Université de Montréal
Récepteurs aux estrogènes: rôle sur la prolifération, la
migration, les MAPKs et CD4O/CD4OL des cellules
endothéliales et musculaires lisses porcines
par
Pedro Miguel Geraldes
Département des sciences biomédicales Faculté de médecine
Thèse présentée à la Faculté des études supérieures envue de l’obtention du grade de Philosophiae Doctor (PhD)
en sciences biomédicales
Septembre 2006
© Pedro Miguel Geraldes, 2006
(
Université
de Montréal
Direction des bibliothèques
AVIS
L’auteur a autorisé l’Université de Montréal â reproduire et diffuser, en totalité ou en partie, par quelque moyen que ce soit et sur quelque support que ce soit, et exclusivement à des fins non lucratives d’enseignement et de recherche, des copies de ce mémoire ou de cette thèse.
L’auteur et les coauteurs le cas échéant conservent la propriété du droit d’auteur et des droits moraux qui protègent ce document. Ni la thèse ou le mémoire, ni des extraits substantiels de ce document, ne doivent être imprimés ou autrement reproduits sans l’autorisation de l’auteur.
Afin de se conformer à la Loi canadienne sur la protection des renseignements personnels, quelques formulaires secondaires, coordonnées ou signatures intégrées au texte ont pu être enlevés de ce document. Bien que cela ait pu affecter la pagination, il n’y a aucun contenu manquant. NOTICE
The author of this thesis or dissertation has granted a nonexciusive license allowing Université de Montréal to reproduce and publish the document, in part or in whole, and in any format, solely for noncommercial educational and research purposes.
The author and co-authors if applicable retain copyright ownership and moral rights in this document. Neither the whote thesis or dissertation, nor substantial extracts from it, may be printed or otherwise reproduced without the author’s permission.
In compliance with the Canadian Privacy Act some supporting forms, contact information or signatures may have been removed from the document. While this may affect the document page count, it does flot represent any Ioss of content from the document.
Cette thèse intitulée
Récepteurs aux estrogènes: rôle sur la prolifération, la migration, les MAPKs et CD4O/CD4OL des cellules endothéliales et musculaires lisses porcines
présentée par: Pedro Miguel Geraldes
a été évaluée par un jury composé des personnes suivantes:
Dr Christian Deschepper, président-rapporteur DrJean-François Tanguay, directeur de recherche
Dr Louis Paul Perrault, membre du jury Dr Vincent Giguère, examinateur externe Dr Gilles Soulez, représentant du doyen de la FES
In
Résumé
Avant le développement des endoprothèses enrobées de médicaments, la resténose représentait la première limitation à l’angioplastie coronarienne transiuminal pertutanée apparaissant dans 20% à 40% des patients. La pathogenèse de la resténose est un processus complexe et multifactoriel impliquant une lésion de la paroi endothéliale ou dysfonction endothéliale, l’agrégation des plaquettes et des leucocytes, la production et sécrétion de cytokines pro-inflammatoires, la prolifération et la migration des cellules musculaires lisses (CML), la synthèse de matrice extracellulaire et le remodelage vasculaire. Il a été suggéré que la thérapie de remplacement hormonal protégeait les femmes ménopausées contre les maladies cardiovasculaires. Toutefois, les études randomisées ont installé un doute sur les effets cardioprotecteurs de l’hormonothérapie. Des études dans le laboratoire du Dr Tanguay ont démontré que la livraison locale de 17-bêta-estradiol (l7f3E) prévient significativement la formation de resténose favorisant la réendothélialisation tout en réduisant l’infiltration de cellules proinflammatoires au site de lésion à la suite d’une angioplastie ou l’implantation d’une endoprothèse coronarienne. Les objectifs de ma thèse de doctorat étaient de mieux comprendre les mécanismes intracellulaires et le rôle exact de chaque récepteur aux estrogènes (RE), utilisant un traitrnent aux antisens, sur la prolifération, la migration et l’activité de la p42144 et p38 mitogen-activatedprotein kinase (MAPK) des cellules endothéliales (CE) et CML de porc. Au cours de ma formation, nous avons démontré que le traitement d’une dose physiologique de 17f3E (10.8 M) inhibe la phosphorylation de la p42144 et p38 MAPK, la prolifération et la migration des CML stimulée au facteur de croissance dérivés des plaquettes-BB, un mécanisme dépendant des REj3. Contrairement aux CML, le l7E, via l’interaction des REa, stimule l’activité de la p42144 et p38 MAPK, la prolifération et la migration des CE de porc. Certaines conditions proinflammatoires, des cytokines telles que l’interféron gamma (TFN-y) induisent l’expression des marqueurs CD4O et CD4OL à la surface des CE. Le potentiel pro- et anti inflammatoire des estrogènes est bien connu mais les mécanismes d’action par lesquels ces effets sont induits et le rôle des RE restent à déterminer. Nos résultats démontrent une augmentation de l’expression de CD4O et CD4OL respectivement, suite à un traitement de 24 heures à l’IFN-y (1000 U) en culture sur des CE. Un prétraitement au 1713E inhibe à
93% l’effet de l’IFN-y.
À
l’aide d’antisens sélectifs, nous avons montré qu’une baisse de l’expression du REa réduit jusqu’à 74% la capacité du l7E à prévenir l’expression de CD4O et CD4OL induit par l’TFN-y. De plus, le l7f3E via l’activation du REŒ inhibe l’expression du signal transducer and activator of transcription 1 (Stati), une kinase impliquée dans la voie de signalisation du récepteur de l’IFN-y et prévient l’adhésion des neutrophiles à la surface des CE. En conclusion, nos résultats suggèrent que la capacité des estrogènes à prévenir la resténose, tout en favorisant la guérison vasculaire, serait en partie liée à l’inhibition de la prolifération et la migration des CML via son REf3 et par l’activation de ces mêmes mécanismes chez les CE via son REa. De plus, nos données démontrent que le 1713E, en se liant à son REa, possède un effet anti-inflammatoire en inhibant l’activité de Stati, l’augmentation de l’expression de CD4O et CD4OL et l’adhésion des neutrophiles induitparl’IfNy au niveau des CE.Mots clés : Resténose, hormones, récepteur aux estrogènes, cellules musculaires lisses vasculaires, cellules endothéliales, MAPK, inflammation, neutrophiles, CD4O/CD4OL.
