRÉVIATIONS
ADN ... acide désoxyribonucléique ARN ... acide ribonucléique
ARN
m ...
ARN messagerA TP ... adénosine triphosphate bp ... paire de base
Br-U ... bro1110uridine °C ... degré Celcius
CTP ... cytosine triphosphate
DMEM ... "Dulbecco 's D10dified Eagle medium" DNase ... desoxyribonucléase
dNTPs ... desoxynucléotides triphosphate OTT ... dithiothréitol
EDT A ... acide tétraacétique éthylènediamine GTP ... guanosine triphosphate
h ... heure
Hepes ... acide N-2-hydroxyéthylpipérazine-N' -éthanesulfonique kDa ... kilodalton
hnRNP ... "heterogenous nuclear ribonucleoprotein" M ... molaire
mg ... milligramme min ... minute
ml ... millilitre
mM ... millimolaire NP40 ... Nonidet-40 nt ... nucléotides
NTPs ... ribonucléotides triphosphate
PAGE ... électrophorèse en gel de polyacrylamide PCR ... "Polymerase chain reaction"
pmol ... picomole
pré-ARNm ... pré-ARN messager Py ... pyrimidine
ARNase ... ribonucléase
RT ... transcription inverse SDS ... sodium dodécyl sulfate
snRNA ... petit acide ribonucléique nucléaire snRNP ... petite ribonucléoprotéine nucléaire UTP ... uridine triphosphate
UV ... ultra-violet µg ... microgramme µI ... microlitre
3' Cas ... site 3' d 'épissage alternatif du pré-ARN m de p53 3' Reg ... site 3' d'épissage régulier du pré-ARNm de p53
RÉSUMÉ
Épissage alternatif dans la région carbosy-terminale du gène suppresseur de tumeun p53
Par Mélanie Laverdière Département de biochimie
Université de Sherbrooke
Mémoire présenté à la Faculté de médecine en we de l'obtention du grade de maître ès sciences (M.Sc.) en biochimie
Le 15 mai 2000
Un événement d'épissage alternatif se produit dans l'extrémité carboxy-terminale du gène suppresseur de tumeurs p53 de souris entre le site d'épissage 5' de l'exon IO et deux sites d'épissage 3' alternatifs de l'exon 11, 3'Cas et 3'Reg. L'analyse des proportions d' ARNm p53Cas et p53Reg produits dans différentes lignées cellulaires de diverses espèces a permis d'établir que le patron d'épissage alternatif varie selon les espèces. En effet, le site d'épissage 3'Cas est mieux utilisé dans le gène p53 de souris que dans le gène p53 de rat. Quant à l'utilisation du site d'épissage 3 'Cas du gène p53 humain, elle n'a pas été détectée dans les lignées cellulaires analysées. La variation observée entre le patron d'épissage du gène p53 de souris et celui de rat ne semble pas être attribuable aux séquences des signaux d'épissage. Cependant, elle pourrait être due à
la présence d'éléments modulateurs d'épissage dans le gène p53 ou bien à des variations dans les niveaux de facteurs trans s'associant à ces éléments régulateurs d'épissage. Par
contre, l'absence d'utilisation du site d'épissage 3'Cas dans le gène p53 humain est probablement due à la faiblesse de ce site d'épissage, le rendant non fonctionnel. L'ensemble de ces travaux suggère donc que l'épissage alternatif dans la région
carboxy-terminale du gène p53 est régulé de tàçon différente selon les espèces puisque les mécanismes pour contrôler l'utilisation des sites d'épissage alternatifs sont divergents.
À l'aide d'un système d'épissage in vitro, nous avons pu démontrer qu'un élément inhibiteur d'épissage, l'élément AH, est situé dans l'exon 11 du pré-ARNm p53 de souris et est responsable de l'inhibition de l'utilisation du site d'épissage J'Reg, situé en amont. Nous avons aussi démontré que l'élément AH peut inhiber l'épissage dans un contexte hétérologue, suggérant que cet élément fonctionne de façon autonome. Ces travaux ont aussi permis de déterminer que l'inhibition de l'épissage par l'élément AH se fait en bloquant la formation du complexe d'épissage de façon précoce. Les expériences de rétention sur gel ont montré qu'un facteur nucléaire se lie de façon spécifique à l'élément AH, puisque cette interaction peut être inhibée par un ARN contenant la séquence de l'élément AH minimal et qu'un ARN compétiteur non spécifique n'a pas cet effet. Les essais d'épissage in vitro en présence d'ARN compétiteurs spécifique et non spécifique de l'élément AH ont démontré qu'un facteur
trans est probablement responsable de l'activité inhibitrice de la séquence AH sur
l'utilisation d'un site d'épissage 3'. Des essais de réticulation aux UV, de NorthWestem et de chromatographie d'affinité ont démontré qu'un facteur nucléaire ayant un poids moléculaire apparent de 64 k.Da se lie de façon spécifique à l'élément AH. Des analyses par spectrométrie de masse et d'immunobuvardage de type Western ont identifié ce facteur comme étant lhnRNP K.
Mots-clés: épissage akematif, gène suppresseur de tumeurs p53, régulation
INTRODUCTION
L'expression des gènes doit être finement régulée afin de permettre aux cellules eucaryotes de maintenir une croissance contrôlée et d'atteindre un haut niveau de spécialisation lors de la différenciation. Cette régulation peut se faire à divers niveaux, soit lors de la transcription des gènes, de la maturation des pré-ARNm ou bien au niveau du transport, de la stabilité et de la traduction de ces ARNm. L'épissage est une étape de la maturation des ARN qui consiste en l'excision des introns d'un pré-ARNm. Cette étape peut amener une grande source de diversité au niveau des ARN messagers, puisque lorsqu'un pré-ARNm est épissé alternativement, il y production de plusieW'S ARNm différents possédant différentes combinaisons d'exons ou bien différentes régions 5' et 3' non traduites. Des modifications dans les régions 5' et 3' non traduites peuvent entraîner une modulation du niveau de traduction et de la stabilité des ARNm. De plus, conséquemment à l'utilisation différentielle de site d'épissage, il peut y avoir production de différentes protéines ayant des activités physiologiques et biochimiques distinctes. L'épissage alternatif peut être régulé dans le cycle cellulaire, de façon tissu-spécifique ou de façon développementale, ce qui démontre l'importance et l'implication de ce processus au sein d'une cellule.
Épissage constitutif
L 'épissage, processus par lequel les introns des pré-ARNm sont excisés, a été mis en évidence en 1977 (Berget et al., 1977; Chow et Broker, 1978). L'épissage permet l'excision précise et reproductible des introns, les séquences intercalées flanquant chacun
des exons, et la réunion des différents exons. C'est en 1985 que l'on a débuté la caractérisation du spliceosome, complexe responsable du processus d'épissage, qui contient plus de cinquante protéines dont cinq petites ribonucléoprotéines nucléaires (snRNP Ul, U2, U4, US et U6) (Brody et Abelson, 1985; Burge et al., 1999; Frendewey
et Keller, 1985; Grabowski et al., 1985; Newman, 1998; Will et L~ 1997).
Pour qu'il y ait reconnaissance précise des introns, plusieurs régions à l'intérieur du pré-ARNm agissent comme signaux d'épissage. La reconnaissance de l'intron à
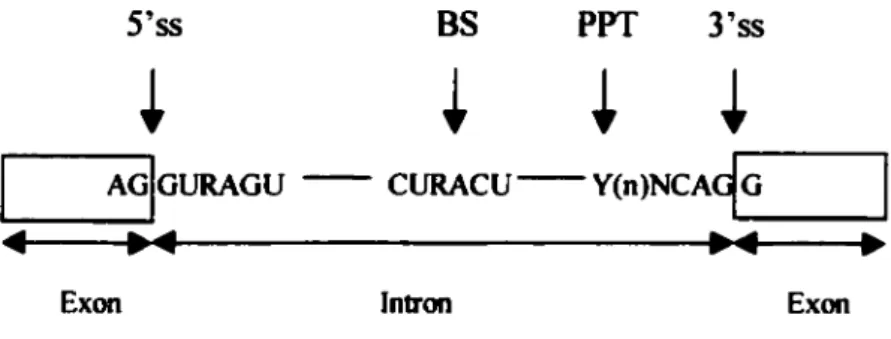
exciser se fait via un site d'épissage 5' et un site d'épissage 3', deux séquences bien conservées chez les eucaryotes supérieurs. Un site consensus a été dérivé pour chacun de ces signaux, soit la séquence intronique AG/GURAGU pour le site d'épissage 5' et CAG/G pour le site d'épissage 3'. L'intron à enlever doit aussi contenir un site de branchement normalement situé de 18 à 40 nucléotides en amont du site d'épissage 3'. Le site de branchement correspond à une adénosine conservée, située dans la région consensus (CURACU). La région du site d'épissage 3' doit aussi comporter une séquence riche en pyrimidines située entre le site de branchement et le site d'épissage 3' (voir figure 1) (Adams et al., 1996; Blencowe, 2000).
