Étude de l'adaptation des microorganismes aux
environnements complexes par l'utilisation de
code-barres moléculaires
Thèse
Clara Bleuven
Doctorat en biologie
Philosophiæ doctor (Ph. D.)
Québec, Canada
Étude de l’adaptation des microorganismes
aux environnements complexes par
l’utilisation de code-barres moléculaires
Thèse
Clara Bleuven
Sous la direction de :
Résumé
La plupart des êtres vivants habitent des environnements dont les facteurs biotiques et abiotiques varient selon une échelle spatiale ou temporelle. Cette hétérogénéité environnementale joue un rôle important sur les processus adaptatifs des organismes. Dans ce contexte, les microorganismes sont des modèles avantageux pour tester les théories évolutives et sont également des sujets d’intérêt en écologie. L’objectif principal de cette thèse est d’apporter un éclairage sur l’adaptation des microorganismes aux environnements complexes, expérimentaux et naturels. D’abord, nous avons rassemblé dans une revue de littérature les connaissances acquises concernant les mécanismes moléculaires adaptatifs des microorganismes aux variations environnementales. Les stratégies adaptatives sont associées à la prédictibilité des variations environnementales et comprennent notamment la mémoire, l’anticipation cellulaire, la stratégie de minimisation des risques et le sauvetage évolutif. Ce chapitre a mis en avant l’opportunité d’appliquer des techniques d’ingénierie moléculaire et des expériences en conditions contrôlées pour étudier l’adaptation des microorganismes à leur environnement naturel et l’effet des interactions biotiques sur leur fitness. Ainsi, pour répondre à cet objectif, nous avons optimisé la mesure de la fitness en expérience de compétition chez l’espèce de levure bourgeonnante non domestiquée Saccharomyces paradoxus en marquant plus de 500 souches regroupant les principales lignées Nord-Américaines SpB, SpC et SpC*, par des code-barres moléculaires uniques. L’insertion n’ayant pas eu d’impact sur la croissance des souches, nous avons démontré que cette collection est fonctionnelle pour la méthode de Bar-seq et permet de détecter la fitness des souche individuelles ainsi que la fitness moyenne des lignées dans différentes conditions expérimentales. Par la suite, afin d’étudier l’adaptation locale chez S. paradoxus et l’effet des facteurs biotiques sur la fitness des lignées SpB, SpC et SpC*, nous avons réalisé une expérience de compétition avec les souches à code-barre de la collection dans un sol naturel.. Notre étude montre un effet de la communauté microbienne sur la différence de fitness relative entre les lignées ainsi qu’une potentielle adaptation locale chez SpC dont les souches locales ont une meilleure fitness que les souches distantes du site d’échantillonnage du sol. Ainsi, ces travaux de doctorat démontrent l’importance de réaliser des expériences dans des microcosmes naturels afin d’appréhender les facteurs influençant potentiellement l’histoire évolutive des microorganismes, comme les interactions biotiques et l’adaptation locale chez S. paradoxus.
Abstract
Most organisms inhabit environments whose biotic and abiotic factors vary on a spatial or temporal scale. This environmental heterogeneity plays an important role in the evolutionary and adaptive processes. In this context, microorganisms are relevant models for testing evolutionary theories and are also subjects of interest in ecology. The main objective of this thesis is to shed light on the adaptation of microorganisms to complex, experimental and natural environments. As a first step, we gathered in a literature review the knowledge gained about the adaptive molecular mechanisms of microorganisms to environmental variations. The adaptive mechanisms are closely associated with the predictability of environmental changes and comprise memory, cellular anticipation, bet-hedging and evolutionary rescue. This chapter highlighted the opportunity to apply molecular engineering techniques and experiments under controlled conditions to study the adaptation of microorganisms to their natural environment and the effect of biotic interactions on their fitness. Thus, to meet this objective, we have optimized the measure of fitness in competitive experience in a non-domesticated yeast species, Saccharomyces paradoxus, by tagging with unique molecular barcodes more than 500 strains within the North American lineages SpB, SpC and SpC*. This collection has been shown to be functional for the Bar-seq method and allows to measure the fitness of individual strain as well as the average fitness of the lineages in different experimental conditions. Finally, in order to study the local adaptation in S. paradoxus and the effect of biotic interactions on the fitness of the SpB, SpC and SpC* lineages, we performed a competition experiment with the barcoded strains in a natural soil. We quantified the relative fitness of S. paradoxus strains and lineages and assessed the effect of the microbial community using control soil whose community was partially eliminated. To test the local adaptation of strains, the effect of their localization and their substrate on their fitness was analyzed. Our study shows i) a potential local adaptation in SpC whose local strains have a better fitness than the distant strains and ii) an effect of the microbial community on the relative fitness variation between the lineages. Thus, this thesis demonstrates the importance of performing experiments in natural microcosms in order to understand the which factors potentially influence the evolutionary history of microorganisms, such as biotic interactions and local adaptation in S. paradoxus.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures ... viii
Figures ... viii
Figures supplémentaires ... viii
Liste des tableaux ... ix
Tableaux ... ix
Tableaux supplémentaires ... ix
Liste des abréviations et symboles ... x
Remerciements ... xi
Avant-propos ... xiii
Introduction ... 1
L’adaptation des organismes à leur environnement ... 1
Évolution et adaptation, un historique ... 1
La notion de fitness ... 3
La variation environnementale et l’adaptation ... 4
Les microorganismes comme modèles d’étude en évolution et écologie ... 9
Les systèmes microbiens ... 9
Méthodes d’étude en écologie et évolution microbienne ... 10
La complexité des milieux naturels microbiens ... 17
Le modèle Saccharomyces paradoxus ... 21
Le genre Saccharomyces ... 21
L’espèce non-domestiquée S. paradoxus ... 24
Objectifs de la thèse ... 28
1
CHAPITRE 1 Molecular and cellular bases of adaptation to a changing
environment in microorganisms ... 32
1.1
Résumé ... 33
1.2
Abstract ... 34
1.3
Introduction ... 35
1.4
Adaptive strategies depend on the rate and predictability of changes ... 35
1.5.1
Negative pleiotropy and specialists ... 37
1.5.2
Positive pleiotropy and generalists ... 39
1.6
Mechanisms of anticipation and memory ... 40
1.6.1
Anticipation and associative learning ... 40
1.6.2
Memory ... 41
1.7
Stochasticity of events and cell fate decisions ... 43
1.8
The race against extinction ... 45
1.9
Conclusions and future perspectives ... 47
1.10
Acknowledgements ... 48
1.11
Figure ... 49
1.12
References ... 50
2
CHAPITRE 2 A collection of barcoded natural isolates of Saccharomyces
paradoxus to study microbial evolutionary ecology ... 59
2.1
Résumé ... 60
2.2
Abstract ... 61
2.3
Introduction ... 62
2.4
Material And Methods ... 64
2.4.1
Barcode and resistance cassette amplification ... 64
2.4.2