V
Abstract
Before the development of drug eluting stents, restenosis formation was the primary limitation of percutaneous transiuminal coronary angioplasty and stenting occurring in 20% to 40% of patients. Restenosis is a complex process involving endothelial dysfunction, platelet and neutrophil aggregation, inflammatory cytokine production and secretion, vascular srnooth muscle celi (SMC) proliferation and migration, extracellular mati-ix synthesis and vascular remodeling. Epidemiological studies over the past years suggested a protective effect of hormonal replacement therapy (HRT). In contrast, randomized clinical trials did flot provide conclusive data that HRT confers cardioprotection against coronary artery disease in postmenopausal women. Our iaboratory demonstrated that a local delivery of 1 7-beta-estradiol (17 f3E) during porcine coronary angioplasty and stent implantation reduced restenosis while improving the reendothelialization process and inhibiting infiltration of inflammatory cells. The objectives of the present thesis were to better understand the intracellular mechanisms of 1713E and the role of each estrogen receptor (ER) using an antisense gene therapy approach to smdy porcine SMCs and endothelial ccli (EC) proliferation, migration and p42144 and p38 mitogen-activated protein kinase (MAPK) activation. Treatment of porcine SMCs with platelet-derived growth factor-BB induces p38 and p42144 MAPK activation which lead to SMC migration and proliferation. These effects are prevented by a pretreatment with a physiological dose of 1713E (1OE8 M). The inhibitory effects of 1713E on SMCs are abrogated by the downregulation of ER13 protein expression with selective ER13 mRNA antisense oligomers, whereas the downregulation of ERa has no effect. On the opposite, treatment with 1713E promotes porcine EC proliferation, migration and p42144 and p38 MAPK phosphorylation. These effects are ERa-dependent as defined by antisense gene therapy. Interferon gamma (LFN y) has been shown to induce CD4O and CD4OL expression on ECs which lead to neutrophil adhesion onto ECs. The pro- and anti-inflammatory effects of estrogens are well recognized but the role of estrogens and their receptors (ERa, ER13) on the regulation of CD4O and CD4OL expression on ECs remains undefined. Treatment of cultured porcine EC with ITN-y for 24 hours enhances CD4O and CD4OL expression. We demonstrated that treatment of porcine ECs with 1713E for 24 hours prevented these events on CD4O/CD4OL
expression. Treatment of EC with antisense oligomers targeting ERŒ mRNA reduced by up to 74% the ability of 173E to prevent the IFN-’y-induced CD4O arid CD4OL protein expression. The IFN-y activation pathway of CD4O is known to involve the phosphorylation of the signal transducer and activator of transcription 1 (Stati). We also demonstrated that 1 7f3E, through an interaction with ERa, abrogated Stati phosphorylation and neutrophil adhesion onto EC thus interfering with LFN-y-mediated effects. In conclusion, our resuits suggest that, in porcine SMC, 17J3E reduces p42144 and p38 MAPK activity through ERJ3 stimulation, whereas in contrast, in EC, 17f3E induces p42/44 and p38 MAPK tlwough ERŒ activation. Indeed, treatment with 1 7f3E prevents IFN-y-mediated Stati phosphorylation, CD4O and CD4OL protein expression and neutrophil adhesion onto ECs through ERŒ activation. Estrogens may contribute to the vascular healing process and to the prevention of restenosis by inhibiting the inftammatory process and improving reendothelialization through ERa activation, and by decreasing SMC migration and proliferation through ER stimulation.
Keywords : Restenosis, hormones, estrogen receptors, vascular smooth muscle celis, endothelial cells, MAPK, inflammation, neutrophils, CD4O/CD4OL.
vii
Table des matières
Introduction 1
1.1 L’athérosclérose 2
1.1.1 Processus de formation de l’athérosclérose 2
1.1.1.1 La dys fonction endothéliale 2
1.1.1.2 L’incorporation des LDL 4
1.1.1.3 Recrutement et infiltration des macrophages 5 1.1.1.4 Production de cytokines et prolifération des CML 6
1.1.1.5 Instabilité et rupture de la plaque $
1.1.1.6 Les mécanismes de rupture de la plaque 10
1.1.2 Les traitements 11 1.1.2.1 L’angioplastie coronarienne 12 1.1.2.2 L’endoprothèse coronarienne 12 1.1.2.3 Autres techniques 13 1.1.2.4 Les inconvénients 13 1.2 Laresténose 14 1.2.1 Dénudation endothéliale 14
1.2.2 Agrégation des plaquettes et formation de thrombus 15
1.2.2.1 Le pÏatelet-derivedgrowthfactor (PDGF) 15
1.2.2.2 Le transforming growth factor-beta (TGF-13) 16 1.2.2.3 LepÏasminogen activator inhibitor-] (PAT-1) 16
1.2.2.4 Lefacteurtissulaire 17
1.2.2.5 La glycoprotéine llb/IIIa 17
1.2.2.6 Le facteur d’activation plaquettaire 17
1.2.3 La migration et la prolifération des CML et synthèse de MEC 18 1.2.3.1 Implication du cycle cellulaire dans la prolifération des CML 18 1.2.3.2 Les rnitogen-activatedprotein kinases (MAPK5) 20
1.2.3.2.1 ERK-1etERK-2 21
1.2.3.2.2 p38MAPKetJNK 21
1.2.4 Le remodelage vasculaire 22
1.2.5.1 Labrachythérapie .22
1.2.5.2 Lathérapie génique 23
1.2.5.3 Les endoprothèses enduits de médicament (drug eluting stent) 24
1.2.5.4 Réendothélialisation 25
1.3 L’inflammation 26
1.3.1 Les cytokines 26
1.3.1.1 Le facteur de tumeur nécrotique alpha (TNF-Œ) 26
1.3.1.2 L’interféron(WN) 27
1.3.1.2.1 Les récepteurs de l’IfN 2$
1.3.1.2.2 La voie de signalisation de JAKISTAT 29
1.3.1.2.2.1 Les protéines JAK 29
1.3.1.2.2.2 Les facteurs de transcription STAT 30
1.3.1.3 Le CD4O/CD4OL 30