5'ss BS PPT 3'ss
+
+ + +
AGIGURAGU -- cURAcu--v(n)NcAtjo
4 .. 4 .. 4
Ex on lntron Ex on
Figure 1 : Séquences du pré-ARNm nécessaires au processus d'épissage (Blencowe, 2000). Le site consensus 5' d'épissage (5'ss), le site de branchement (BS), la séquence riche en pyrimidines (PPT) et le site consensus 3' d'épissage (3'ss) sont montrés.
Le complexe d'épissage est une machinerie dynamique et doit se réassembler après chaque réaction d'excision d'un intron (Burge et al., 1999; Ne~ 1998). La
formation du spliceosome se fait d'une manière très ordonnée et sa composition change au fil de sa formation (Staley et Guthrie, 1998). L'assemblage débute avec la liaison du snRNP Ul au niveau du site d'épissage
5'
par un appariement de bases entre la séquence consensus du site5'
et le snRNA Ul contenu dans le snRNP Ul (Staknis et Reed, 1994). Ensuite, il y a liaison de la séquence riche en pyrimidines du site d'épissage 3' par le facteur U2AF65, un facteur d'épissage associé au recrutement du snRNP U2 (Black et al.,1995; Chabot et Steitz, 1987; Reed, 1996). Il a été démontré que la liaison du snRNP UI
à un Site d'épissage 5' favorise la liaison du facteur U2Af65 à un site d'épissage 3' en
amont via des interactions par dessus l'exon (Hoffinan et Grabowski, 1992; Michaud et Reed, 1993). Tiré de ces évidences, le modèle de définition de l'exon suggère que l'unité exonique est la première reconnue sur un pré-ARNm (Cunningham et al., 1995; Robberson et al., 1990; Staknis et Reed, 1994). Tout récemment, le rôle de la sous-unité U2AF35 du facteur U2AF a été bien décrit par trois groupes qui ont démontré que cette
sous-unité se lierait au site d'épissage 3'. En effet, le facteur U2AF35 contient un
domaine de liaison séquence-spécifique à I' ARN qui reconnait la séquence consensus d'un site 3' (AG/G). Cette interaction serait nécessaire dans les cas d'épissage où la
séquence riche en pyrimidines serait non conforme au consensus et aurait peu d'affmité pour la sous-unité U2AF65 (Guth et al., 1999; Merendino et al., 1999; Wu et al., 1999;
Zorio et Blumenthal, 1999). Plusieurs autres facteurs se lient à la région du site d'épissage 3' tels les hnRNP l/PTB, hnRNP Al, hnRNP C et hnRNP D (Ashiya et
Grabowsk~ 1997; Chabot et al., 1985; Côté et al., 1995; Garcia-Blanco el al., 1989;
Gerke et Steitz, 1986; Gozani et al., 1994; Lin et Patton, 1995; Tazi el al., 1986).
La liaison du snRNP Ul et du facteur U2AF sur le pré-ARNm forme le complexe E. La formation de ce complexe est nécessaire pour l'engagement du pré-ARNm dans la voie d'épissage et aussi pour le recrutement du snRNP U2 via le facteur U2AF65• La liaison du snRNP U2 se fait au niveau du site de branchement et ce, par une interaction ARN-ARN entre la séquence du site de branchement et le snRNA U2. La liaison du snRNP U2 permet la formation du pré-complexe d'épissage A. L'assemblage du complexe A requiert aussi un site d'épissage 3' et une séquence riche en pyrimidine fonctionnels (Roscigno et al., 1993). Cette étape et toutes les étapes suivantes dans la formation du spliceosome sont dépendantes de I' A TP (Krainer et al., 1984; Sawa et al.,
1988). L'association des snRNP U4, U5 et U6 avec le complexe A sous forme du tri-snRNP U4/U6-U5 permet l'assemblage du complexe d'épissage B. Ce complexe est par la suite converti par des réarrangement conformationnels en complexe C (voir figure 2).
5'ss BP 3'ss
~E_x_rn_1!
_l~j
GUAAGU--- A-~Py Aoj~i
_E_xo_11_2___,l---~ ~ [ .. ·.
---~
~---A- (\À~ll
~vl
L ___
I U2 Complexe: A UMJ6-U5 ComplexeB +[
L
J
En plus des snRNPs, d'autres facteurs faisant partie de la famille des protéines SR
' 11 1995;
contiennent un motif de reconnaissance à l' ARN (RRM : RNA recognition motif) et sont nommées ainsi puisqu'elles possèdent un domaine en carbox.y-terminal riche en résidus sérine (S) et arginine (R) (Zahler et al., 1993). Il a été démontré que des protéines de cette famille sont nécessaires pour l'assemblage du complexe A (Fu, 1993). Le domaine RS leur permet d'interagir avec la sous-unité U2AF35 et avec une protéine de 70 kDa contenue dans le snRNP Ul (Kohtz et ai., 1994; Wu et Maniatis, 1993). En interagissant avec le snRNP Ul, les protéines SR ont probablement un rôle de premier ordre dans le recrutement de ce facteur au site
5'
d'épissage pour former le complexe d'engagement (Jamison et al., 1995; Kohtz et al., 1994). La capacité d'interagir simultanément avec le snRNP UI et U2AF35 suggère un modèle dans lequel les protéines SR permettraient lacommunication directe entre le site d'épissage 5' et le site d'épissage 3' (Wu et Maniatis, 1993 ). Récemment, un nouveau modèle a été proposé pour le fonctionnement des protéines SR dans la formation du complexe d'épissage. Contrairement au modèle décrit plus haut, ce modèle propose que les protéines SR permettraient linteraction entre différents composants du spliceosome via un co-activateur d'épissage comme SRml60/300 contenant SRml60, une protéine apparentée aux protéines SR associée à la matrice nucléaire et un antigène de la matrice nucléaire de 300 kDa (Blencowe, 2000; Blencowe et al., 2000; Eldridge et ai., 1999).
L'excision des introns se fait via deux réactions de transestérification (Burge et
al., 1999). Pour la première réaction, le groupement 2'0H de l'adénosine du site de
branchement attaque le groupement phosphate au site d'épissage 5'. Cette étape permet la libération de l'exon en 5' et la formation d'un intermédiaire sous forme de lasso contenant l' intron et I' exon en 3 '. Suite à des réarrangements du spliceosome,
l'extrémité 3'0H libre de l'exon en 5' attaque le groupement phosphate au site d'épissage 3', libérant ainsi les deux exons qui seront ensuite réunis et l'intron sous forme de lasso (Adams et al., 1996; Burge et al., 1999; Sharp, 1994).
Épissage alternatif
Un pré-ARNm épissé alternativement permet la production de différents isoformes d'une protéine.