Fusion PCR ... 65
2.4.3
Barcodes insertion ... 66
2.4.4
Barcoded strains phenotypic analysis ... 67
2.4.5
Barcoded strain competition assay ... 68
2.4.6
Barcode Sequencing ... 70
2.4.7
Barcode sequence analysis and quantification ... 70
2.5
Results ... 71
2.5.1
Transformation and integration of the barcodes in the S. paradoxus strains .. 71
2.5.2
Comparison of the growth between the barcode and parental strains ... 72
2.5.3
Competition assay ... 72
2.6
Discussion ... 74
2.7
Acknowledgements ... 76
2.8
Conflict of Interest ... 76
2.9
Data Accessibility ... 76
2.10
Author Contributions ... 76
2.11
References ... 76
2.12
Tables ... 79
2.13
Figures ... 80
2.14
Supplementary data ... 86
2.14.1
Supplementary tables ... 86
2.14.2
Supplementary figures ... 92
2.14.3
Supplementary references ... 93
3
CHAPITRE 3 An active soil microbial community influences the fitness of
natural yeast populations ... 94
3.1
Résumé ... 95
3.2
Abstract ... 96
3.3
Introduction ... 97
3.4
Material and methods ... 99
3.4.1
Soil sampling ... 99
3.4.2
Yeast Pool preparation ... 100
3.4.3
Competition experiment in natural and irradiated soils ... 100
3.4.4
Metabarcoding amplicon sequencing and analysis ... 101
3.4.5
Yeast Barcode sequencing and analysis ... 102
3.5
Results ... 103
3.5.1
Identification of the experimental soil microbial community ... 104
3.5.2
Effect of the microbial community on relative fitness ... 105
3.5.3
Geographical and substrate of origin influence on relative fitness ... 106
3.6
Discussion ... 106
3.7
Acknowledgements ... 110
3.8
Conflict of interest ... 110
3.9
Data availability ... 110
3.10
Author Contributions ... 110
3.11
References ... 110
3.12
Figures ... 115
3.13
Supplementary material ... 120
3.13.1
Supplementary figures ... 120
3.13.2
Supplementary tables ... 127
3.13.3
Supplementary File ... 131
3.13.4
ITS and 16S amplicon sequencing ... 131
3.13.5
OTU assignation protocol ... 132
3.13.6
High-Throughput barcode sequencing ... 132
4
Conclusion ... 133
Mécanismes moléculaires de l’adaptation aux environnements variables ... 133
Collection de levures marquées par code-barres moléculaires ... 135
Adaptation locale et effet des interactions biotiques ... 136
Perspectives ... 137
Adaptation de S. paradoxus aux conditions fluctuantes de l’environnement ... 137
Adaptation de S. paradoxus à son environnement naturel local ... 139
L’adaptation des organismes dans le contexte du changement climatique ... 141
Bibliographie ... 143
Annexe A : Glossaire ... 155
Définitions ... 155
Liste des figures
Figures
Figure I 1. Décomposition de la variation environnementale. ... 6
Figure I 2. Critères pour identifier l’adaptation locale. ... 8
Figure I 3. Représentation du système Nord-Américain de S. paradoxus. ... 25
Figure 2-1. The S. paradoxus population structure and geographical distribution in North America. ... 80
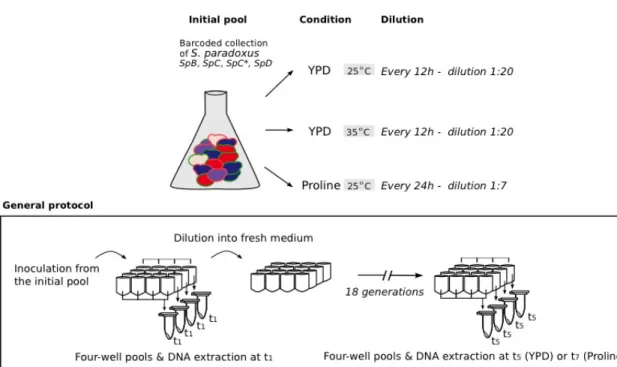
Figure 2-2. Integration protocol to barcode natural S. paradoxus strains. ... 81
Figure 2-3.Competition assay using the barcoded strains. ... 82
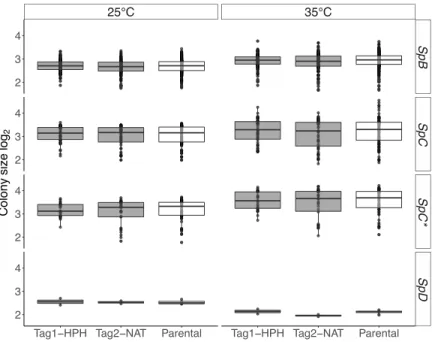
Figure 2-4. Barcoded strains show growth similar to that of parental strains on solid medium. ... 83
Figure 2-5.Relative fitness of strains and lineages of S. paradoxus assessed by barcode sequencing. ... 84
Figure 2-6.Genotype-by-environment interaction for fitness. ... 85
Figure 3-1.Experimental design. ... 115
Figure 3-2. Persistence of S. paradoxus strains in the experimental soils. ... 116
Figure 3-3. Diversity of the microbial communities in the experimental soils at the phylum level. ... 117
Figure 3-4. Relative fitness of the S. paradoxus strains in natural and irradiated soils. .. 118
Figure 3-5. Relation between latitude and substrate of origin with the relative fitness of S. paradoxus SpB and SpC lineages. ... 119
Figures supplémentaires
Figure S 2-1. Correlation of read counts among replicates (Pearson’s r = 0.94-0.99, p-value <0.01). ... 92Figure S 3-1. Sampling site. ... 120
Figure S 3-2. Comparison of dilution plating between the two soils, natural (room-temperature for 72h) and irradiated (gamma-irradiation for 72h). ... 121
Figure S 3-3. Persistence of S. paradoxus strains in the experimental soils. ... 122
Figure S 3-4. Persistence state of the barcoded strains throughout the competition assay in soil. ... 123
Figure S 3-5. Data filtering to limit the experimental noise du to indexing amplification. . 124
Figure S 3-6. Diversity of microbial communities in the experimental soils at the genus level. ... 125
Figure S 3-7. Comparison between of relative fitness in laboratory conditions and on natural susbtrates. ... 126
Liste des tableaux
Tableaux
Tableau I 1. Résumé des interactions écologiques entre espèces de microorganismes. . 18
Tableau 2-1. Number of S. paradoxus barcoded strains in the collection. ... 79
Tableaux supplémentaires
Table S 2-1. List of barcoded strains. ... 86Table S 2-2. List of oligonucleotides used in this study. ... 86
Table S 2-3. Summary of barcoding efficiency during the rounds of transformation. ... 87
Table S 2-4. PCR cycles of the barcoding protocol ... 88
Table S 2-5. Comparison of colony size (log2) between the HPH, NAT modules barcoded strains and their parental strains on solid YPD media. ... 89
Table S 2-6. Number of strains used in the analysis of the competitive assay. ... 90
Table S 2-7. Pairwise comparisons between the fitness of the S. paradoxus lineages in each condition. ... 91
Table S 3-1.Characteristics of the strains used in this study: names, barcode sequence, lineage, geographical location, substrate and distance from sampling site. ... 127
Table S 3-2. Raw CFU counts of S. paradoxus barcoded strains at Day 0 and Day 21 of the experiment. ... 127
Table S 3-3. Oligonucleotides used in this study. ... 128
Table S 3-4. Description of the OTUs detected in the experimental natural and irradiated soils. ... 129
Table S 3-5. Read counts for each S. paradoxus barcoded strain. ... 129
Table S 3-6. Analysis of variance (ANOVA) of the S. paradoxus lineages relative fitness. ... 130
Table S 3-7. Pairwise comparisons between the fitness of the S. paradoxus strains of each latitude using Kruskal-Wallis and Dunn post-hoc tests. ... 131
Liste des abréviations et symboles
% Pourcentage
°C Degré Celsius
°F Degré Farenheit
AEC Avant l’ère commune
ANOVA Analyse de variance (Analysis of variance)
bp Paire de bases (base pair)
CFU Unité formant colonie (Colony Forming Unit) DDPCR « Digital Droplet PCR »
DNA / ADN Acide désoxyribonucléique
GWAS Étude d’association pangénomique (Genome Wide Association Study)
h Heure
HPH Hybromycine B
LB Milieu de culture « Lysogeny Broth »
log Logarithme mg, µg Milligramme, microgramme min Minute(s) mL, µL Millilitre, microlitre mm, µm Millimètre, micromètre mM, µM Millimolaire, micromolaire NAT Nourséothricine
OD Densité optique (Optical Density)
PCR Réaction en chaîne par polymérase (Polymerase Chain Reaction)
pH Puissance Hydrogène
QTL Locus de caractères quantitatifs (Quantitative Trait Loci) RNA / ARN Acide ribonucléique
sd Écart-type
SpA Saccharomyces paradoxus lignée A SpB Saccharomyces paradoxus lignée B SpC Saccharomyces paradoxus lignée C SpC* Saccharomyces paradoxus lignée C* SpD Saccharomyces paradoxus groupe D
t Temps
Remerciements
Parce qu’il faut un village pour réussir un doctorat, je tiens à remercier toutes les personnes qui ont joué un rôle de près ou de loin, conscient ou inconscient sur l’aboutissement de ce projet de recherche.