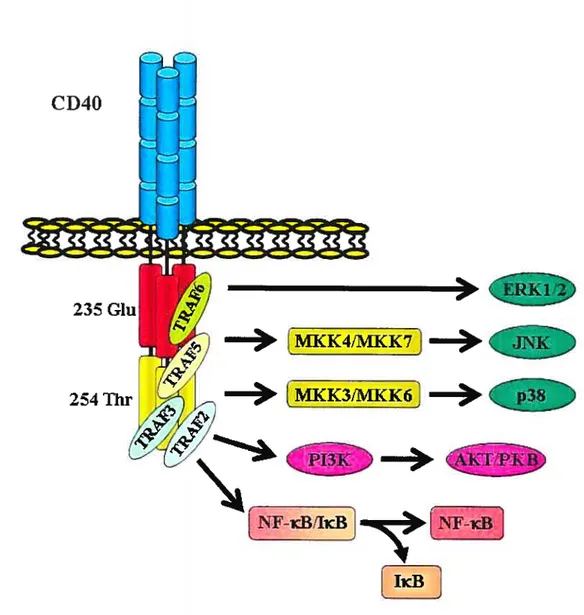
1.3.1.3.1 La signalisation de CD4O 31
1.3.1.3.2 Le rôle de CD4O/CD4OL dans l’athérosclérose et laresténose 32
1.3.2 Les molécules d’adhésion 33
1.3.2.1 Les sélectines 34
1.3.2.1.1 LaL-sélectine 35
1.3.2.1.2 LaE-sélectine 36
1.3.2.1.3 LaP-sélectine 36
1.3.2.2 La superfamille des immunoglobulines —CAM 37
1.3.2.2.1 La molécule d’adhésion intercellulaire (ICAM) 37 1.3.2.2.2 La molécule d’adhésion des cellules vasculaires (VCAM) 37
1.3.2.3 Les intégrines 3$
1.3.2.4 Les cadhérines 38
2 Les hormones stéroïdiennes 39
2.1 Letyped’hormone 39
2.1.1 Les estrogènes 40
2.1.2 La progestérone 41
2.1.3 L’hormonothérapie 41
ix
2.2 Le rôle des estrogènes et de leurs récepteurs 45
2.2.1 LastnicturedesRE 46
2.2.2 Les isoformes des REΠet REf3 48
2.2.3 La régulation des RE 49
2.2.4 La phosphorylation des RE 50
2.2.5 Les mécanismes d’action des RE 50
2.2.5.1 Les coactivateurs et corépresseurs 51
2.2.5.2 Les mécanismes régulateurs des estrogènes et des RE 52
2.2.5.2.1 Les canaux ioniques 52
2.2.5.2.2 Les MAPKs 53
2.2.5.2.3 Laphosphoinositide-3 kinase 54
2.2.5.2.4 Les récepteurs couplés aux protéines G 55
2.2.5.2.5 Les autres récepteurs membranaires 55
2.2.5.3 Les effets des estrogènes sur la paroi vasculaire artérielle 56
2.2.5.3.1 Le profil lipidique 56
2.2.5.3.2 La fonction vasculaire 57
2.2.5.3.3 Les effets dépendants de l’endothélium 57
2.2.5.3.3.1 Les mécanismes génomiques des estrogènes sur le NO 57 2.2.5.3.3.2 Les mécanismes nongénomiques des estrogènes sur le NO 58 2.2.5.3.4 Les effets indépendants de l’endothélium 59 2.2.5.3.5 Les effets des estrogènes sur le système de coagulation 59 2.2.5.3.6 Les estrogènes et les marqueurs inflammatoires 61
2.2.5.3.6.1 LeNFiB 61 2.2.5.3.6.2 Les CAI\4 61 2.2.5.3.6.3 LeTNF-Œ 62 2.2.5.3.6.4 LeMCP-1 62 2.2.5.3.6.5 L’IL-l, 1’IL-6 et le TGF- 63 2.2.5.3.6.6 La protéine réactive C (CRP) 63
2.2.5.3.7 Les estrogènes et l’hypertension artérielle systémique 64
2.2.5.3.7.1 Le caractère hémodynamique 64
2.2.5.3.7.3 Les influences sur les facteurs circulants (ANG II, ET-l et AMP cyclique) 65
2.2.5.3.8 Les estrogènes et la croissance cellulaire, le remodelage vasculaire et l’ischémie 66
2.2.5.3.9 Les estrogènes et l’angiogénèse 67
2.2.6 Les modulateurs sélectifs des récepteurs aux estrogènes (SERMs) 6$
3 Problématique 71 3.1 Butduprojetdethèse 72 4 Article#1 73 4.1 Abstract 74 4.1.1 Condensed abstract 74 4.2 Introduction 75
4.3 Materials and Methods 76
4.3.1 Cell Culture 76
4.3.2 Mitogenic Assay 76
4.3.3 Chemotactic Assay 77
4.3.4 Western Blot Analysis of p38 and p42144 MAPK Phosphorylation 77
4.3.5 Statistical Analysis 79
4.4 Resuits 79
4.4.1 Effects of 1713E on PSMC Proliferation 79
4.4.2 Effects of 1713E on PSMC Migration 79
4.4.3 Effects of 1713E on PSMC p42144 and p38 MAPK Phosphorylation 80
4.4.4 Effects of 1713E on PAEC Proliferation $1
4.4.5 Effects of 1713E on PAEC Migration 81
4.4.6 Effects of 1713E on PAEC p42144 and p38 MAPK Phosphorylation 81
4.5 Discussion 82
4.5.1 Anti-mitogenic and Anti-chemotactic Effects of 1713E in PSMC $3 4.5.2 1713E Promotes Reendothelialization by Increasing the Proliferation and the
Migration ofPAEC 84
4.6 References 85
xi
5 Article #2. 96
5.1 Abstract .97
5.2 Introduction. 98
5.3 Materials and Methods 99
5.3.1 CeÏl Culture 99
5.3.2 Antisense Oligonucleotide Gene Therapy 99
5.3.3 Western Blot Analyses of ERΠand ERI3 Expression, p42144 and p38 MAPK
Phosphorylation 100
5.3.4 Mitogenic Assay 102
5.3.5 Chemotactic Assay 102
5.3.6 Statistical Analysis 103
5.4 Resuits 103
5.4.1 Modulation ofERΠand ERf3 Protein Expression by Antisense Oligonucleotide
Gene Therapy 103
5.4.2 Contribution ofERa and ER13 on PSMC Proliferation 104 5.4.3 Anti-Chemotactic Effect of 1713E on PSMC: Role ofERa and ERI3 105 5.4.4 Role of ERa and ERI3 on p42144 and p38 MAPK Phosphorylation in PSMC
105
5.4.5 Contribution ofERa and ERI3 on PAEC Proliferation 106 5.4.6 Anti-Chemotactic Effects of 1713E on PAEC: Role of ERa and ER13 mRNA
106
5.4.7 Role of ERa and ER13 on p42144 and p38 MAPK Phosphorylation in PAEC 107
5.5 Discussion 107
5.5.1 Regulation of ERa and ER13 Protein Expression by Antisense Gene Therapy 10$
5.5.2 Biological Activities of 1713E Are Mediated Through ER13 in PSMC 10$ 5.5.3 ERa Activation by 1713E Induces MAPK Phosphorylation in PAEC 109
5.6 Acknowledgements 110
5.7 References 110
6 Article#3. 123
6.1 Abstract 124
6.2 Introduction 125
6.3 Materials and Methods 126
6.3.1 Ceil culture 126
6.3.2 Antisense oligonucleotide gene therapy 126 6.3.3 Analyses ofCD4O and CD4OL expression 127
6.3.4 Analyses ofCD4O mRNA levels 12$
6.3.5 Neutrophil isolation and purification 122
6.3.6 Neutrophil adhesion assay 129
6.3.7 Statistical Analysis 129
6.4 Results 130
6.4.1 Effect of 1713E on CD4O and CD4OL mRNA and Protein Expression 130 6.4.2 Role ofERa and ER13 on CD4O and CD4OL Expression 130 6.4.3 Effect ofERa on IFN-y Signalling Pathway 131
6.4.4 1713E Inhibits NeutrophilAdhesion 131
6.5 Discussion 133
6.6 Acknowledgements 136
6.7 References 137
6.8 Figure Legends 141
7 Discussion 149
7.1 La régulation de la prolifération et de la migration des cellules musculaires lisses et
endothéliales par le 1713E Rôle de la p42/44 et p38 MAPK 150 7.2 La caractérisation du rôle des REcL et RE13 dans les mécanismes de régulation de la p42144 et p38 MAPK, de la prolifération et de la migration des cellules musculaires lisses
et endothéliales de porc 153
7.3 Les effets du 1713E sur la phosphorylation de Statl, l’expression protéique de CD4O et CD4OL et l’adhésion des neutrophiles induits par 1’1FN-y au niveau des CE: implication