De
plus, des événements d'épissage alternatif dans les régions5' et 3' non codantes d'un pré-ARNm peuvent amener une modulation dans la stabilité et dans le niveau de traduction de 1' ARNm, sans toutefois en changer la séquence codante (Burge et al., 1999; Lopez, 1998). Les premiers travaux sur la fréquence d'événements d'épissage alternatif estimaient que plus d'un gène sur vingt subit un épissage alternatif (Sharp, 1994). Tout récemment, de nouveaux travaux ont suggéré qu'environ 35% des gènes sont épissés alternativement. De plus, la majorité des événements d'épissage alternatif se produiraient dans la région 5' non traduite (Hanke et al., 1999; Mironov et
al., 1999). Des informations sur les gènes épissés alternativement, leurs produits et leur
patron d'expression, peuvent être trouvés dans une banque de données consacrée à
l'épissage alternatif (ASDB) (URL http://cbcg.nersc.gov/asdb) (Dralyuk et al., 2000; Gelfand et al., 1999). L'épissage alternatif peut être régulé dans le cycle cellulaire, de façon tissu-spécifique ou de façon développementale. Ce processus est donc complémentaire à la régulation de l'expression génique au niveau de l'activité du promoteur (Hodges et Bernstein, 1994; Jamison et al., 1995). L 'épissage alternatif de certains pré-ARNm peut former un nombre impressionnant d'isoformes. L'épissage alternatif amène une très grande source de diversité lors de la maturation d'un
pré-ARNm. Par exemple, l'épissage alternatif du pré-ARNm de la troponine T permet de former théoriquement jusqu'à 64 isoformes de la protéine. L'épissage alternatif du pré-ARNm de la glycoprotéine de surface CD44 est un autre exemple. En théorie, la somme des différentes combinaisons des 10 exons épissés alternativement excède mille différents isoformes. Cependant, au moins cent isoformes ont été identifiés jusqu'à maintenant (Goodison et al., 1999; Gunthert, 1996; Sherman et al., 1996). La famille de protéines des neurexines sont un exemple encore plus éloquent. Des milliers d'isoformes de neurexines sont générés par l'épissage alternatif de six pré-ARNm transcrits à partir de trois gènes (Missler et al., 1998; Missler et Sudhof, 1998; Ullrich et al., 1995).
Les différentes protéines générées par épissage alternatif possèdent de grandes similarités mais varient aussi dans des régions spécifiques, ce qui permet une modulation précise de la fonction de la protéine (Lopez, 1998; Smith et al., 1989). Par exemple, l'épissage alternatif à l'intérieur du domaine de liaison à l'ADN du pré-ARNm de wtl (tumeur de Wilm's) génère des protéines se distinguant par leur activité de liaison spécifique à l' ADN (Bickmore et al., 1992). Aussi, l'utilisation différentielle de sites d'épissage alternatifs dans le pré-ARNm de fosB peut mener à la production d'un activateur ou d'un répresseur de la transcription (Foulkes et Sassone-Corsi, 1992; Mumberg et al., 1991). Un exemple particulièrement intéressant est celui du gène suppresseur de tumeurs INK4a. Un événement d'épissage alternatif dans ce pré-ARNm génère, à partir de différents cadres de lecture, deux protéines, pl6 et pl~, qui sont d'une grande importance dans la régulation du cycle cellulaire (Kamijo et al., 1997).
Mécanisme de sélection des sites d'épissage
Les mécanismes de sélection des sites d'épissage alternatifs impliquent des éléments reliés à la séquence du pré-ARNm (cis) et aussi à des facteurs protéiques (trans). Dans certains cas, la sélection des sites d'épissage est influencée par des
structures secondaires à l'intérieur du pré-ARNm (Blanchette et Chabot, 1997; Clouet et
al., 1991; Sirand-Pugnet et al., 1995). L'encombrement stérique entre les sites
d'épissage ou la localisation du site de branchement peut aussi jouer un rôle dans la sélection des sites d'épissage (Goux-Pelletan et al., 1990; Smith et Nadal-Ginard, 1989). Le site de branchement ainsi que la séquence riche en pyrimidines, qui sont nécessaires
à l'efficacité d'épissage, peuvent aussi influencer la sélection des sites d'épissage. En effet, ces signaux d'épissage sont souvent sous-optimaux dans les régions épissées alternativement, ce qui procure une certaine flexibilité à ce processus quant à la sélection des sites d'épissage (Dirksen et al., 1995; Katz et Skalka, 1990; Mullen et al., 1991; O'Reilly et al., 1995; Si et al., 1997; Staffa et Cochrane, 1994; Zheng et al., 1999).
Séquences stimulatrices d'épissage
Les séquences introniques ou exoniques entourant les sites d'épissage peuvent moduler le choix des sites d'épissage utilisés. Ainsi, des séquences peuvent influencer de façon positive la sélection des sites d'épissage d'où leur nom d'éléments stimulateurs d'épissage. Des éléments exoniques stimulateurs d'épissage ont été mis en évidence par exemple dans la régulation du pré-ARNm doublesex (dsx) de la drosophile. En effet, les protéines Transformer (Tra) et Transformer 2 (Tra2) régulent de façon positive la
sélection d'un site d'épissage dans le pré-ARNm dsx en liant spécifiquement l'élément
répété dsx, localisé dans l'exon en aval d'un site d'épissage 3' utilisé chez la femelle seulement (Tian et Maniatis, 1994). Plusieurs éléments introniques stimulateurs d'épissage ont aussi été bien caractérisés. Par exemple, il a été démontré que l'inclusion de l'exon NI du gène c-src est attribuable à la présence d'un élément régulateur intronique qui est lié spécifiquement par le facteur KSRP (Black, 1992; Modafferi et Black, 1997).
Des exemples très bien caractérisés d'éléments exoniques stimulateurs d'épissage sont les séquences riches en purines qui sont retrouvées dans plusieurs exons de pré-ARNm (Dirksen et al., 1994; Lavigueur et al., 1993; Sun et al., 1993; Watakabe et al.,
1993). Les éléments modulateurs riches en purines sont présents dans des exons épissés de façon constitutive ou alternative (Adams et al., 1996; Black, 1995; Chabot, 1996; Manley et Tacke, 1996; Schaal et Maniatis, 1999; Valcârcel et Green, 1996). Il a été démontré que plusieurs de ces éléments stimulateurs d'épissage sont liés par des membres de la famille des protéines SR (Adams et al., 1996; Chabot, 1996; Côté et al.,
1999; Cramer et al., 1999; Lavigueur et al., 1993; Manley et Tacke, 1996; Nagel et al.,
1998; Ramchatesingh et al., 1995; Sun el al., 1993; Valcârcel et Green, 1996; Zheng el
al., 1997). De plus, ce groupe de protéines reconnaissent aussi d'autres stimulateurs
d'épissage dont la séquence consensus est divergente des séquences riches en purines (Cavaloc et al., 1999; Liu et al., 1998; Schaal et Maniatis, 1999). La liaison des protéines SR à ces éléments stimulateurs d'épissage favorise l'assemblage du spliceosome et peut aussi stimuler la liaison de U2AF à la séquence riche en pyrimidines (Staknis et Reed, 1994; Wang et al., 1995). Par contre, il faut noter que ce ne sont pas toutes les séquences
riches en purines qui possèdent cette activité de stimulation (Ramchatesingh et al., 1995; Tanaka et al., 1994). De façon intéressante, un groupe a démontré qu'une séquence exonique riche en purines ayant une activité stimulatrice peut avoir une activité inhibitrice lorsque positionnée dans
un
intron (Kanopka et al., 1996).Séquences inhibitrices d'épissage
Des séquences régulatrices introniques et exoniques peuvent aussi influencer de façon négative l'utilisation d'un site d'épissage. Par exemple, l'épissage d'un site 3' du pré-ARNm transformer est régulé négativement par la protéine Sxl qui lie la région riche en pyrimidines et empêche la liaison du facteur constitutif U2AF65 à cette même région
(Valcarcel et al., 1993). Un autre cas est l'inhibition de l'épissage du troisième intron du pré-ARNm de l'élément P de la drosophile, résultant de la liaison de PSI (P-element somatic inhibitor) à
un
élément de régulation négatif dans l'exon 3 (Siebel et al., l 995; Siebel et al., 1994). Toujours chez la drosophile, la protéine PTB liant la séquence riche en pyrimidines est impliquée dans la régulation négative de plusieurs gènes en compétitionnant pour le même site de liaison que le facteur d'épissage constitutif U2AF65 (Ashiya et Grabowski. 1997; Chan et Black, 1997; Southby et al., 1999; Zhang et al.,1999).