Tout d’abord, merci à toi, Christian pour cet extraordinaire aventure. Tu m’as offert cette opportunité de rejoindre ton laboratoire après notre rencontre en Été 2014. Je me souviendrais toujours de cette journée décisive, des marmottes du campus et de ma découverte du système S. paradoxus. Merci pour la confiance que tu as m’a accordée. Merci d’avoir su me guider patiemment à travers les méandres de ces 4 années de doctorat.
Je tiens à remercier les membres de mon comité d’encadrement : Julie Turgeon et Michel Frenette. Merci d’avoir été présent pendant toutes ces années, Merci pour vos questions, vos remarques et conseils qui m’ont fait progresser dans la mise en place de mes projets. Merci aux professeurs Louis Bernier et James Anderson d’avoir accepté de réviser et évaluer ma thèse.
Je remercie également Isabelle Gagnon-Arsenault et Alexandre Dubé, sans qui rien n’aurait été possible. Merci de m’avoir appris tout ce que je sais à présent sur les levures et les robots. Merci pour votre soutien technique mais aussi votre écoute et votre collaboration.
Merci à Guillaume N. pour ta précieuse collaboration et pour tes conseils oculaires qui ont définitivement changé ma vie/vue !
Merci à tous les membres du laboratoire, ça été une expérience non pas que scientifique mais incroyablement humaine de réaliser mon doctorat à vos côtés. Merci particulier à Caroline, Véro, Carla et Angel. Votre enthousiasme, bienveillance et bonne humeur m’ont beaucoup apporté tout au long de ces années. Je tiens également à remercier mes relecteurs : Damien, JB, Pauline.
Merci au membre du Comité Étudiant de l’IBIS. L’équipe a changé au cours des années mais il y a toujours eu une belle dynamique, du rire et toujours des évènements réussis. Ça été une chouette expérience à vos côtés !
Un merci particulier à Hélène V. Chacune a suivi sa route mais les souvenirs des moments passés ensemble resteront. Idem pour la fine équipe, Éléonore, Axelle, Souhir et Hélène H. Rien n’aurait été pareil sans vous.
Merci à Maeva, Claire, et les tous les membres du laboratoire Bernatchez avec qui j’ai partagé de chouettes moments, merci de m’avoir embarquée dans vos aventures. Merci pour votre soutien et votre joie de vivre.
Merci à mes amies, Anaïs, Kelly, pour ces skypes pendant des heures, ces bulles d’air et de rires.
Merci à ma famille et mes parents, évidemment. Maman, Papa, merci d’avoir toujours été à l’écoute, de m’avoir soutenu et encouragée quelque soit la direction choisie. Après ces 10 années d’études, entre vers de terre, cténophores et levures, je touche au but. Merci pour ce que vous m’avez transmis et offert : cette liberté d’étudier et d’aimer apprendre. Malgré la distance, pas toujours évidente, ces 4 années ont été remplies de beaux souvenirs et l’avenir nous réserve encore de belles aventures à travers le monde.
Merci à Plume, Watson et Simone pour l’indispensable zoothérapie.
Et enfin, last but not least, merci à toi, Jean-Marc. Il n’existe pas de mots qui rendrait suffisamment justice à ton rôle dans l’achèvement de cette thèse ni pour rendre compte de ton soutien. Merci pour tout, ta patience, ton rire et ton amour indéfectible, même aux moments les plus sombres de la rédaction.
Avant-propos
La thèse présentée ici est composée publications mises en page conformément avec les règles de préparation de thèses de la Facultés des études supérieures et postdoctorales (FESP) de l’Université Laval. L’ouvrage inclut une introduction générale en français, 3 articles rédigés en anglais présentés aux chapitres 1, 2, et 3, ainsi qu’une conclusion générale en français.
• Le Chapitre 1 est publié depuis le 26 Octobre 2016 sous la référence : Bleuven, Clara, and Christian R. Landry. 2016. "Molecular and cellular bases of adaptation to a changing environment in microorganisms." Proc. R. Soc. B 283 (1841):20161458. doi: 10.1098/rspb.2016.1458.
J’ai écrit la revue de littérature en collaboration avec Christian R. Landry.
• Le Chapitre 2 est publié depuis le 19 Décembre 2018 sous la référence : Bleuven, C., A. K. Dubé, G. Q. Nguyen, I. Gagnon-Arsenault, H. Martin, and C. R. Landry. 2018. "A collection of barcoded natural isolates of Saccharomyces paradoxus to study microbial evolutionary ecology." MicrobiologyOpen :e773. doi: 10.1002/mbo3.773.
Alexandre K. Dubé, Isabelle Gagnon-Arsenault, and Christian R. Landry ont conçu le projet ; Alexandre K. Dubé, Isabelle Gagnon-Arsenault et moi avons acquis les données avec l’aide de Guillaume Q. Nguyen ; J’ai analysé et interprété les résultats avec le support de Guillaume Q. Nguyen et Hélène Martin ; J’ai rédigé le manuscrit avec la collaboration de Guillaume Q. Nguyen et Christian R. Landry. Tous les auteurs ont approuvé la version finale.
• Le Chapitre 3 est en préparation pour une soumission à The ISME Journal : Bleuven, C., G. Q. Nguyen, P., C. Després, M., Filteau, and C. R. Landry. 2018. "Quantifying wild yeast fitness in a natural soil to test for local adaptation and for the role of microbial interactions."
J’ai conçu le projet avec Christian R. Landry ; J’ai acquis les données ; J’ai analysé et interprété les résultats en collaboration avec Guillaume Q. Nguyen, et Philippe
C. Després ; J’ai rédigé le manuscrit avec la collaboration de Guillaume Q. Nguyen et Christian R. Landry.
Les travaux de recherche de la thèse ont été présentés sous forme d’affiches et de présentations orales à des journées scientifiques de l’Institut de Biologie Intégrative et des Systèmes (IBIS, Québec, Canada), du Département de Biologie de l’Université Laval (Québec, Canada) et du regroupement québécois de recherche sur la fonction, l’ingénierie et les applications des protéines (PROTÉO) (Québec, Canada).
Mes recherches ont été possibles en partie grâce à l’appui financier du Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG). J’ai eu la chance d’être la récipiendaire d’une bourse de « Leadership et Développement drable » de l’Université Laval pour 3 années de recherche, de 2015 à 2017 et d’une bourse pour études supérieures de PROTÉO d’une durée d’un an en 2015.
En parallèle, j’ai apporté ma contribution au sein du Comité Étudiant de l’IBIS en tant que membre et ensuite co-représentante pendant 3 années consécutives (2016-2018). Je me suis fortement impliquée dans l’organisation d’évènements scientifiques comme la Journée de l’IBIS qui se déroule chaque année et des évènements non-scientifiques comme la Journée Carrière de l’IBIS en 2017.