duREΠ15$
Conclusions 166
xiii
Liste des tableaux
Tableau I: Résumé des études observationnelles et randomisées en prévention primaire et secondaire sur les effets de la thérapie de remplacement hormonale 42 Tableau II: Les différentes voies d’activation des RE 51
xv
Liste des figures
Figure 1: Processus initial de la formation de la plaque athéromateuse 5 Figure 2: Progression de la plaque athéromateuse et formation de la chape fibreux $ Figure 3: Processus de rupture de la plaque et de la formation d’un thrombus 10 Figure 4: Formation de la resténose à la suite d’une ACTP ou l’implantation d’une
endoprothèse vasculaire 14
Figure 5: Déroulement du cycle cellulaire 19
Figure 6: La cascade des MAPKs 20
Figure 7: Blocage de la traduction par les antisens 24
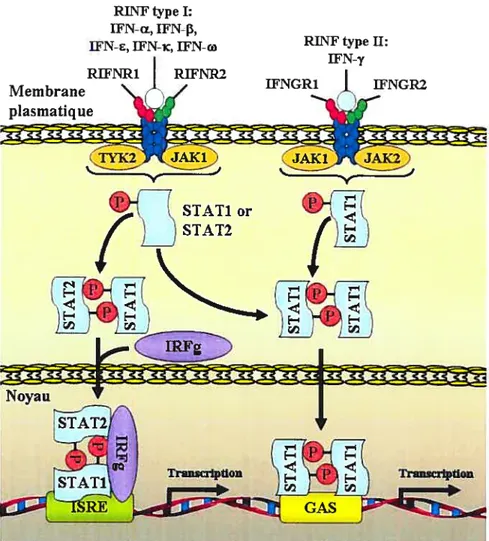
Figure 8: Récepteurs à l’IFN et activation classique des protéines JAKJSTAT 29 Figure 9: Forme trimérique du récepteur CD4O et ses voies de signalisation 32
Figure 10: Famille des molécules d’adhésion 34
Figure 11: Concentrations plasmatiques des hormones ovariennes 40 Figure 12: Structure des récepteurs aux estrogènes alpha et bêta 47 Figure 13: Représentation des voies de signalisation et d’activation des estrogènes 52
Article #1
Figure 1: Effects of 1713E and ER antagonists on PSMC proliferation 90 Figure 2: Effects of 1713E and ER antagonists on PSMC migration 91 Figure 3: Effects of 1713E and ER antagonists on PSMC p42144 and p38 MAPK activity. 92 Figure 4: Effects of 1713E and ER antagonists on PAEC proliferation 93 Figure 5: Effects of 1713E and ER antagonists on PAEC migration 94 Figure 6: Effects of 1713E and ER antagonists on PAEC p42144 and p38 MAPK activity. 95
Article #2
Figure 1: Antisense regulation of ERa and FR13 expression on PSMC and PAEC 116 Figure 2: Contribution of ERΠand ER13 on PSMC proliferation 117 Figure 3: Contribution of ERa and ER13 on PSMC migration 118 Figure 4: Contribution ofERa and FR13 on p42144 and p38 MAPK activation in PSMC 119
Figure 5: Contribution of ERΠand ERj3 on PAEC proliferation . 120
Figure 6: Contribution ofERΠand ERf3 on PAEC migration 121 Figure 7: Contribution of ERΠand ERf3 on p42144 and p38 MAPK activation in PAEC 122
Article #3
Figure 1: Effect of 1713E on CD4O and CD4OL expression induced by IFN-y 143 Figure 2: Effect of 1713E on CD4O and CD4OL mRNA expression induced by ffN-y in
PAEC 144
Figure 3: Contribution of ERΠand ER13 on the regulation of CD4O and CD4OL expression
inPAEC 145
Figure 4: Role of 1713E and ER on Jaki, Jak2 and Stati phosphorylation induced by WN-y 146 Figure 5: Contribution ofICAM, CD4O and CD4OL on neutrophil adhesion on PAEC... 147 Figure 6: Effects of 1713E on neutrophil adhesion on PAEC 14$
xvii
Liste des abréviations
17E = 17-bêta-estradiol
ACTP= angioplastie coronarienne translurninale percutanée
ADN= acide désoxyribonucléique
AF = activation factor
ANG II= angiotensine II
ARN = acide ribonucléique
BKca-=canaux potassiques et voltage-dépendants
CAM ccli adhesion moÏecule CE= cellules endothéliales
CEE= conjugé d’éthinylestradiol
CML= cellules musculaires lisses
CRP = C reactiveprotein
eNOS =endothelial nitric oxide synthase
ERK extracellular signai-regulated kinase ET-1 endothéline- 1
FM femmes ménopausées HDL=high density lipoprotein
HSP =heat shockprotein
IFN-y= interféron-gamma
ICAM intercellular adhesion molecule IL interleukine
JAK =janus kinase
JNK=c-juil N-terminal kinase
LDL=low density lipoprotein
LPS =lipopolysaccliaride
MAPKs = mitogen-activatedprotein kinases
MCP- 1 = macrophage chemoattractant protein-]
MCV = maladies cardiovasculaires
MEC = matrice extracellulaire
MPA= médroxyprogestérone
NADPH = nicotinamide adenine dinucleotide phosphate reduced
NFKB = nuclearfactor kappa B
NO nitric oxide
ODN= oligodésoxynucléotides
PAT-l =plasminogen activator inhibitor-]
PDGF =plateÏet-derived growth factor
PI3K =phosphoinositide-3 kinase PRG=progestérone
RE =récepteur aux estrogènes
RIFN=récepteur de l’interféron
SERM=selective estrogen receptor modulators
STAT=signal transducer and activator oftranscription
TGF- transforming growthfactor-beta TNF-Π= tumor necrosisfactor-alpha
t-PA =tissue plasminogen activator
TRE= thérapie de remplacement hormonale
VCAM= vascular ceil adhesion molecuÏe
VEGF=vascular endothelial growthfactor
xix
« Douter de tout ou tout croire sont deux solutions également commodes, qui l’une et l’autre nous dispensent de refléchir » Henri Foincaré
Remerciements
Au cours de ces années, j’ai côtoyé plusieurs personnes qui m’ont aidé, enduré, influencé et supporté tout au long de mon cheminement. Pour cela et pour tout ce qu’ils ont apporté, j’aimerais les remercier.
Tout d’abord, je remercie mon directeur, Dr Jean-françois Tanguay, un mentor exceptionnel. Vous avez cru en moi et en mon potentiel. Vous m’avez également formé, instruit, encouragé et supporté au cours de toutes ces années. Vous avez été un modèle et un exemple à suivre. Je vous dois beaucoup. Merci mille fois.
Je remercie les membres de monjury (Dr Christian Deschepper, Dr Jean-François Tanguay, Dr Louis Paul Perrault et Dr Vincent Giguère) d’avoir accepté et pris le temps de réviser ma thèse de doctorat.
Je remercie grandement le Dr Martin G. Sirois qui, sans avoir été officiellement mon codirecteur, a quand même agi de la sorte. J’ai énormément appris par vos judicieux conseils. Vous m’avez accueilli dans votre laboratoire comme si j’étais l’un de vos étudiants et pour cela, je n’oublirai jamais.