Les éléments inhibiteurs ou atténuateurs d'épissage sont retrouvés dans les gènes cellulaires (Blanchette et Chabot, 1999; Carstens et al., 1998; Chan et Black, 1995; Chew
et al., 2000; Del Gatto et Breathnach, 1995; Del Gatto et al., 1996; Gooding et al., 1994;
1994; Amendt et al., 1995; Arrigo et
Beemc>n,
1988; Berberich et Stoltzfus, 1991; Gontarek et al., 1993; McNally et al., 1991; Nemeroff et al., 1992; Wentz et al., 1997;Zheng et al., 1996). Les éléments inhibiteurs d' épissage constituent une famille de
régulateurs qui croit très rapidement (voir Tableau 1).
Séquence Gène Non-Virale UGUGGG 13-tropomyosine UAAGUGUUCUGAGCU a-tropomyosine CAAG fibonectine UAAG FGF-2R AGACAGAAUCAGCACCAGUGCUCAUGGAGAAAAUUGGACC C044 CUAGUAAACUUAUUCUUACGUCUUUCCUGUGUUGCCCUCCAGCUUUUAUCUCUGAGAUGGUCUUCUUUCAG fg~ CACUGAUGUGGAUGUCGAUUCCAUCAAAAUUGCCUUGGGAAAGCCCACAGGGGCAAGUUUCCAGGUACAGGGUG fibronectine
CCAAGUCAAAAUUUAC DNA ligase Ill
Virale AGAUCCUAGACUAGAGCCCU AGAUCCAUUCGAUUGUGAA GGCUCCCCC CCCGAAGGAAUAGUGGAGAGAGAGACAGAGACAGAUGGAUUCGA VIH-1 tat2 VIH-1 tat3 BPV-1 VIH-1 tat-rev
Tableau 1: Séquences des éléments exoniques inhibiteurs d'épissage identifiés jusqu'à maintenant
(Chew et al., 2000).
Jusqu'à maintenant, un certain nombre de facteurs reconnaissant ces éléments régulateurs ont été identifiés. Trois de ces éléments inhibiteurs d'épissage sont liés par des snRNPs. En effet, l'élément de régulation négatif d'épissage (NRS) dans le transcrit tardif du virus du sarcome de Rous (RSV) est lié par le snRNP Ul et le snRNP Ul l (Cook et McNally, 1998; Gontarek et al., 1993; McNally et McNally, 1999). Le snRNP
1999), tandis que le snRNP Ul peut s'associer à un pseudo sites• d'épissage pour inhiber l'épissage du troisième intron dans le pré-ARNm P-element de la drosophile (Siebel et
al., 1992). La caractérisation des complexes assemblés sur les éléments inhibiteurs
d'épissage des pré-ARNm du virus du papillome bovin (BPV-1) et du virus du sarcome de Rous (RSV) a aussi montré que des protéines SR se retrouvent dans ces complexes (McNally et McNally, 1996; Zheng et al., 1998). Les mécanismes par lesquels les snRNPs et les protéines SR pourraient agir de façon négative sur la sélection des sites d'épissage restent à établir.
D'autres facteurs liant les éléments inhibiteurs d'épissage ont été caractérisé au cours des dernières années. L'élément inhibiteur d'épissage de l'exon 7 du pré-ARNm
f3-tropomyosine de rat est lié spécifiquement par le hnRNP H, indiquant que ce hnRNP participe à l'exclusion de l'exon 7 dans des cellules non musculaires et dans la régulation de l'épissage alternatif de ce gène (Chenet al., 1999). D'autres exemples de régulation négative impliquent aussi des hnRNPs. Les inhibiteurs d' épissage de1'
exon 2 du gène tat du virus du VIH et de l'exon K-SAM du gène FGF-R2 sont liés plus spécifiquement par le hnRNP Al comparativement à un ARN correspondant ne contenant pas la séquence inhibitrice. Ces résultats suggèrent que le hnRNP Al peut fonctionner comme facteur inhibiteur d'épissage et que des éléments exoniques inhibiteurs d'épissage peuvent fonctionner en recrutant le hnRNP Al (Caputi et al., 1999; Del Gatto-Konc?.ak et al.,1999). Il a aussi été démontré que l'élément inhibiteur d'épissage de l'exon 2 du VIH requiert les protéines hnRNP Al, A2, BI et 82 (Caputi et al., 1999). Tous les différents facteurs mentionnés ici interagissent avec des éléments inhibiteurs d'épissage mais leur
interaction potentielle avec la machinerie d'épissage pour inhiber ce processus reste à établir.
Dans plusieurs gènes, les éléments inhibiteurs sont juxtaposés à des éléments stimulateurs d'épissage, ce qui suggère que la sélection des sites d'épissage est sous le contrôle de plusieurs éléments qui agissent de façon positive et négative (Amendt et al.,
1994; Carstens et al., 1998; Del Gatto et Breathnach. 1995; Kan et Green, 1999; Konig
et al., 1998; Staffa et al., 1997; Staffa et Cochrane, 1995). Le niveau d'utilisation d'un
site d'épissage est donc le résultat d'une modulation par plusieurs éléments cis.
Gène suppresseur de tumeurs p53
Le gène suppresseur de tumeurs p53 possède un rôle de premier ordre dans l'oncogénèse puisqu'il est muté dans près de 50% des tumeurs (Greenblatt et al., 1994: Hollstein et al., 1996). La protéine p53 est impliquée dans plusieurs processus cellulaires comme la transition de la phase G 1 à la phase S, la transition de la phase G2 à la phase M du cycle cellulaire, l'induction de l'apoptose et une voie de réparation de l'ADN (Giaccia et Kastan, 1998; Gottlieb et Oren, 1996; Ko et Prives, 1996; Levine, 1997). Suite à un stress cellulaire comme par exemple un dommage à 1' ADN ou en des conditions d'hypoxie, le niveau de la protéine p53 augmente puisque la stabilité de la protéine est accrue (Kastan, 1996; Lane et Hall, 1997; Prives, 1998). Conséquemment à cette hausse, p53 pourra conduire à un arrêt du cycle cellulaire ou induire l'apoptose (Lane et Hall, 1997; Prives, 1998; Sherr, 1998). La protéine p53 peut agir en tant
qu'activateur ou répresseur de la transcription de plusieurs gènes. En effet, les gènes p21, mdm-2, GADD45, cyclin G, bax, IGF-BP3 sont des exemples de gènes dont la
transcription est induite par p53. La transcription des gènes c-fos, c-jun, IL-6, Rb et Bcl-2 est quant à elle réprimée par p53 (Prives, 1998). Une autre des caractéristiques de p53 est que cette protéine peut promouvoir le réappariement de I' ADN et de I' ARN (Oberosler et al., 1993; Wu et al., 1995).
Le gène p53 contient 11 exons encodant une protéine de 390 acides aminés (aa)
chez la souris et de 393 aa chez l'humain. Cette protéine contient quatre domaines distincts : une région activatrice de transcription en amino-terminal (aa 1-42), un domaine riche en proline impliqué dans l'arrêt du cycle cellulaire et dans l'induction de l'apoptose (aa 61-94), un domaine central de liaison séquence-spécifique à l'ADN (aa
102-292) et une région carboxy-terminale possédant plusieurs fonctions (aa 287-393). La région carboxy-terminale de p53 contient un domaine de tétramérisation. un domaine de réappariement de I' ADN et de 1' ARN et une région responsable du repérage de
1' ADN endommagé (Giaccia et Kast~ 1998). Il a aussi été suggéré que ce domaine
régule de façon allostérique l'activité de liaison séquence-spécifique à I' ADN de p53 (Giaccia et Kast~ 1998; Ko et Prives, 1996; Levine, 1997).