Introduction
L’adaptation des organismes à leur environnement
Évolution et adaptation, un historique
La diversité des organismes et leur adaptation à des environnements aussi divers qu’extrêmes est un objet de fascination et de recherche chez les biologistes. L’histoire de la biologie évolutive illustre cet attrait. Dès l’antiquité grecque, Aristote (Ier siècle av. JC)
développa une classification naturelle qui perdurera jusqu’à Carl von Linné (1707-1778), naturaliste suédois (Buican and Grimoult 2011). L’œuvre de ce dernier repose sur la mise en place d’une nomenclature binomiale pour l’inventaire du vivant, qui caractérise un organisme par une terminologie latine du genre suivi de l’espèce. Cette classification des espèces devint un langage international qui servit de base taxonomique aux réflexions théoriques. Jusqu’au XIXème siècle, la théorie du fixisme personnifiée par Georges Cuvier (1769-1832) s’appuyait sur l’harmonie naturelle pour justifier l’ordre naturel et son origine divine. En opposition, le transformisme proposé par Jean-Baptiste de Lamarck (744-1829), naturaliste français, évoque la variation des espèces au cours du temps, notamment par l’hérédité des caractères acquis et par la tendance à la complexification des organismes au cours du temps, en fonction de la « loi d’usage et de non-usage » des organes (Buican and Grimoult 2011).
La théorie formulée par Alfred Russel Wallace dans l’article « On The Tendency of Varieties to Depart Indefinitely from the Original Type » de 1858 et par Charles Darwin, qu’il énonce dans L’Origine des espèces (1859) apporte une nouvelle idée : celle de la sélection naturelle comme moteur de l’évolution. Le concept central d’adaptation qui en découle peut être défini comme le résultat d’une évolution par sélection naturelle. Lors de cette évolution, les caractères pouvant être transmis à la descendance et conférant une meilleure chance de survie, et/ou de reproduction augmentent en fréquence au sein d’une population. Les éléments cruciaux de la théorie darwiniste sont : la variation de traits entre les individus, l’aspect héritable de ces traits et la survie et reproduction différentielle des individus (Brandon 1978). Ce processus de sélection favorise les traits avantageux dans des environnements donnés soumis à des contraintes écologiques spécifiques (Bock 1980). La théorie de sélection naturelle de Darwin fut testée expérimentalement pour la première fois par William H. Dallinger à la fin du XIXème siècle. Pendant sept ans, W. H.
Dallinger fit croître des eucaryotes unicellulaires dans un incubateur dont la température augmentait régulièrement (jusqu’à l’explosion). Au fil des expériences, les microorganismes avaient augmenté leur limite de tolérance de 60°C à 70°C et n’étaient plus capable de croître à leur température ancestrale de 20°C (Dallinger 1887). Ces résultats illustrent un exemple d’adaptation par sélection naturelle et de compromis1 évolutif dans lequel l’adaptation à une augmentation de la température est associée à une perte de la capacité de croissance à la température ancestrale.
La découverte des lois de l’hérédité par Mendel (1865), des processus de mutation, de recombinaison génétique ainsi que les travaux en génétique des populations ont amené la théorie synthétique de l’évolution, dont Theodosius Dobzhansky, Julian Huxley et Ernst Mayr sont les principaux fondateurs. Cette théorie, proposée dans les années 1930-1940, explore les bases génétiques de l’évolution et propose que ces bases soient micromutationnelles : des mutations à faible effets sont favorisées et accumulées par sélection naturelle (Orr 2005). Grâce à l’émergence de la biologie moléculaire et de la génétique contemporaine, cette théorie a été enrichie et complétée, notamment grâce à la théorie neutraliste introduite par Moto Kimura en 1968.
Aujourd’hui, il est admis que l’évolution des organismes est soumise à différentes forces : la dérive génétique, la mutation, le flux de gènes et la sélection naturelle, cette dernière étant la force responsable de l’évolution adaptative. Depuis peu, une synthèse étendue de l’évolution (EES, extended evolutionary synthesis) est proposée par certains chercheurs qui souhaitent enrichir la théorie de l’évolution et mettre en avant le rôle de certains processus dans les changements évolutifs (Laland et al. 2015): les biais développementaux (la manière dont le développement influe sur l’émergence de la variation), la plasticité (la modification des traits selon l’environnement), la construction de
niche (la modification de l’environnement par les organismes) et la transmission
extra-génétique (la transmission épiextra-génétique et comportementale entre les générations). La nécessité d’une extension de la théorie de l’évolution et l’importance de ces processus est controversée (Laland et al. 2014). Les chercheurs en désaccord avec l’EES modèrent l’importance de ces processus déjà bien documentés en les désignant comme des additions non indispensables aux processus évolutifs fondamentaux (mutation, dérive, sélection, flux de gènes). Ces chercheurs préconisent d’approfondir les recherches afin de
mieux comprendre l’importance de ces processus et en évoquent d’autres comme l’épistasie et la variation génétique cryptique comme étant également des pistes d’intérêt pour de futures avancées en biologie évolutive.
La notion de fitness
La sélection naturelle étant le mécanisme fondamental par lequel les adaptations émergent, elle ne peut pas agir sans différence de fitness2 entre les individus. H. Allen Orr définit la fitness comme la capacité des organismes, populations ou espèces à survivre et à se reproduire dans l’environnement dans lequel ils se trouvent (Orr 2009). La variation génétique qui existe entre des individus peut prendre plusieurs formes : polymorphismes nucléotidiques ponctuels (SNPs), insertions, délétions ou encore variation du nombre de copies de gènes (CNVs). Cette variation peut dans certains cas être impliquée dans la variation phénotypique des individus et ce, à plusieurs niveaux d’organisation biologique : fonctions et interactions protéiques, fonctionnement cellulaire, physiologie, morphologie, comportement (Dalziel, Rogers, and Schulte 2009). Ainsi, la fitness est la variable qui mesure la performance des organismes dans leur environnement.
Malgré ce rôle central en biologie évolutive, définir et mesurer la fitness reste problématique. Comme le précise H. Allen Orr (2009) « Il est parfois plus facile de réaliser des expériences qui mesurent la fitness plutôt que de penser à sa signification. En d’autres termes, la difficulté est plus conceptuelle qu’empirique ». Mesurer et intégrer la fitness d’un organisme sur toute sa durée de vie est complexe, car elle ne peut pas être inférée à partir d’une mesure unique dans le temps. De plus, la fitness est considérée comme une propension, le potentiel d’un individu à survivre et à se reproduire plutôt que le nombre réel de descendants qu’aura cet individu (Wagner 2010).
La fitness peut se calculer mathématiquement, de façon absolue ou relative. La fitness absolue (W) correspond à la fitness attendue totale d’un génotype, qui peut être supérieure ou égale à 0. La fitness relative (w) est la fitness absolue normalisée, le plus souvent divisée par la fitness absolue du génotype présentant la plus grande fitness. Ces mesures mathématiques permettent de mettre en place des équations de sélection qui prédisent le changement de la fréquence des allèles sous sélection naturelle.
2Le terme fitness peut être traduit en français par « valeur adaptative » ou « valeur sélective » mais le terme anglais sera conservé tout au long de cette thèse de doctorat (Heams et al. 2009).