Je remercie le Dre Isabelle Cloutier qui s’est jointe à l’équipe du Dr Tanguay en début de l’année 2004. Vous avez amené mon apprentissage et ma formation au doctorat à un niveau nettement supérieur.
Je remercie les membres de mon laboratoire pour leur aide précieuse et constante. Merci à Pascale Geoffroy, assistante de recherche hors pair, à Sandra Gilligan et Julie Lebel pour leur aide au niveau des techniques animales, à Dominique Lauzier et Véronique Philibert pour leur rigueur et leur aide au niveau des techniques d’histologie et d’immunohistochimie, à Louis-Robert Villeneuve pour son aide et ses précieux conseils pour les techniques d’immunofluorescence et à Chantal Sauvé pour son support au niveau administratif.
Je remercie le Dr Aiigelo Calderone, le Dr Yahye Merlu, le Dr Eric Thorin et le Dr Chantai Lambert pour leur collaboration, support, conseils et contributions à l’achèvement de mon doctorat.
Je remercie également Dr Pascal Bematchez, Stéphanie Gagnon, Dre Roselyne Villiard, Dr Sofiane Hadjadj et Caroline Lemieux pour leur soutient technique et leurs précieux conseils.
xxi
Je remercie le Fonds de la Recherche en Santé du Québec en partenariat avec la Fondation des Maladies du Coeur du Québec et le réseau cardiovasculaire pour leur support financier.
Je remercie mes amis pour leur compréhension. Un merci tout à fait spécial à deux personnes, Eve-Amélye Morel et Émilie Gauthier pour leur compréhension, leur tolérance, leur sacrifice et leur soutien.
À
vous deux, vous avez toute ma gratitude. Vous m’avez influencé et contribué à faire de moi une meilleure personne.Et finalement, je remercie mes parents pour leur dévouement, leur patience et leur support.
À
leur façon, ils ont su faire tout ce qu’il fallait sans poser aucune question, sans rien demander en retour.Avant-propos
Les diverses recherches dans le passé nous ont permis d’affirmer que l’athérosclérose n’est pas un simple processus d’accumulation de matières graisseuses obstruant la lumière artérielle. Il est maintenant bien connu que l’athérosclérose est une forme d’inflammation chronique impliquant plusieurs médiateurs cellulaires et menant ultimement au développement de lésions complexes. Les maladies cardiovasculaires représentent la principale cause de mortalité chez les femmes ménopausées des pays industrialisés. Les études observations ont suggéré que le traitement hormonal réduisait d’environ 50% les incidences de maladies cardiovasculaires. Cependant, les données d’études randomisées ont indiqué que la thérapie par remplacement hormonal ne présentait aucun effet bénéfique en prévention primaire ou secondaire après un événement cardiaque. Cette disparité entre les données des études cliniques randomisées et celles des études in vitro et in vivo démontre clairement que les mécanismes de régulation des estrogènes au niveau vasculaire sont encore mal compris. Notre laboratoire s’est grandement intéressé aux estrogènes et à leurs effets cardioprotecteurs. Par le passé, nous avons démontré que la livraison locale d’estrogènes réduisait la resténose tout en favorisant la guérison des vaisseaux suite à une blessure coronarienne dans un modèle porcin. Il est évident que les estrogènes agissaient à plusieurs niveaux afin de prévenir la formation de resténose. Toutefois, les mécanismes d’actions par lesquels les estrogènes et ses récepteurs aux estrogènes induisent leurs effets demeurent peu élucidés. Mes travaux de recherche explorent de façon approfondie les rôles précis des récepteurs aux estrogènes au niveau de la prolifération et de la migration des cellules musculaires lisses et endothéliales. De plus, nous avons étudié et caractérisé le rôle des récepteurs aux estrogènes au niveau de certaines cascades de signalisation intracellulaire chez les cellules vasculaires porcines. Finalement, nous explorons les effets des récepteurs aux estrogènes dans l’expression de cytokines proinflammatoires ainsi que leurs impacts sur l’adhésion des neutrophiles à la surface des cellules endothéliales. Une meilleure compréhension du rôle des estrogènes et de ses récepteurs au niveau des mécanismes de guérison vasculaire permettrait de contribuer au développement de nouvelles approches thérapeutiques afin de diagnostiquer et de traiter plus efficacement les maladies cardiovasculaires.
Introduction
Une maladie cardiovasculaire (MCV) est un trouble ou une lésion touchant l’appareil cardiovasculaire, qu’il s’agisse du coeur ou des vaisseaux sanguins. Au Canada, en 2001, environ le tiers (33%) de tous les décès étaient attribuables aux maladies cardiovasculaires.1 Bien que les taux de mortalité aient diminués depuis les années 90, le nombre absolu de décès chez les hommes et les femmes est resté stable depuis le milieu des années 90. Les problèmes cardiaques les plus courants sont la maladie coronarienne athérosclérotique se manifestant par l’angine de poitrine, l’infarctus aigu du myocarde ou la mort subite, les arythmies, les valvulopathies, les atteintes myocardiques, l’insuffisance cardiaque, les péricardites, et les malformations cardiaques congénitales. Plusieurs facteurs de risque traditionnels tels que le tabagisme, l’hypercholestérolémie, l’hypertension artérielle, le diabète et l’obésité augmentent les risques d’événements cardiaques.’