Épissage altenatif du gène suppresseur de tumeun pS3
Un événement d'épissage alternatif a été décrit dans le pré-ARNm p53 murin entre le site d'épissage 5' de l'exon 10 et deux sites d'épissage 3' de l'exon 11 (voir Figure 3)
(Arai et al., 1986; Kulesz-Martin et al., 1994). Le site 3' alternatif, appellé 3'Cas, est situé 96 nucléotides en amont du site 3'Reg. Un codon d'arrêt est inséré dans l'ARNm lors de l'utilisation du site d'épissage 3'Cas et mène à la production d'une protéine de 381 aa. La protéine p53Cas diffère de la protéine p53Reg par le fait que les 26 derniers
aa de p53Reg sont remplacés par 17 aa de p53Cas dont on ne connaît pas encore la fonction (Kulesz-Martin et al., 1994).
5'ss 3'Cas 3'Reg
Exon 10 Exon 1 la Exon 1 lb
Figure 3. Représentatioo de l'événement d'épissage alternatif dans l'extrémité carboxy-terminale du gène suppresseur de tumeurs p53 murin.
Les deux protéines produites suite à l'épissage alternatif, p53Cas et p53Reg, possèdent des activités biochimiques différentes et sont fonctionnellement distinctes. La première différence se situe au niveau de leur activité de liaison séquence-spécifique à
l' ADN. La protéine p53Cas peut lier l' ADN de façon constitutive alors que la protéine p53Reg doit être préalablement activée afin de pouvoir lier l'ADN (Bayle et al., 1995; Wolkowicz et al., 1995; Wu et al., 1994). Une autre distinction entre les deux isoformes est que p53Reg peut réapparier des régions d' ADN et d' ARN simples brins tandis que p53Cas ne possède pas cette activité (Wu et al., 1995). De plus, l'induction de l'apoptose via p53Cas ou p53Reg ne se fait pas selon la même cinétique; la protéine
p53Cas induirait plutôt une apoptose atténuée comparativement à la protéine p53Reg (Almog
et al.,
1997). La protéine p53Cas a été détecté autant dans des lignées cellulaires de souris que dans des tissus murins normaux (Han et Martin, 1992; Kulesz-Martinet al.,
1994). Il a aussi été démontré que I' isoforme protéique p53Cas est exprimé préférentiellement durant la phase G2 du cycle cellulaire contrairement à !'isoforme p53Reg, qui est exprimé de façon préférentielle en phase Gl (Kulesz-Martinet al.,
1994).
Au cours des dernières années. deux protéines p73 et p63 ont été associées à la famille du gène p53. Ces deux protéines possèdent une bonne homologie avec p53 et elles peuvent activer les promoteurs répondant à p53 et induire l'apoptose. Contrairement à p53, p63 et p73 ne semblent pas être fréquemment mutés dans les
cancers humains étudiés jusqu'à présent.
De
plus, plusieurs isoformes de p63 et p73 sont formés par épissage alternatif et l'expression de ces deux gènes semble être restreinte à certains tissus (Kaelin, 1999).Les travaux que j'ai effectués dans le cadre de ma maîtrise ont porté sur l'épissage alternatif du gène suppresseur de tumeurs p53. La première partie de mes recherches a porté sur la caractérisation du patron d'épissage alternatif de l'extrémité carboxy-terminale de p53 de différentes espèces et lignées cellulaires. Par la suite, les différences observées dans les diverses espèces étudiées (souris, rat et humain) de même qu'à l'intérieur des lignées cellulaires d'une même espèce nous ont dirigé vers l'analyse des bases moléculaires responsables de ces variations.
La deuxième partie de mes travaux de recherche a porté sur la caractérisation d'un élément inhibiteur d'épissage situé dans l'exon l l du gène p53. À l'aide d'un système d'épissage in vitro, nous avons pu démontrer qu'une séquence située dans l'exon l l de
p53 est responsable de l'inhibition de l'utilisation du site d'épissage 3'Reg, situé en amont. Ces travaux ont démontré que cette inhibition se fait en bloquant la formation du complexe d'épissage de façon précoce, soit au niveau des complexes A et B. Des essais d'épissage in vitro et de rétention sur gel en compétition ont aussi permis de déterminer
que l'inhibition se fait via un facteur nucléaire qui se lie spécifiquement à l'élément inhibiteur. Des essais d'épissage in vitro en présence d' ARN compétiteurs spécifique et
non spécifique de l'élément AH ont démontré qu'un facteur trans est probablement
responsable de l'activité inhibitrice de la séquence AH sur l'utilisation d'un site d'épissage 3'. Des expériences de réticulation aux UV, de NorthWestem et de chromatographie d'affmité ont toutes indiqué qu'un facteur nucléaire d'un poids moléculaire apparent de 64 k.Da lie spécifiquement l'élément inhibiteur d'épissage du gène p53. Une analyse par spectrométrie de masse ainsi que qu'une analyse par immunobuvardage de type Western du facteur de 64 k.Da ont identifié cette protéine comme étant hnRNP K.
RÉSULTATS
CHAPITREI: Alternative splicing in the carboxy-terminal region reveals species specificity in the regulation of the p53 tumor suppressor gene.
Article publié dans Nucleic Acids Research (2000), Vol. 28, No. 6, 1489-1497.
CHAPITRER: Characterization of a splicing silencer element modulating the utiliz.ation of exon eleven 3' splice site in the p53 tumor suppressor gene.
CHAPITRE 1
Alternative splicing in the carbosy-terminal region reveals species specificity in the regulation of the p53 tumor suppressor gene.
Laverdière, M. 1, Beaudoin, J.1 and Alain Lavigueur2
Département de Biochimie, Faculté de Médecine, Université de Sherbrooke, Sherbrooke, Québec, JIH 5N4 Canada
1 These authors contributed equally to this paper
2 Corresponding author
1-819-564-5284 1-819-564-5340 (fax)
e-mail: alavigue@courrier.usherb.ca
Running Head: alternative splicing of p53
ABSTRACT
Alternative splicing occurs in the carboxy-terminal region of the p53 tumor suppressor gene between two alternative 3' splice sites in intron 1 O. This alternative splicing event bas been detected in murine cells, but not in rat or human tissues. In this paper, we have characterized the pattern of p53 alternative splicing in cell lines from five different species. Our results confirm that p53 alternative splicing is species-specific, being detected only in cell fines of rodent origin. Using transient transfection assays, we have established that the rat p53 gene undergoes efficient alternative splicing in both mouse
and rat cell lines, thus demonstrating that it bas ail the necessary
cis-acting
sequences tobe altematively spliced. ln contrast, we were unable to detect any usage of the human alternative 3' splice site under the same experimental conditions. Thus, the low levels or absence of altematively spliced p53 mRNA in rat and human cell lines seems to be the result of different mechanisms. Our results support the hypothesis that there are species-specific mechanisms implicated in the regulation of p53 activity.
INTRODUCTION
The p53 tumor suppressor gene plays a very important role
in
oncogenesis, as it is mutated in nearly 500/o of most types of tumors (1,2). The p53 protein is involved inG l/S and G2/M transitions of the cell cycle, in a pathway responsible for DNA damage repair and in the induction of apoptosis (3-6). After various forms of cellular stress such as DNA damage or hypoxia, p53 protein levels are greatly increased, mainly by protein stabilization (7-9). FoUowing these augmented levels, p53 will send signais to downstream effectors which will ultimately lead to cell cycle arrest or apoptosis (8-10). The p53 protein can positively or negatively regulate the transcription of several cellular genes and binds directly to cellular and viral proteins ( 5). Furthermore, p53 is able to directly recognize damaged DNA (11-13), and to promote reannealing of both DNA and RNA (14,15).
The p53 protein contains four functional domains, an amino-terminal transcriptional activation region (aa residues 1-42), a proline-rich domain (aa residues 61-94) involved in growth suppression and apoptosis, a central sequence-specific DNA-binding domain (aa residues 102-292), and a carboxy-terminal region (aa residues 287-393). The carboxy-terminal region can be subdivided into a flexible linker, a tetramerization domain, and a DNA and RNA annealing domain which can also bind single-stranded DNA without any sequence specificity (6). The carboxy-terminal region of p53 plays a direct role in sensing damaged DNA and it bas been suggested that this region allosterically regulates p53 binding activity, possibly by masking the DNA-binding region located in the central part of the molecule (3,5,6).