Pour mesurer la fitness expérimentalement, la plupart des études se focalisent sur des éléments mesurables corrélés à la fitness (« proxy ») ayant un effet sur la fitness, par exemple le nombre de fleurs ou graines produites chez les plantes (Leimu and Fischer 2008) ou le taux de survie entre différents cycles de vie chez les animaux (Fraser et al. 2011). Les expériences de « compétition » sont également un moyen de quantifier les fitness relatives des génotypes. Différents génotypes ou populations sont mis en culture dans un même environnement et les changements de leurs fréquences relatives au cours du temps sont considérés comme une mesure de leurs fitness relatives. Cette méthode est particulièrement utilisée avec les microorganismes : le taux de croissance d’une population microbienne est utilisé comme proxy de la fitness et l’expérience peut être conduite jusqu’à plusieurs centaines voire milliers de générations, avec de nombreuses répétitions et dans des conditions contrôlables. La fitness relative est alors calculée comme le ratio entre le taux de croissance du génotype ciblé et celui de son compétiteur (Elena and Lenski 2003, Wiser and Lenski 2015). Différents types de marqueurs peuvent être utilisés pour distinguer chaque génotype, tels que des marqueurs conférant une résistance à des antibiotiques, ou une séquence nucléotidique spécifique à chaque génotype (Nislow et al. 2015b, Rodriguez-Verdugo et al. 2014). La fitness peut également être mesurée à l’échelle d’une population : par exemple, dans le cas d’une population clonale initiale, cette mesure permet de traquer l’émergence de mutations bénéfiques qui vont affecter cette fitness par leur augmentation en fréquence au court du temps (Long et al. 2015).
La variation environnementale et l’adaptation
L’adaptation des organismes aux conditions ambiantes dépend des facteurs environnementaux, lesquels peuvent être séparés en deux catégories: (1) les facteurs abiotiques qui composent l’environnement physico-chimique et (2) les facteurs biotiques, c’est à dire les interactions entre les organismes qui partagent un même espace spatio-temporel. Les pinsons de l’archipel des Galápagos, rendus célèbres par les travaux fondateurs de Darwin, sont des exemples classiques d’adaptation évolutive. Bien que divergeant d’un ancêtre commun, ces espèces présentent une diversité de tailles et de formes de becs qui résulte de leur adaptation aux différentes ressources qu’ils exploitent, formant autant de niches écologiques distinctes. Par exemple, les espèces aux formes de becs les plus larges se nourrissent de grosses graines qu’ils sont capable de briser, tandis
que les espèces aux becs les plus fins se nourrissent d’insectes ou de petites graines (Grant and Grant 2005).
Lors d’un changement environnemental, les pressions s’exerçant sur les individus sont modifiées et peuvent conduire à l’adaptation de la population à la nouvelle niche. Les mécanismes permettant cette adaptation aux nouvelles contraintes environnementales peuvent être observables à l’échelle d’une seule génération. Par exemple, lors d’une période de sécheresse, la disponibilité des ressources diminue drastiquement. En 2004, la compétition entre deux espèces Geospiza fortis et G. magnirostris pour les graines de Tribulus raréfiées par la sécheresse, a induit une forte sélection en direction des oiseaux capables de se nourrir et donc de survivre. Alors que G. magnirostris présentait un avantage dans la capacité de se procurer les graines de Tribulus, l’alternative possible d’alimentation pour G. fortis était des graines d’autres plantes, plus petites. La réponse évolutive observée a été la diminution de la taille des becs de G. fortis au fil des générations (Grant and Grant 2006). Le séquençage des génomes des individus ayant succombé ou survécu à cette sécheresse a mis en évidence le rôle du gène HMGA2 dans le changement de taille des becs et confirmé l’avantage conféré par un bec plus fin pour la survie des individus (Lamichhaney et al. 2016).
La variation environnementale peut être décomposée en deux types : la variation spatiale et la variation temporelle, dont les échelles varient en fonction de la durée de vie des organismes (Kassen 2002) (Figure I 1). Les niches écologiques des espèces, ou des génotypes, se forment en partie en réponse à l’hétérogénéité environnementale, qui correspond aux variations des pressions de sélection en fonction de l’espace et du temps (Kassen 2002). De manière générale, les niches évoluent en réponse à la quantité de variation endurée. Ainsi les organismes écologiques spécialistes émergent dans les environnements spatio-temporellement homogènes, tandis que les organismes
généralistes, dont l’amplitude de la niche est généralement plus étendue par rapport à
celle des spécialistes, émergent dans les environnements hétérogènes en fonction de la fréquence des changements (Kassen 2002). La question de l’émergence des types écologiques spécialistes ou généralistes, largement documentée autant théoriquement qu’expérimentalement, sera abordée dans le chapitre1.
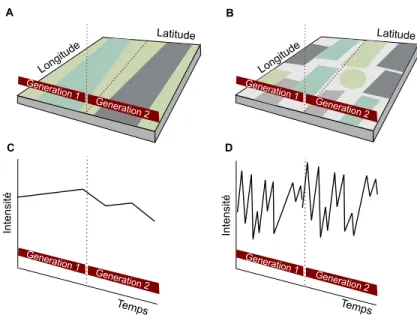
Figure I 1. Décomposition de la variation environnementale.
(A) La variation spatiale est à large échelle ou « grain grossier » par rapport au temps de génération, les différentes couleurs représentent les différents environnements. (B) La variation spatiale est à fine échelle ou « grain fin » par rapport au temps de génération. (C) La variation temporelle est à large échelle ou « grain grossier »par rapport au temps de génération. (D) La variation temporelle est à fine échelle ou « grain fin » par rapport au temps de génération. Figure d’après Kassen (2002).
L’hétérogénéité spatiale génère de la sélection divergente car différentes conditions environnementales peuvent favoriser préférentiellement différents génotypes et ainsi favoriser l’adaptation locale (Figure I 2 A). L’adaptation locale se définie comme le fait qu’une population au sein d’une métapopulation possède une meilleure fitness dans son
habitat que celle d’autres populations issues d’habitats distincts (Kawecki and Ebert
2004). Un habitat se définissant ici comme l’ensemble des conditions environnementales locales dans lesquelles vit un génotype.
La variation de la fitness peut être décomposée entre les effets du génotype, de l’environnement et de l’interaction entre chaque génotype et environnement :
𝐹𝑖𝑡𝑛𝑒𝑠𝑠 = 𝐺é𝑛𝑜𝑡𝑦𝑝𝑒 + 𝐸𝑛𝑣𝑖𝑟𝑜𝑛𝑛𝑒𝑚𝑒𝑛𝑡 + (𝐺é𝑛𝑜𝑡𝑦𝑝𝑒 × 𝐸𝑛𝑣𝑖𝑟𝑜𝑛𝑛𝑒𝑚𝑒𝑛𝑡)
Dans ce contexte, l’interaction 𝐺𝑒𝑛𝑜𝑡𝑦𝑝𝑒 × 𝐸𝑛𝑣𝑖𝑟𝑜𝑛𝑛𝑒𝑚𝑒𝑛𝑡 (G x E) pour la fitness est une conséquence et donc un prérequis pour l’identification de l’adaptation local (Figure I 2 B).
Generat ion 1 Generat ion 2 Generat ion 1 Generat ion 2 A B Latitude Longit ude Latitude Longit ude Generat ion 1 Generat ion 2 Generat ion 1 Generat ion 2 C D Int ensi té Int ensi té Temps Temps
Ainsi, certaines conditions sont requises à l’adaptation locale :
o Les flux de gènes entre les populations doivent être limités, afin de favoriser la différenciation génétique nécessaire à l’émergence de l’adaptation locale.
o La sélection doit être relativement stable au court du temps car comme vu précédemment, la variation environnementale temporelle favorise l’émergence de génotypes généralistes.
o Une adaptation locale suppose que la meilleure performance dans l’environnement local ait une base génétique et ne soit pas exclusivement le résultat de la plasticité.
L’adaptation locale a été étudiée chez de nombreux organismes, principalement chez les plantes (Lind et al. 2018, Leimu and Fischer 2008) et les animaux (Hereford 2009, Fraser et al. 2011). Par exemple, chez Arabidospis thaliana, Agren et Schemske (2012) ont démontré une différenciation adaptative par des expériences de transplantations entre des populations européennes et identifié le trait de tolérance au gel comme pouvant contribuer à cette différenciation. Les auteurs ont comparé pendant 5 ans la fitness de populations locales et non locales dans des sites situés en Suède et en Italie, représentant les limites de l’air de distribution de l’espèce (Agren and Schemske 2012). La fitness totale a été quantifiée par le nombre de graines produites par fruit ou par jeune plant. À chaque site, la fitness des populations locales est meilleure que les populations non locales, suggérant que les populations sont localement adaptées à leur environnement. La tolérance au gel augmente avec la latitude d’origine des populations, ce trait est donc considéré comme adaptatif et important dans l’adaptation locale le long du gradient climatique.