1.1 L’athérosclérose
Pendant longtemps, l’athérosclérose a été vue comme un processus par lequel des éléments composant les vaisseaux sanguins s’assemblent pour obstruer la lumière artérielle menant ainsi aux MCVs. L’athéroclérose pouvait ainsi prendre plusieurs années a être formée. Les travaux récents à l’aide de recherches cliniques et épidémiologiques, de recherches fondamentales et le développement de nouvelles techniques de biologie moléculaire ont permis de clarifier que l’athérosclérose est en fait un phénomène complexe impliquant de nombreux facteurs. Les connaissances sur la pathogénie et l’étiologie de la formation de l’athérosclérose ont été marquées par deux grands courants de pensée. La mise en évidence du rôle du cholestérol par Ariitschkow et Chalatow (1914) a permis d’émettre l’hypothèse selon laquelle l’infiltration lipidique est le facteur déclenchant la formation de la plaque athéromateuse.2 L’athérosclérose est également considérée comme une forme d’inflammation chronique résultant d’une interaction entre les lipoprotéines modifiées, les macrophages dérivés des monocytes, les lymphocytes T et les cellules endothéliales et musculaires lisses de la paroi artérielle. Ce processus inflammatoire, décrit par Russel Ross, peut ultimement mener au développement de lésions plus complexes ou de plaques athéromateuses avancées dont les plus fréquentes sont l’insuffisance coronaire qui se manifeste par l’angine de poitrine et l’infarctus du myocarde, conséquences de l’atteinte des artères coronaires, les accidents vasculaires cérébraux liés aux lésions des vaisseaux à destinée cérébrale et l’artérite des membres inférieurs lorsque les artères fémorales sont obstruées.3
1.1.1 Processus de formation de l’athérosclérose
1.1.1.1 La dysfonction endothéliale
Dans des conditions physiologiques normales, I’endothélium forme une couche semi-perméable entre les éléments circulants du sang et la paroi du vaisseau sanguin. L’endothélium est maintenant considéré comme étant le plus gros organe endocrinien ayant un impact direct sur le développement de l’athérosclérose. Il sécrète des substances
3
vasoactives afin de réguler le tonus artérielle, de maintenir une surface antithrombotique, de moduler les réponses inflammatoires et d’inhiber la prolifération des cellules musculaires lisses (CML) vasculaires sous jacentes.4 La principale substance protectrice synthétisée par l’endothélium est le monoxyde d’azote (NO), synthétisé par les synthétases de NO. Lorsque produit, le NO cause une vasorelaxation des CML et inhibe l’agrégation des plaquettes. De plus, il stimule la prolifération des cellules endothéliales (CE), permettant ainsi la réparation des blessures à l’endothélium. Le NO est aussi un puissant anti-oxydant qui élimine les espèces réactives d’oxygène, dont l’anion superoxyde, qui peuvent endommager l’endothélium. Par contre, la production concomitante dans un même lieu de NO et d’anion superoxyde s’avère très dommageable en donnant naissance au peroxynitrite, un précurseur de radicaux libres. Ainsi, l’endothélium agit sur le tonus vasculaire à l’aide du NO, de la prostacycline et du facteur hyperpolarisant dérivé de I’endothélium. Il équilibre l’effet vasodilatateur de ces substances à l’aide de peptides vasoconstricteurs, tels que l’endothéline (ET-1) et l’angiotensine II (ANG II). La balance des effets de ces substances dicte donc le tonus vasculaire. Il a été longtemps considéré que les nombreuses conditions physiopathologiques observées chez les humains menant à la formation de l’athérosclérose débutaient par une réponse à une blessure qui provoquait une dénudation endothéliale. Toutefois, cette hypothèse s’est raffinée par l’introduction du concept de dysfonction endothéliale plutôt que d’une dénudation résultant d’une blessure artérielle ou mort des cellules endothéliales.5
La dysfonction endothéliale est donc un débalancement des facteurs homéostatiques de l’endothélium. Cet état est caractérisé par une diminution de la biodisponibilité du NO, par un tonus vasculaire anormal, par une activation du système de coagulation et par conséquant une augmentation de la croissance des CML.6 La diminution de la biodisponibilité du NO est une conséquence centrale car, en plus de la diminution des effets protecteurs du NO, elle entraîne l’augmentation du stress oxydatif qui favorise alors l’oxydation des low densitv lipoproteins (LDL). La dysfonction endothéliale est le processus permissif menant à l’état inflammatoire. Plusieurs facteurs contribuent à la dysfonction endothéliale et au développement de l’athérosclérose: l’hypercholestérolémie, le diabète de type 1 et 2, l’hypertension artérielle, l’élévation et l’oxydation des LDL, les
radicaux libres causés par le tabagisme, certaines cytokines, les facteurs génétiques, l’élévation des concentrations d’homocystéine plasmatique, les infections par différents microorganismes tels que les herpesvirus, le virus d’immunodéficience humaine et chlamydia pneumoniae ou la combinaison de ces facteurs.3
1.1.1.2
L’incorporation des LDL
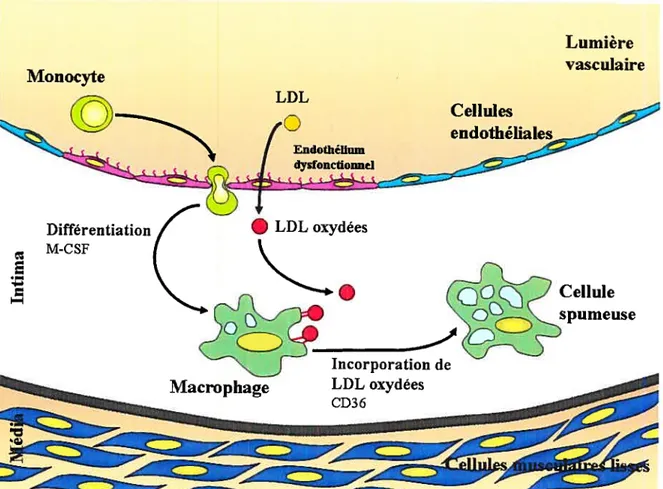
Les LDL traversent la paroi des vaisseaux sanguins ce qui leur permettent d’interagir avec les CE. Lors de leur migration, les LDL peuvent demeurer prisonnières de protéoglycans sécrétés par les CE au niveau de l’intima des artères. Cette accumulation se produit principalement à des sites privilégiés où l’on retrouve des conditions hémodynamiques et mécaniques particulières telles que les branches et les bifurcations où les niveaux de turbulence sont élevés et les forces de cisaillement sont faibles. L’accumulation de LDL dans l’espace sous-endothélial conduit à la formation de masses lipidiques. Subséquemment, l’oxydation des LDL représente une étape essentielle au processus athérosclérotique.7 Bien que les quantités de LDL oxydées circulants soient très faibles, les quantités présentes dans les plaques athéromateuses sont élevées.8 L’oxydation des LDL est provoquée par contact avec les CML et les macrophages, ou de façon chimique par des radicaux libres et produits de la cigarette.9 L’oxydation des LDL est ensuite reconnue par le système immunitaire comme étant une substance étrangère et les macrophages recrutés dans l’espace subendothélial au niveau de la lésion peuvent alors intemaliser les LDL oxydées devenant par le fait même des cellules spumeuses (Figure
5
Monocyte
Lumière vasculaire
figure 1: Processus initial de la formation de la plaque athéromateuse
Les LDL sont sujets à une oxydation et constituent un facteur de dysfonction endothéliale. L’expression de molécules d’adhésion favorise l’adhésion des monocytes à la paroi qui migrent dans l’espace sous-endothélial pour ensuite incorporer à l’aide de leurs récepteurs les LDL oxydées (adapté d’après Glass C.K. h1)•
1.1.1.3 Recrutement et infiltration des macrophages
Les altérations du flot sanguin à certains sites artériels spécifiques tels que les branches se division et les bifurcations, où les forces de cisaillement sont minimales et les turbulences sont maximales, élèvent l’expression des molécules d’adhésion, la migration et l’accumulation des monocytes et des lymphocytes T.3 Les monocytes adhérents pénètrent dans la paroi vasculaire par les jonctions interendothéliales sous l’effet de facteurs chimiotactiques tels que le macrophage chemoattractantprotein-] (MCP-l). Cette protéine
est fortement exprimée par les macrophages et les CML dans la plaque d’athérosclérose LDL Cellules Endothékum M-CSF z
.
Cellule spumeuse Incorporation de LDL oxydées CD3 6-œ
- —-humaine. Des études effectuées chez les souris déficientes pour le MCP-l ou pour son récepteur, ont démontré que ces souris développent très peu de lésions athérosclérotiques suggérant leur rôle important dans cette pathologie.’2’13 La transformation des macrophages en cellules spumeuses débute par l’accumulation d’une grande quantité de LDL oxydées dans leur cytoplasme par l’intermédiaire des récepteurs piégeurs ou scavengers tSR-A, CD36 et CD68).’4 Les monocytes et les macrophages ont la capacité de se multiplier. Ainsi, le macrophage cotony stimuÏatingfactor (M-CSF) contribue au processus de formation de la plaque athéromateuse en favorisant la multiplication et la différentiation des monocytes/macrophages.” 5 Les macrophages, en association avec les LDL oxydées, forment donc une couche de stries lipidiques présente dès la vingtaine, lésion très précoce dans le développement de l’athérosclérose.