An alternative splicing event bas been described in the murine p53 gene between exon 10 and two alternative 3' splice sites, one located at the border of exon 11 (3'R) and the other (3'Cas) located 96 nucleotides upstream of the regular 3' splice site (16-18). The presence of a stop codon in the 96 nucleotide insert from intron IO results in the production of a protein of 381 residues in which the fast 26 amino acids of the regular p53 (p53R)
are
missing andare
replaced by a 17 amino acid sequence of unknown function, unique to the altematively spliced p53 (p53Cas) (18).p53R and p53Cas gene products have distinct biochemical activities and are functionally different. The p53Cas protein bas constitutive sequence-specific DNA binding whereas the p53R protein requires activation for efficient DNA binding (19-21). While p53R bas RNA and DNA annealing activities, p53Cas lacks these activities (15).
It bas also been reported that murine p53Cas induces apoptosis in myeloid cells, albeit with kinetics that are different from the p53R isoform (22). Finally, although both pSJ isoforms can inhibit growth of cells lacking endogenous p53, endogenous pSJR and pSJCas proteins respond to DNA damage with different kinetics of nuclear accumulation and efficiencies ofbinding to a p53 consensus DNA sequence (23).
pSJCas RNA has been detected in both murine cells lines and normal mouse tissues (17, 18). Using a p53 antibody specific for the alternatively spliced form of p53, it bas been shown that the p53Cas protein is preferentially expressed during the G2 phase of the cell cycle in contrast to the regular pSJ protein which is preferentially expressed in G 1 (18). ln addition, p53Cas is differentially expressed in proliferating epidermal cells when compared to differentiating cultures ( 18) or at different stages of keratinocyte
differentiation (24). The timing of p53Cas and p53R RNA and protein synthesis during the cell cycle could be important in p53 biochemical functions which are mediated by interaction between two protein products of the wild-type p53 gene (21 ). The marked
increase in DNA-binding activity of p53Cas relative to p53R implies that a potential mechanism for activating p53 function in a cellular context could be by increasing the relative amount of p53Cas to p53R.
Recently, a more complex alternative splicing pattern generating four different isoforms in the carboxy-terminal region of the p73 gene, a protein having substantial functional and structural similarity to p53, bas also been described in the human p 73 gene (25,26). Another closely related member of the p53 family, named p63, also exhibits a complex pattern of alternative splicing that is conserved between the murine and the
human
genes in both the amino and carboxy-terminal regions (27).In this paper, we have characterized the alternative splicing pattern in the carboxy-terminal region of the p53 gene from different cell fines and species and have used transient transfection assays to study the molecular basis for the difference observed in the alternative splicing pattern of carboxy-terminal region of the rat, murine and human p53 genes.
MATERIAL AND METHODS
DNA sequences
DNA sequence have been deposited in GenBank under the following accession numbers. Mouse: AF190269; rat; AF190270; hamster: AF190271; gorilla: AF190272.
Cell lines
Ail the cell lines were grown in DMEM supplemented with 10% foetal bovine serum and an antibiotic-antimycotic mix ( 1 OOU/ml penicillin G, 1 OOµg/ml streptomycin, 0.25 µg/ml
Fungizone) (Gibco), with the exception of the HeLa cell line which was grown in RPMI 1640 and the CHO cell line which was grown in alpha-MEM. Both media were supplemented with 10% foetal bovine serum and an antibiotic/antimycotic mix ( 1 OOU/ml
penicillin G, IOOµg/ml streptomycin, 0.25 µg/ml Fungizone) (Gibco).
Probes for RNase protections
Probe MT7C-E9. The murine probe MT7C-E9 corresponding to the mouse p53Cas cDNA was generated by in vitro transcription on a PCR template containing the T7 RNA polymerase promoter region (28). The PCR template was generated by amplification of a
p53Cas cDNA with oligonucleotides E9Sl
(5'AACCACTTGATG-GAGAGTA TITCACC3 ') and T7Cas (5'T AA TACGACTCACT AT AGGGAAG-GCTTGGAAGGCTCT AGGCTGGAGG3). The p53Cas cDNA was obtained by reverse transcription of cytoplasmic mRNA purified from the N2A cell line. Probe MT7E-E9.
in vitro transcription on a PCR template containing the T7 RNA polymerase promoter region (28). The PCR template was generated by amplification of a p53R cDNA with oligonucleotides E9Sl (S'AACCACTIGATGGAGAGTATITCACC3') and T7El1Al (S'T AATACGACTCACTATAGGGTCAGTCTGAGTCAGGCCC3'). The p53R cDNA was obtained by reverse transcription of cytoplasmic mRNA purified from the N2A cell line. Probe HT7E-Pul. The human probe HT7E-Pul was generated by in vitro
transcription on a PCR template containing the T7 RNA polymerase promoter region (28). The PCR template was generated by amplification of the plasrnid pxp53 with the oligonucleotides T7E l lAl (S'T AATACGACTCACTATAGGGTCAGTCTGAGTCAG-GCCC3') and HPulnSl (5'GGTAAGGGAAGATTACGAGAC3'). Probes Mp53L and Rp53L. The mouse probe Mp53L and the rat probe Rp53L were generated by in vitro
transcription with T3 RNA polymerase on the plasmids pTEP Mp53L and pTEP Rp53L digested with Nhel. An 181 nt BamHI-Nhel fragment from phage lambda was inserted between the p53 minigene and the SV40 polyadenylation signal of pTEP Mp53 to generate pTEP Mp53L and ofpTEP Rp53 to generate pTEP Rp53L. Probe Hp53B. The human probe Hp53B was generated by in vitro transcription with the T3 RNA polymerase on the plasmid pTEP Hp53 digested with Bglll.
RNase protection of endogenous p53 mRNA Crom cell lines
Extraction of cytoplasmic RNA was done using the NP40/urea method as described previously (28). RNA hybridiz.ation was perfonned essentially as described previously (29). Hybridiz.ation was perfonned overnight at 55°C with the mouse probe MT7C-E9 for the mouse, rat and hamster cell lines and with the human probe HT7E-Pul for the human and monkey cell lines. Following hybridiz.ation, the RNA was digested with 50 U
of RNase Tl and O.OSµg of RNase A at 30°C for l hour. The mouse Mf7C-E9 and hurnan HT7E-Pul RNA probes were synthesiz.ed by in vitro transcription witb T7 RNA polymerase and radiolabelled with 32P-UTP as described previously (28).
RT-PCR analysis of RNA from cell Unes
Cytoplasmic RNA from cell lines was extracted witb the NP40/urea method (28) Reverse transcription with AMV reverse transcriptase was done at 42°C for 30 minutes, using the El IA7 oligonucleotide primer (5'ACTITCTTAGCCATTGTTTT3') for mouse, hamster and rat cell lines, or the MPuA2 oligonucleotide primer (5'AGACTGGCCCTTCTTGGTCTTCAGG3') for human and monkey cell Iines. Following denaturation at 94°C for l min, PCR was performed for 30 cycles (94°C for l min, 65°C for l min), with a final incubation at 72°C for 2 min, using the primers El OS l (5'GTCTCGAGCGCTTCGAGATGTTC3') and MPuA2. PCR products were
run
on a5% polyacrylamide gel with 0.5X TBE.
PCR amplification of pSJ exon 10-exon Il region
The mouse p53 exon l 0-exon 11 region was amplified by PCR from the plasmid pM53R-18 containing the p53 gene from the CB7 cell line (30) using the oligonucleotides EIOSI (5'GTCTCGAGCGCTTCGAGATGTTC3') and El IAI (5'GGAAGCTTCAGTCTGAGTCAGG3'). PCR was performed for 30 cycles (94°C for l min, 55°C for l min). The human p53 exon 10-exon 11 region was amplified by PCR from the plasmid pxp53 containing the human p53 gene from human foetal liver (kindly provided by Dr. L. Crawford, ICRF, London) using the oligonucleotides ElOSland El IAJ. PCR was performed for 30 cycles (94°C for l min, 55°C for l min). The gorilla
p53 exon 10-exon 11 region was amplified by PCR with oligonucleotides EIOS3 (5'TGTICCGAGAGCTGAATGAGGCCT3') and El lAl, using gorilla genomic DNA (kindly provided by
Dr.