Dans la revue de littérature de Hereford qui reprend les études de l’adaptation local en milieu naturel, l’adaptation locale apparaît comme être un processus commun : dans plus de 50% des études testant l’adaptation locale, celle-ci a été confirmée (Hereford 2009). Dans ces cas, la fitness des populations locales est plus élevée de 45% en moyenne par rapport aux populations non-locales. Parmi les études citées, seules 3 concernent des microorganismes. Pourtant, ces organismes très ubiquitaires sont fondamentaux dans le fonctionnement des écosystèmes et représentent des modèles d’études particulièrement adapté aux questions de biologie évolutive et écologie.
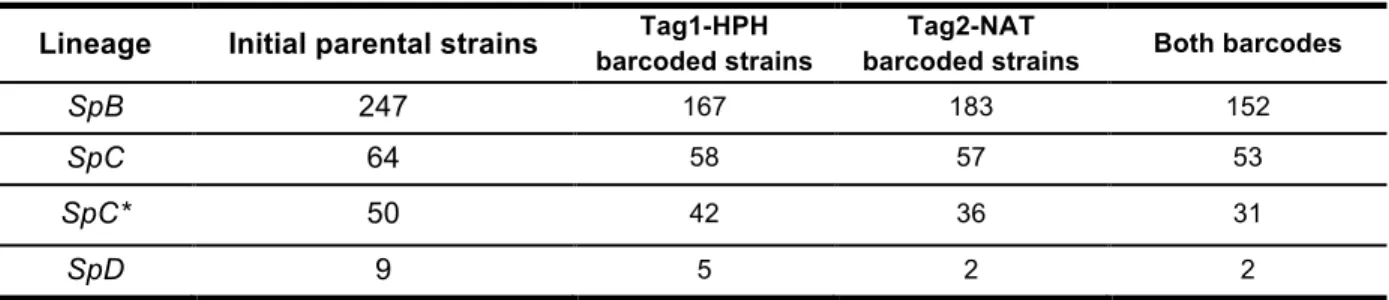
Figure I 2. Critères pour identifier l’adaptation locale.
(A) La sélection divergente à la suite d’un changement environnemental favorise l’adaptation de populations (en bleu et rose) issues d’une métapopulation (gris) aux nouvelles conditions locales. Le génotype 1 provient de l’environnement 1 (E1) (en bleu) et le génotype 2 de l’environnement 2 (E2) (en rose). (B) i.) L’interaction G × E est significative avec un changement de rang des fitness. Les génotypes sont adaptés selon le critère « home vs. away » : la fitness des génotype est meilleure dans leur environnement local, et selon le critère « local vs. foreign » : le génotype local a une fitness plus élevée par rapport à la fitness du génotype non-local, dans son environnement . ii.) L’interaction G × E est significative mais sans changement de rang des fitness entre les deux génotypes. Les génotypes présentent une adaptation du type « home vs. away » car leur fitness est meilleure dans leur environnement que dans l’autre. Cette condition n’est pas suffisante pour conclure quant à une adaptation locale. iii.) L’interaction G × E est significative avec un changement de rang des fitness. Chaque génotype a une meilleure fitness dans l’environnement non-local comparé à son environnement local, c’est une maladaptation. iv.) Pas d’interaction 𝐺𝑒𝑛𝑜𝑡𝑦𝑝𝑒 × 𝐸𝑛𝑣𝑖𝑟𝑜𝑛𝑛𝑒𝑚𝑒𝑛𝑡 (G × E), les génotypes ne sont pas adaptés localement. Figure selon Kraemer and Boynton (2016) selon les critères de Kawecki and Ebert (2004).
Les microorganismes comme modèles d’étude en évolution et
écologie
Les systèmes microbiens
La plupart des exemples cités en écologie illustrent largement les processus évolutifs chez les plantes et les animaux. Or, les microorganismes gagnent une place importante dans ces domaines de la biologie grâce aux nombreux intérêts et avantages qu’ils offrent comme modèles pour tester les théories et les concepts en écologie et évolution (Jessup et al. 2004, Rainey et al. 2000).
Les microorganismes sont des organismes microscopiques très divers, représentés par tous les domaines du vivant : bactéries, archées et eucaryotes parmi lesquels des champignons, des algues et les protistes. Ils sont définis comme ubiquitaires, car présents dans la plupart des environnements terrestres comme les sols, les lacs, les océans jusqu’aux plus extrêmes comme les fosses hydrothermales. Ils sont également associés aux plantes, aux animaux et à d’autres microorganismes avec lesquels ils entretiennent des relations de symbiose ou de parasitisme, par exemple. Alors que les microorganismes représentent une part importante de la biomasse, un gramme de sol pouvant contenir 1010
bactéries, leur grande diversité taxonomique reste encore peu documentée. Il est estimé que 1012 espèces de microorganismes incluant bactéries, archées et eucaryotes existent
sur Terre (Locey and Lennon 2016).
Les microorganismes jouent un rôle prépondérant dans les processus biochimiques indispensables (Falkowski, Fenchel, and Delong 2008): les réactions que catalysent les microorganismes vont procurer des services comme la décomposition, la production primaire, le cycle des nutriments ou encore la régulation du climat (Bodelier 2011). En plus de leurs services écosystémiques, les microorganismes ont également un rôle important en santé humaine, par exemple, en étant utilisés pour la production d’antibiotiques, mais aussi un rôle économique notamment en agro-alimentaire où ils sont utilisés dans différents processus alimentaires comme la vinification, le brassage, la fabrication de fromages ou encore dans l’industrie biotechnologique (Gonçalves et al. 2016, Wolfe et al. 2014, Narancic and O'Connor 2017).
L’utilisation des microorganismes comme modèles en écologie et en évolution est aujourd’hui largement répandue grâce à de nombreuses caractéristiques avantageuses (Jessup et al. 2004). En effet, les microorganismes ont un temps de génération court et de grandes tailles de populations qui offrent un potentiel d’évolution plus rapide que les plantes ou animaux. Ces propriétés offrent l’opportunité de tester des théories évolutives en laboratoire. Par exemple, l’évolution de la spéciation par adaptation divergente et l’isolement reproducteur ont été démontrés expérimentalement avec des populations de levures évoluées en laboratoire (Dettman et al. 2007). Malgré le fait que toutes les questions évolutives ou écologiques ne soient pas abordables avec les microorganismes, ils présentent certaines similarités avec les macroorganismes qui permettent d’aborder des questions générales sur l’évolution du vivant. Certains microorganismes sont capables de reproduction sexuée, ils présentent des interactions sociales riches, par exemple la coopération et la communication, (West et al. 2006) et peuvent être soumis au processus de vieillissement (Ackermann et al. 2007).
Méthodes d’étude en écologie et évolution microbienne
La plupart des méthodes d’étude en écologie et en évolution peuvent être appliquées aussi bien aux animaux et aux plantes qu’aux microorganismes. Par exemple, l’évolution expérimentale est une méthode largement utilisée avec les espèces modèles comme la mouche, Drosophila melanogaster ou la plante, Arabidopsis thaliana (Kawecki et al. 2012).