1.1.1.4 Production de cytoldnes et prolifération des CML
L’infiltration des macrophages dans la plaque athéromateuse a pour effet d’engendrer une réaction inflammatoire chronique. Cette inflammation s’accompagne d’une libération de certains médiateurs solubles, cytokines proinflammatoires telles que le tumor necrosis factor-atpha (TNF-Œ), l’interleuldne-l (IL-l), l’IL-6, l’IL-8 1’IL-12 et l’interféron gamma (TFN-y) ainsi que les molécules immunorégulatrices membranaires CD4O et son ligand, le CD4OL.” 16 Ces cytokines sont synthétisées par les lymphocytes T cytotoxiques (TH1). Les cytokines proinflammatoires de la plaque peuvent aussi intervenir dans les complications thrombotiques associées à l’athérosclérose. Les propriétés antithrombotiques des CE sont profondément altérées par l’IL-l ou le TNF-Œ, qui augmentent l’activité procoagulante du facteur tissulaire, diminuent l’expression de la thrombomoduline et suppriment l’activité anticoagulante du système thrombomoduline protéine C17 18 De plus, l’activation des plaquettes permet la formation de l’acide arachidonique qui est transformée en prostaglandines telles que la thromboxane A2, l’un des plus puissants vasoconstricteurs, et amplifie ainsi la réponse inflammatoire. Ces cytokines modifient également les propriétés fibrinolytiques des CE en réduisant la production du tissue plasminogen activator (tPA) et en augmentant la production du ptasm inogen activator inhibitor-] (PAl-1).‘$ Toute réponse inflanimatoire s’ accompagne
7
de production de cytokines anti-inflammatoires dont l’IL-4, l’IL-lO, l’IL-13 et le transforming growth factor-beta (TGF-f3). Ces cytokines sont synthétisées par les lymphocytes T auxiliaires (TH2).’9 Tous ces médiateurs ont la capacité d’induire l’expression des molécules d’adhésion et de moduler l’activité des CML. Ainsi, des facteurs de croissance tels que le platelet-derived growth factor (PDGF) et le facteur de croissance de la famille de l’insuline (insitÏin-like growth Jactor) sécrétés par les macrophages stimulent la prolifération et la migration des CML.3’ ‘ La multiplication des CML est accompagnée de la synthèse de matrice extracellulaire (MEC) soit le collagène, les protéoglycans et l’élastine. De plus, les macrophages sécrètent des enzymes protéolytiques, les matrix metalloproteinases (MMP) qui sont responsables de la dégradation des protéines de la MEC.2° De plus, la dégradation par les MMP des limitantes élastiques des vaisseaux favorisent la migration des CML à la paroi subendothéliale contribuant à la formation de la plaque au stade initial. L’activité des MMP peut être réduite par des inhibiteurs tissulaires des MMP synthétisés de façon constitutive par les CML. Toutefois, dans les plaques athéromateuses, l’infiltration importante de cellules inflammatoires accroît l’expression de l’IL-l, du TNF-a et l’TFN-y, favorise l’expression des MMP et diminue la production des protéines de la MEC sans modifier le taux d’expression des inhibiteurs tissulaires des MMP. La prolifération des CML et la synthèse de la MEC forment ainsi la chape fibreuse (Figure 2).
Les macrophages interagissent avec les lymphocytes impliqués dans le processus inflammatoire. Les CML stimulées par les macrophages et les lymphocytes migrent de la
média vers l’intima, prolifèrent et synthétisent de la matrice extracellulaire formant la chape fibreuse (adapté d’après Glass C.K. ‘i).
1.1.1.5 Instabilité et rupture de la plaque
En réponse à une inflammation chronique, la mort des CML par apoptose, la
protéolyse ainsi que la dégradation et l’érosion de la chape fibreuse provoquent l’érosion et la rupture de la plaque athéromateuse. De nombreuses données angiographiques, angioscopiques, d’exploration chirurgicale et d’autopsies ont démontré que l’occlusion abrupte des artères coronaires menant aux syndromes coronariens aigus est, dans 70 à 80%
Lumière vasculaire
Fïgure 2: Progression de la plaque athéromateuse et formation de la chape fibreux
9
alors très thrombogénique puisqu’elle expose aux éléments du sang circulant des substances pro-coagulantes telles que le collagène et la fibrine ce qui engendre des cascades d’activation, d’adhésion et d’agrégation plaquettaire. Différents facteurs extrinsèques (anomalies plaquettaires) et intrinsèques (libération de l’ADP, le Thromboxane A2, les acides gras et le facteur tissulaire) par les plaquettes peuvent favoriser la thrombose Les pathologistes ont par conséquent décrit que les plaques vulnérables sont associées à une coque fibreuse mince, à un centre lipidique riche en LDL et à une abondance de macrophages et autres cellules inflammatoires. Une telle plaque est susceptible de rupture parfois due à des causes extrinsèques telles qu’une poussée hypertensive ou une élévation de la pression artérielle suite à un effort physique important (on parle alors de rupture passive), mais surtout pour causes intrinsèques à la plaque telles que la réponse inflammatoire (on parle ici de rupture active). Le concept de plaque instable fait donc son apparition dans les années 90 (Figure 3)24
La formation d’un corps nécrotique riche en lipides composé de cellules spumeuses et de MEC. Les macrophages sécrètent également des MMP qui contribuent à amincir la chape fibreuse. La rupture de la plaque provoque l’activation des plaquettes et la formation d’un thrombus (adapté d’après Glass C.K. h1)•
1.1.1.6 Les mécanismes de rupture de la plaque
Le pourcentage relatif occupé par le centre lipidique dans les lésions athérosclérotiques est l’une des principales différences anatomiques entre les plaques stables et instables. En effet, le centre lipidique de la plaque instable peut représenter jusqu’à 64% du volume total en comparaison à 14% pour la plaque stable.25 De plus, les plaques instables se caractérisent par des changements phénotypiques des CML contenant deux fois plus de granules sécrétoires.26 La libération par les macrophages de certaines Figure 3: Processus de rupture de la plaque et de la formation d’un thrombus
11
cytokines pro-inflammatoires telles que le TNF-a engendrant l’apoptose des CML contribue à rendre la plaque de plus en plus instable. Les macrophages prolifèrent et produisent également de l’WN-y qui a pour effet d’inhiber la prolifération des CML. Cette suppression de la prolifération des CML entraîne une réduction de la synthèse de MEC. Ce phénomène s’amplifie par la dégradation de la MEC qui se fait essentiellement par les MMPs relâchées par les macrophages. Les LDL oxydées participent aussi à la surexpression des MMPs par les macrophages. D’autres molécules telles que IL-l et IL-8 sont susceptibles aussi de dissoudre la chape fibreuse.27 En plus de sa dégradation, l’inhibition de la synthèse de MEC est le résultat de la perte de fonctionnalité et de l’apoptose de CML induites par le TNF-Œ et l’WN-y.28’29 La plaque instable se caractérise donc par une prédominance de macrophages et de cellules inflammatoires et par la réduction de CML et de la quantité de MEC. IYautres facteurs sont passibles de déstabiliser ou au contraire de « stabiliser » une plaque. Ainsi l’IL-l, l’IL-7, 1’IL-8, le TNf-a, PET-l, la thrombine, 1’ANG II sont des molécules proinflammatoires, alors que l’IL-lO, l’IL-13 et le NO sont des facteurs protecteurs.27’30-32 La rupture de la plaque augmente de façon massive l’adhérence des plaquettes et des leucocytes et mène éventuellement à l’occlusion totale du vaisseau. Cette réduction graduelle de la lumière du vaisseau engendre de l’angine de poitrine pouvant mener à des infarctus du myocarde. L’occlusion d’une artère coronarienne entraine une nécrose du muscle myocardique suite à l’absence d’oxygénation. La mort des cellules myocardiques provoquent la libération d’enzymes telles que le TNF-Œ pour ainsi induire la mort des cellules avoisinant. Certains infarctus sévères peuvent mener à la mort subite du patient.