G. Boissonneault, Université de Sherbrooke). PCR was performed for 40 cycles (94°C for 1 min, 50°C for 1 min, 72°C for 1 min). The rat p53 exon 10-exon 11 region was amplified by PCR with oligonucleotides E 1 OS4 (5'CGCTICGAGATGTICCGGGAGCTGAAT3') and Rp53Al (5'GGACTAGCAT-TGTCTIGTCAGC3 '), using genomic DNA extracted from rat kidneys. PCR was performed for 40 cycles (94°C for 1 min, 60°C for 1 min, 72°C for 1.5 min). The hamster p53 intronlO-exon 11 region was amplified by PCR with oligonucleotides Puln (5'GGTGAAAGGGAGGATAAACTGA3') and EIIAI, using genomic DNA extracted from hamster kidneys. PCR was performed for 40 cycles (94°C for l min, 60°C for 1 min, 72°C for 1.5 min).DNA sequence analysis of PCR products
Ali the DNA sequences were determined using the Pharmacia sequencing kit (Pharmacia)
and using 35S-ATP. Oligonucleotides EIOSI and El IAI contain respectively Xhol and
Hindlll restriction sites that were used to clone the PCR fragments into the plasmid pBluescript KS+ (Stratagene). The mouse p53 sequence (C87) was determined from two independent plasmids (pMp53) generated from cloning of DNA fragments derived from two independent PCR reactions. The PCR fragments were digested with Xhol and Hindlll and cloned into the plasmid pBluescript KS+ digested with Xhol and Hindlll.
The sequence was determined by using the universal and reverse primers (Pharmacia) with the pMp53 plasmid and a derivative plasmid obtained by deletion of an internai
generated from cloning of a DNA fragment obtained by PCR. The DNA from the PCR reaction was digested with Xhol and HindIII, and cloned into the plasmid pBluescript KS+ digested with Xhol and Hindlll. The sequence was determined by using the universal and reverse primers with pHp53 and derivative plasmids obtained by EcoRl/EcoRI and BglII/Bamlll internai deletions. The gorilla p53 sequence was determined from a plasmid (pGop53) generated from cloning of a DNA fragment obtained by PCR. The DNA from the PCR reaction was cloned into the plasmid pBluescript SK digested with EcoRV. The sequence of the 3' region was determined by using the reverse primer with pGop53. The rat p53 sequence was determined by direct cycle sequencing of a PCR reaction from the rat p53 gene with the oligonucleotides EIOS4 and El lAl on an Applied Biosystems 373A (University of Calgary Sequencing Services) and also from a plasmid (pRp53) generated from cloning of a DNA fragment obtained by PCR. DNA from the PCR reaction was cloned in the plasmid pBluescript KS+ digested with EcoRV. The sequence was determined by using the oligonucleotides EIOS4 (5'CGCTTCGAGATGTTCCGGGAGCTGAAT3'), El IAI and El IA4 (5'GATCAAGGCTTGGAAGGCTCTAGGCTGGA3') with pRp53. The hamster p53 sequence was determined from a plasmid (pHap53) generated from cloning of a DNA fragment obtained by PCR. A DNA fragment from the PCR reaction was cloned in the plasmid pBluescript KS+ digested with EcoRV. The sequence was determined by using the universal and reverse primers with pHap53.
p53 minigene expression vectors and transient transfection assays
The exon 10-exon 11 region of the mouse, rat and human p53 genes were inserted into the pTEP expression vector, a derivative of pCMVSV containing the cytomegalovirus
(CMV) promoter region and the polyA signal from SV40 virus (kindly provided by Dr.
B. Chabo~ Université de Sherbrooke) (31). In Figure 7A (lanes 1-3), cells were
transfected with 2µg of plasmid DNA and 4µ1 of Fugene (Roche) according to
the
supplier's recommendations. Cytoplasmic RNA was isolated 48 hours after transfection by the NP40/urea method as described previously (28), and RNA samples were subjected to DNaseI digestion. RNA hybridi7.ation was performed essentially as described (29). Cytoplasmic RNA and the MpS3L probe were hybridized at S0°C ovemight. RNase digestion was performed with IOOU ofRNase Tl.In Figure SA (lanes 4 and S), cells were transfected with 2µg ofplasmid DNA and 4µ1 of Fugene (Roche) according to the supplier's recommendations. Cytoplasmic RNA was isolated 24 hours after transfection by the NP40/urea method and subjected to DNasel digestion. RNA was hybridized ovemight at S0°C with the MpS3L probe and digested with IOOU of RNase Tl (lane 4) or with the HpS3B probe and digested with IOOU of RNase Tl and 0.1 µg of RNase A (lane S). In Figure SB, cell were transfected with 2µg of plasmid DNA and 4µ1 of Dosper Liposomal Transfection Reagent (Boehringer) according to the supplier's recommendations. Cytoplasmic RNA was isolated 48 hours after transfection by the NP40/urea method and treated with DNasel. RNA was hybridized ovemight at S0°C with the MT7E-E9 probe, and digested with l OOU of RNase Tl. In Figure SC, cells were transfected with 2µg of plasmid DNA and 4µ1 of Fugene (Roche). Cytoplasmic RNA was isolated 24 hours after transfection by the NP40/urea method and digested with DNasel. RNA was hybridized ovemight at S0°C with the MpS3L probe (lanesl and 2) or with the Rp53L probe (lanes 3 and 4) and digested with IOOU ofRNase Tl.
RESULTS
Analysis of the expression of the p53R and p53Cas isoforms in cell lines.
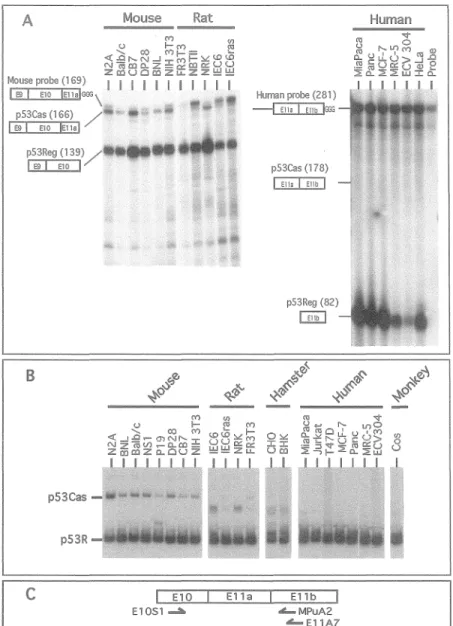
As a first step in the cbaracterization of the alternative splicing event affecting the carboxy-tenninal region of the p53 gene (Figure 1), we analyzed the levels ofp53Cas and
p53R mRNA in cell lines from five different species.
To determine the relative frequency of utilization of the two alternative 3' splice sites of the p53 gene, we analyzed the steady-state levels of mRNA of each type by RNase protection assays and by RT-PCR (see Table 1). As shown by the RNase protection assay depicted in Figure 2A. although the ratio of p53R/p53Cas varied between cell lines, the alternatively spliced p53 mRNA (p53Cas) was readily detected in ail the murine cell lines tested. The quantification of the two murine isoforms by Phosphoimager indicated that the p53Cas is expressed at levels varying between 2 to 26% of the p53R mRNA. The analysis of the expression of the two different p53 isoforms in rat cell lines also showed the presence of low to almost undetectable levels of p53Cas in the FR3T3, IEC6, IEC6ras, NRK and NBID cell fines by RNase protection assay or by RT-PCR with incorporation of radiolabelled 32P-dCTP (Figure 28 and 28). We were unable to detect any p53Cas rnRNA either by RNase protection or by RT-PCR in 1 monkey, 2 hamster, and 7 human cell lines despite the fact that we easily detected the p53R isoform in those cell lines (Figure 2).
1
A
b---
-
--- p53Reg..