L’évolution expérimentale
L’évolution expérimentale consiste à reproduire les processus évolutifs en conditions contrôlées afin d’en étudier les mécanismes (Kawecki et al. 2012, Buckling et al. 2009). Des populations d’organismes, généralement issues du même ancêtre, sont propagées dans des environnements définis et reproductibles pendant des dizaines, des centaines voire des milliers de générations sans intervention de l’expérimentateur sur le processus de sélection naturelle. Les types évolués sont comparés au type ancestral, sur le plan génomique et phénotypique selon les critères ciblés, fournissant des informations sur les processus et l’étendue des changements évolutifs découlant des conditions imposées par l’expérimentateur. Il est ainsi possible d’étudier en laboratoire l’effet des différentes forces évolutives, comme l’étude de la dynamique d’apparition et les effets des mutations (Halligan and Keightley 2009), les mécanismes adaptatifs ainsi que les effets de la dérive et de la migration (Morgan, Gandon, and Buckling 2005). Les technologies de séquençage
donnent en outre accès aux bases moléculaires de l’adaptation chez un nombre croissant de systèmes modèles chez les bactéries (Velicer et al. 2006), les levures (Araya et al. 2010) et les virus (Stern et al. 2017).
Les microorganismes sont largement utilisés en évolution expérimentale car ils offrent des avantages expérimentaux qui permettent la mise en place de systèmes simplifiés (Elena and Lenski 2003):
§ Leur petite taille et leur temps de génération relativement court permettent de produire rapidement des populations de grande taille, ce qui permet de répliquer l’expérience et de la conduire sur plusieurs centaines, voire des milliers de générations. Par exemple, Escherichia coli a un temps de génération de 20 min dans des conditions de laboratoire riche en nutriments avec une taille cellulaire allant de 0,5 à 3 µm, alors que la plupart des cellules eucaryotes ont une taille allant de 10 à 30 µm.
§ Pour certaines espèces, il existe d’abondantes données moléculaires et génomiques ainsi que des techniques de manipulations génétiques précises comme la délétion de gène d’intérêt ou le marquage cellulaire conférant une résistance antibiotique.
§ Ils peuvent être conservés par congélation à-80°C et ensuite réanimés, ce qui permet la comparaison directe entre les types ancestraux et évolués.
L’évolution expérimentale est une technique qui s’est développée rapidement à partir des années 1990, notamment à la suite du travail de Richard Lenski et ses collègues sur l’évolution expérimentale à long-terme chez Escherichia coli qui dure depuis plus de 25 ans et a atteint plus de 65 000 générations (Blount, Lenski, and Losos 2018, Good et al. 2017, Lenski et al. 1991). Douze populations issus d’un ancêtre unique sont cultivées en parallèle dans un milieu artificiel pauvre en glucose renouvelé régulièrement. Des échantillons sont prélevés et congelés toutes les 500 générations afin de conserver une trace viable des bactéries ancestrales à différentes étapes de l’évolution. Il est ensuite possible de comparer les génotypes et phénotypes des bactéries ancestrales et leurs descendants, notamment par des expériences de compétition. Cette expérience à long terme a permis d’étudier la convergence et la divergence évolutive entre populations (Cooper, Rozen, and Lenski 2003, Tenaillon et al. 2012, Blount, Borland, and Lenski 2008), l’évolution du taux de mutation adaptatives (Barrick and Lenski 2013),l‘effet de l’hétérogénéité environnementale sur l’évolution des génotypes (Cooper and Lenski 2010)
ou encore la dynamique adaptative et l’importance du hasard dans le processus adaptatif (Buckling et al. 2009). Par exemple, l’observation du taux d’adaptation sur des milliers de générations montre un changement au court du temps : en 2000 générations, la fitness des populations évoluées a augmenté rapidement de 30% par rapport à la population ancestrale grâce à la fixation de mutations bénéfiques à large effet. Ce taux diminue au cours du temps alors que les mutations bénéfiques se font plus rare et avec des effets plus faibles (Elena and Lenski 2003). Cependant, la trajectoire évolutive des populations évoluées diffère : les taux de mutations ne sont pas identiques, des variations de fitness entre les populations existent dans les conditions de l’expérience mais également dans d’autres environnements (Blount, Borland, and Lenski 2008). L’étude des bases moléculaire par Ostrowki et al (2008) montre un fort parallélisme évolutif par l’accumulation de mutations bénéfiques dans le même gène, qui induisent un effet avantageux dans le même environnement expérimental La variation de fitness entre les populations dans des conditions différentes est due aux effets pléiotropes de ces mutations (Ostrowski, Woods, and Lenski 2008).
D’autres travaux avec Saccharomyces cerevisiae (Ferea et al. 1999), Chlamydomonas reinhardtii (Reboud and Bell 1997) ou Pseudomonas fluorescens (Rainey and Travisiano 1998) ont également beaucoup contribué à la compréhension des processus évolutifs. Par exemple, l’expérience de Paul Rainey et Michel Travisiano (1998) montre le rôle de l’hétérogénéité spatiale dans la diversification des espèces en créant des opportunités écologiques pour différents morphotypes de P. fluorescens. Cette expérience sert encore de modèle pour étudier de nombreuses questions associées à la radiation adaptative, la spécialisation écologique et les bases moléculaires sous-jacentes.
Dans le chapitre 1, nous ferons une revue de littérature sur les mécanismes moléculaires sous-jacent à l’adaptation des microorganismes aux environnements hétérogènes. Les travaux que nous décrirons ont utilisé, pour la plupart, des méthodes d’évolution expérimentale afin d’étudier l’adaptation de populations sous différentes conditions environnementales.
Alors que l’évolution expérimentale donne un aperçu des processus évolutifs et des mécanismes adaptatifs en testant les théories et les prédictions, cette méthode présente des inconvénients. Par exemple, il est souvent souligné que les conditions expérimentales
ne reflètent pas la complexité des environnements naturels. Bien que l’évolution expérimental ne s’applique pas à mimer des systèmes particuliers avec toute leur complexité mais plutôt à mettre en lumière les processus évolutifs généraux, la plupart des expériences se concentrent sur l’environnement abiotique. De plus, l’étude des interactions biotiques se limite souvent à la coévolution de deux espèces. Afin de mieux comprendre l’écologie et l’évolution de microorganismes, l’utilisation de méthodes issues de l’évolution expérimentale combiné à des conditions plus proches des facteurs biotiques et abiotiques naturels est pertinente pour déterminer leur réponse aux pressions de sélection de leur habitat. Par exemple, des microcosmes, tel que des tubes de sol, peuvent être utilisés et répliqués en laboratoire, en contrôlant certains paramètres, comme la température.
Étudier l’adaptation en milieu naturel
Les recherches sur l’adaptation des microorganismes à leurs environnements naturels font surtout référence à leur adaptation locale. L’adaptation locale est étudiée par des expériences appelées de « jardin commun » et de « translocation réciproque » dans lesquelles la fitness des organismes est comparée selon différents critères cités précédemment : un génotype adapté localement aura (1) une meilleure fitness dans son environnement par rapport à des génotypes non-locaux, selon le critère « local vs. foreign » et (2) une meilleure fitness dans son environnement par rapport à un autre, selon le critère « home vs. away » (Kawecki and Ebert 2004) (Figure 2). Dans les expériences de jardin commun, différents génotypes sont placés dans un environnement unique tandis que les expériences de translocation réciproque consistent à comparer ces génotypes dans différents environnements locaux et non-locaux.