--11 b 3'Reg p53CasFigure 1. Alternative splicing of the pSJ tumor suppressor gene. (A) Diagrammatic representation of the pSJ gene structure. (B) Alternative splicing pattern in the carboxy-tenninal region of the pSJ gene with two alternative 3' splice sites (J'Cas and J'R) in intron 10.
Species Cell U•e pSJCas Mause N2A (+) ,_ Balblc (+) 1.2 CB7 (+) 1.2 DP28 (+) 1.2 BNL (+) 1.2 NIH3T3 (+) 1.2 Pl9 (+)2 Rat NBTll (+/-) 1 NRK (+/-) 1.2 IEC6 (+/-) 1.2 IEC6ras (+/-) 1.2 FR3T3 (+/-) 1.2 Hamster CHO (-)
-BHK (-) 2 Monkeycos
(-).:Hum an Mia Paca (-)
·-Pane (-) 1.2 MCF7 (-) 1.2 MRC-5 (-) 1.2 ECV-304 (-) 1.2 He La (-) 1 T47D (-) 2 Jurkat (-) 2
1 : tested by RNase protection. : tested by RT-PCR.
-(+): detected. (+/-): very low levels. (-): not detected
Table 1: Cell lines tested for alternative splicing in the carboxy-terminal region ofp53 between exon JO
A Mo use Rat u ~ <J) '- <X) M~= ~ <(..01'-N--':c:Ml-:.l<D<D ~~ôêi~zff~~~~ Mouseprobe(169) 1 1 1 1 1 1 1 1 1 .1 1 8l 1 E10 lmalGGG \> p53Cas (166) 1 8l 1 E10 lmal p53Reg ( 139) 1 8l 1 E10 1 B p53Cas p53R
c
E10 E11a E10S1 -ii. Human probe (281) ~ p53Cas (178) 1 Ella l Ellb l p53Reg (82) G1LJ El lb .&...MPuA2 .&...E11A7 HumanFigure 2. Analysis of p53 mRNA levels in cell lines. (A) Analysis of the expression of the p53Cas and p53Reg mRNAs in different cell lines by RNase protection assay. The mouse probe (MT7C-E9) and the human probe (H7E-PuI) used are depicted schematically (see Material and Methods). The size of the protected fragments in nucleotides is indicated in parentheses. (B) Analysis of the expression of the p53Cas and p53Reg mRNAs in different cell lines by RT-PCR. The identity of the amplified PCR fragment corresponding to the two p53 isoforms is indicated. (C) Diagrammatic representation of the p53 oligonucleotides used in the RT-PCR analysis shown above. Reverse transcription of cytoplasmic RNA was done using the E11A7 oligonucleotide primer for mouse, hamster or rat RNA, or the MPuA2 oligonucleotide primer for human or monkey RNA. PCR amplification with 32P-dCTP was performed for
Sequence analysis of the p53 eion 10-eion 11 region from different species.
To gain a better understanding of the differences observed in the p53R/p53Cas ratio between the different species analyzed above, we first ana1)7.ed the DNA sequence of the altematively spliced p53 region to see whether
cis-acting
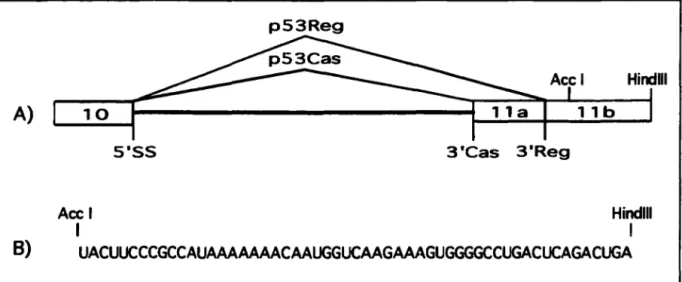
sequences could explain the differences observed. To do so, we amplified by PCR the region between exon 10 and exon 11 from cloned genomic fragments (murine and human p53 genes) or genomic DNA (gorilla, hamster and rat p53 genes) with p53-specific oligonucleotides (Figure 3).The region between exon 10 and exon 11 of the human p53 gene produced a PCR fragment of 1105 base pairs compared to PCR fragments of 778 and 884 base pairs for the murine and rat p53 genes respectively. The hamster p53 gene was amplified with oligonucleotides located in intron 10 and exon 11 giving an amplification product of 293 nucleotides (Figure 3 and Material and Methods).
Sequencing of an exon 10-exon 11 PCR fragment amplified from a plasmid containing part of the human p53 was done to ensure that the amplified p53 fragment used to generate our human pSJ expression vector did not suffer mutations in its 5' and 3' splicing signais during the process of PCR amplification. The human pSJ DNA we obtained was shown to have a
5'
splice site and 3' splicing signais identical to the splicing signais of the published human p53 gene (Genebank accession numbers X54156 and U94788).Mouse 3'Cas 3'Reg
t
t
1 BlO 1 Blla ---1 107 490 96 1 82 1 ElOSl...:.. "'-EllAl 785Hu man 3'Cas 3'Reg
Alu
t
t
BlO t~ 1 !Ula 107 408 286 1 127 1 94 1 82 1 821 El OSl ...:.. "'-EllAl 1105Rat 3'Cas 3'Reg
t
t
BlO
1
Blla1
107 510 1 95 1
E10S4...:i. "'-Rp53Al
884
Hamster 3'Cas 3'Reg
t
t
1
Blla204 1 90 82
Puln...:.. "'-E11A1 293
Figure 3. Diagrammatic representation of the PCR amplification products used for DNA sequencing of the murine, human, rat and hamster p53 exon 10-exon 11 region. The p53-specific oligonucleotides are indicated by arrows. The size of the PCR amplification products are shown below the oligonucleotides.
Similarly, we determined the sequence of the mwine exon 10-exon 11 region from the erythroleukemic cell line CB7 (30). The DNA sequence of the mouse alternatively spliced 3' region was found to be identical to the previously published sequence of this region (16). In addition, the sequence of the exonlO-exon 11 region of the murine p53 gene was determined from genomic DNA isolated from the Balb/c cell line. The sequence of the 5' splice site and the alternatively spliced 3' region of the p53 gene from the CB7 and Balb/c cell lines were found to be identical (M.L. and A.L. unpublished results). The complete DNA sequence of the rat p53 intron 10 and the sequence of the hamster and gorilla p53 3' region of intron 10 was also determined (Figures 4 and 5).
As determined by DNA sequence analysis, the size difference between the human and the rodent p53 genes is due to the presence of an Alu element present in intron 10 of the human gene. While the same region from the gorilla p53 gene was not sequenced completely, the size of the PCR amplification fragment (approximately 1100 bp) and the very close evolutionary distance between these two species is consistent with the presence of an Alu element in intron 10 of the p53 gorilla gene. One peculiar feature of the murine and rat p53 gene is the presence ofTGA\G triplet repeats in intron 10 ofboth genes. Two stretches of ten and fourteen triplets were found in the murine gene whereas a stretch of twenty repeats was present in the rat gene (Figure 4).
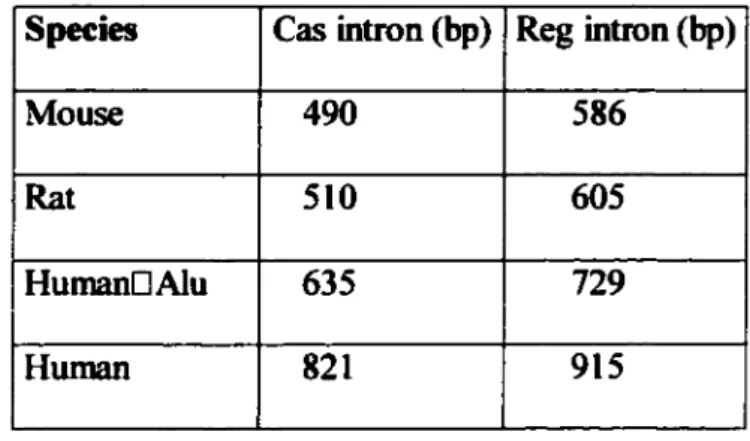
Comparison of the homology between the murine, rat and human p53 intron 10 sequences, showed 78% conservation of nucleotides between the murine and the rat gene,