Ces expériences de translocation réciproque et jardin commun sont difficilement faisables dans les environnements d’origine des microorganismes car l’utilisation de marqueurs génétiques pour suivre les individus au cours du temps est impossible en conditions naturelles, notamment à cause de problématiques éthiques et légales d’introduction des organismes génétiquement modifiés (Francescon 2001), ce qui rend la mesure des fitness particulièrement difficile. Pour pallier cette limite, l’utilisation de microcosmes expérimentaux artificiels, ou proches des conditions naturelles, est répandue. Par exemple, une expérience de translocation réciproque pour tester l’adaptation locale de bactéries Bacillus issues de sols forestiers a été réalisée en laboratoire. À partir de chaque échantillon, des bactéries Bacillus « locales » ont été isolées. Pour créer le microcosme
expérimental, les auteurs ont fait infuser leurs échantillons de sol dans de l’eau distillée. L’extrait obtenu a été stérilisé et du glycérol a été ajouté comme source de carbone. Les bactéries isolées ont été incubée dans chaque extrait de sol et leur croissance mesurée afin d’inférer leur fitness. Parmi les bactéries, certaines ont démontré une adaptation locale à leur environnement par une meilleure fitness dans leur site local et une diminution de cette fitness de 40% partir de 10m de distance du site (Belotte et al. 2003).
Dans ces microcosmes expérimentaux, de nombreux paramètres abiotiques simples sont contrôlables, comme la température ou l’humidité, mais il est également possible d’augmenter la complexité du microcosme avec l’ajout d’une communauté microbienne. Lawrence et al. (2012) ont ainsi étudié l’évolution de la croissance de 5 espèces bactriennes isolées depuis les racines d’un hêtre dans deux conditions : chaque espèce seule, en monoculture, et les 5 espèces ensemble, en polyculture dans une infusion concoctée à partir de feuilles de l’arbre. L’interaction entre les espèces a influencé l’adaptation au nouvel environnement : les espèces divergent dans leur utilisation des ressources selon leur évolution en monoculture ou polyculture et certaines espèces issues de la polyculture se sont adaptées à l’utilisation des déchets produits par les autres. Les auteurs démontrent ainsi l’importance de considérer les interactions biotiques dans la dynamique évolutive des microorganismes (Lawrence et al. 2012b).
Il existe cependant certaines limites à ces expériences. Les microcosmes ne sont pas des répliques fidèles de l’environnement naturel, ce qui implique que certains facteurs écologiques sélectifs peuvent manquer (Kraemer and Boynton 2017). Par exemple, certains microorganismes produisent des molécules extra-cellulaires antagonistes, qui peuvent impacter la fitness des espèces coexistantes et donc modifier la sélection (Kraemer, Soucy, and Kassen 2017). Cet effet se trouve masqué dans un microcosme stérile. Ainsi, si les environnements testés ne sont pas les habitats naturels, les microcosmes utilisés à la place doivent considérer au mieux les différents facteurs biotiques et abiotiques potentiellement impliqués dans la sélection.
Le suivi des changements de la fréquence relative des individus (ou génotypes) permet également d’inférer leur fitness : lorsqu’un génotype local est comparé à un génotype étranger, le génotype local augmente en fréquence s’il est adapté localement. Ce suivi est facilité par le marquage au niveau génomique des microorganismes, par exemple via la méthode de code-barre moléculaire inséré durablement dans le génome (Smith et al.
2009b). Cette méthode sera développée dans les chapitres 2 et 3 où nous développons et utilisons une collection de levures à code-barres dans des expériences de compétition dans différents environnements. Récemment, des méthodes pour mesurer directement la fitness de microorganismes non modifiés génétiquement en milieu naturel ont été développée afin de pallier les limitations des expériences en laboratoire. Boynton et al. (2017) ont ainsi montré que l’utilisation de réaction en chaîne de type « gouttelettes digitales » (DDPCR) qui se base sur un polymorphisme naturel, permet de suivre en temps réel la fréquence relative de levures dans la litière forestière et d’inférer l’influence de l’origine de la souchei sur sa fitness (Boynton et al. 2017). En utilisant une mutation
spontanée comme marqueur, Anderson et al. (2018) ont réalisé une expérience de translocation réciproque, en milieu naturel afin de suivre la persistance des levures résidentes (S. paradoxus) par rapport aux levures transplantée (S. cerevisiae) (Anderson et al. 2018).
Métagénomique et métabarcoding
Staley et Konopka (1985) identifièrent ce qu’ils appellent « great plate-count anomaly » c’est-à-dire l’incapacité actuelle de cultiver en laboratoire la majorité des microorganismes observables par microscopie et étalement (Staley and Konopka 1985, Handelsman 2004). Cependant, aujourd’hui, par le séquençage et l’analyse de l’ADN et de l’ARN contenu dans un environnement donné, il est devenu possible d’identifier les espèces microbiennes présentes ainsi que de mesurer leur diversité et abondance, sans avoir recours à leur culture.
Parmi ces approches, la métagénomique a pour but d’étudier la structure globale de la communauté, qui peut être définie comme sa composition et sa dynamique dans un écosystème spécifique en réponse aux facteurs environnementaux et pressions de sélection (Alves et al. 2018). Dans ce contexte, le séquençage de type « shotgun » est utilisé et consiste à caractériser l’ADN total d’un échantillon. Les résultats représentent les génomes de toutes les espèces de la communauté à un temps donné. La métagénomique fonctionnelle quant à elle, permet d’identifier les gènes qui code pour une fonction d’intérêt, ce qui implique de construire une librairie de milliers de clones avec l’ADN extrait de l’échantillon environnemental afin de tester l’activité de ces clones (Lam et al. 2015). Une autre approche, le métabarcoding se définit comme une méthode automatisée d’identification des espèces présentes au sein d’une communauté, le plus souvent à partir d’un échantillon environnemental (Taberlet et al. 2012). Le séquençage utilisé est appelé
« par amplicon » et consiste à cibler un gène, un marqueur taxonomique à la fois commun à plusieurs espèces mais dont la variabilité est suffisamment discriminante selon le niveau taxonomique d’intérêt à caractériser (phylum, classe, ordre, famille, genre). Par exemple, pour les bactéries, le gène essentiel 16S de l’ARN ribosomique (ARNr) est utilisé pour l’identification des espèces, tandis que la région non codante ITS (« internal transcribed spacer ») de l’ADN ribosomique (ADNr) est utilisée pour les champignons (Abarenkov et al. 2010). L’analyse des séquences obtenues requiert des outils bio-informatiques et se divise en deux parties :
o L’identification des OTUs (Unité Taxonomique Opérationelle d’après l’anglais Operational Taxonomic Unit) qui implique un filtre des séquences et leur assignation à une unité taxonomique. Les OTUs sont formés par le regroupement de séquences en fonction de leur similarité à partir d’un seuil défini et permettent de quantifier la diversité microbienne (Blaxter et al. 2005). En fonction du seuil choisi, les résultats sont à prendre avec précaution. Le seuil généralement utilisé, proposé en 1994, est de 97% d’identité entre les séquences. Cependant, ce seuil est considéré comme trop bas pour caractériser précisément des espèces à cause de la faible résolution génétique apportée par les gènes 16S ADNr (Achtman and Wagner 2008, Edgar 2018). Il existe des espèces différentes décrites comme ayant une identité de séquence supérieure à 98,7% (Achtman and Wagner 2008).
o L’analyse statistique des données de OTU qui inclut la caractérisation de la diversité alpha (diversité au sein des échantillons) et beta (diversité entre les échantillons par des statistiques multivariées).
Outre de révéler l’étendue de la diversité microbienne existante dans les mers, sols et être-vivants, les données des séquençages à large échelle permettent également l’identification des réseaux d’interactions biotiques au sein des communautés (Faust and Raes 2012), l’étude de la variabilité des communautés au cours du temps (Voigt et al. 2015) ou encore l’identification des réseaux métaboliques existant entre les microorganismes (Tringe et al. 2005). Dans le chapitre 3, le séquençage des gènes 16S et régions ITS de l’ADNr permettront de caractériser la diversité taxonomique des communautés bactériennes et fongiques présentes dans les sols expérimentaux utilisés comme microcosmes en laboratoire.