1.1.2
Les traitements
La progression de la plaque aboutissant à une réduction du calibre de l’artère fait donc apparaître de l’angor stable (angine de poitrine dont la plaque est stable, sans thrombose ni rupture de la plaque) et les syndromes coronariens aigus qui sont provoqués par la rupture de la plaque, une étape déterminante dans l’évolution de l’athérosclérose. Sur le plan thérapeutique, l’utilisation d’agents anti-plaquettaires, de l’aspirine, du clopidogrel, d’antagonistes des récepteurs anti-intégrines plaquettaires de la glycoprotéine IIb/Illa et
d’agents thérapeutiques neutralisant la thrombine vise à contrôler la formation du thrombus coronarien.33 Cependant, l’emploi d’agents anti-thrombotiques ne suffit pas à revasculariser le coeur. Afin de rétablir le flot sanguin, les chirurgies cardiovasculaires avaient traditionnellement recours aux pontages coronariens qui consistent à la greffe de veines prélevées dans les jambes ou d’artères prises dans la cavité thoracique et anastomosées distalement aux artères coronariennes sténosées. Toutefois, des méthodes moins invasives telle que 1’ angioplastie coronarienne transiuminale percutanée (ACTP) et l’implantation d’endoprothèse apparues depuis presque une trentaine d’années ont révolutionné le traitement de la maladie coronarienne athérosclérotique..
1.1.2.1 L’angioplastie coronarienne
L’ACTP se pratique au moyen d’une sonde munie d’un ballonnet qui, lorsque gonflé au site d’obstruction, permet de comprimer la masse lipidique athéromateuse de façon radiaire et de dilater la paroi du vaisseau. Le ballon est monté par voie artérielle jusqu’au lieu du rétrécissement par l’intermédiaire d’une sonde introduite en pénétrant une artère périphérique comme l’artère fémorale au niveau de l’aine ou l’artère radiale au niveau du poignet. Cette technique établie depuis 1978 comporte toutefois certaines limitations.34 En effet, dans plusieurs cas, la dilatation par ballon seulement demeure insuffisante pour permettre une bonne revascularisation principalement lorsqu’il y a durcissement (calcification) de la plaque. De plus, le recul élastique suite à F étirement initial du vaisseau et la guérison tissulaire peut provoquer un remodelage vasculaire et mener au rétrécissement de l’artère plutôt qu’à l’élargissement du vaisseau.35
1.1.2.2 L’endoprothèse coronarienne
Afin de palier au phénomène de recul élastique et à la vasoconstriction du vaisseau à la suite d’une ACTP, l’introduction d’une endoprothèse coronarienne métallique, un tube expansible formé d’un filet en différents matériaux biocompatibles, permet d’élargir les artères coronaires rétrécies. Cette endoprothèse est délivrée à l’extrémité d’un cathéter porteur, puis déployée à la suite du gonflement du ballonnet auquel elle est fixée. Elle est ensuite libérée dans la lumière vasculaire par le dégonflement et le retrait du ballonnet.36
13
1.1.2.3 Autres techniques
D’autres techniques telles que l’utilisation du laser et du «Rotablator» ont fait leur apparition comme outils adjuvants permettant d’ouvrir et de dilater des lésions plus complexes. Le laser consiste à émettre de courtes pulsations de photons qui pulvérisent la plaque athéromateuse alors que le «Rotablator» est un appareil de traitement qui, au lieu de déplacer et écraser la plaque d’athérome, la supprime à l’aide d’une fraise revême de micro-cristaux de diamants tournant à très haute vitesse. Ces deux techniques sont donc une solution complémentaire au ballonnet de l’angioplastie des artères calcifiées ou à l’hyperplasie néointimale plus ou moins égale à l’intérieur d’une endoprotèse.37’38
1.1.2.4 Les inconvénients
L’utilisation de I’angioplastie et de l’endoprothèse coronarienne provoque des blessures à la paroi déclenchant divers processus de guérison vasculaire. II peut donc y avoir formation d’une nouvelle masse cellulaire composée principalement de CML et de MEC communément appelée resténose. La resténose qui survient dans 20 à 30% des cas 6 mois après l’intervention demeure la première cause d’échec de l’angioplastie et l’implantation des endoprothèses coronariennes métalliques (Figure4)•39
Artère partiellement obstruée Angioplastie avec ou sans endoprothèse Rétablissement de flot sanguin Formation de la resténose 6 mois après lintervention
A
E
Figure 4: Formation de la resténose à la suite d’une ACTP ou l’implantation d’une endoprothèse vasculaire
1.2
La resténose
La resténose est une réponse d’adaptation à la suite d’une lésion artérielle survenant lors d’une ACTP avec ou sans mise en place d’endoprothèse. La pathogenèse de la resténose est un processus complexe et multifactoriel impliquant une atteinte endothéliale, l’agrégation des plaquettes et des leucocytes, la production de cytokines inflammatoires, la prolifération et la migration des CML, la synthèse de MEC et le remodelage vasculaire.40
1.2.1 Dénudation endothéliale
La dénudation endothéliale de la paroi artérielle est considérée comme le premier événement qui survient à la suite d’une angioplastie ou de l’implantation d’une
15
endoprothèse. La perte d’inhibition de contact produite par la dénudation des CE provoque leur réplication de la partie proximale vers la partie distale du segment affecté. Les CE produisent alors des facteurs de croissance tels que le PDGF, TGF-f3 et le facteur de croissance des fibroblastes qui permettent aux CML de migrer, de proliférer et de synthétiser de la MEC. Ce processus se perpétue jusqu’à environ 1 mois chez un modèle porcin et 6 mois chez l’humain afin d’obtenir une réendothélialisation complète du segment.41
1.2.2 Agrégation des plaquettes et formation de thrombus
À
la suite de la blessure artérielle, les plaquettes s’activent et adhèrent rapidement au site de lésion. Plusieurs agents anti-plaquettaires ont été utilisés afin de prévenir la formation de la resténose sans que les études cliniques d’aient pu démontrer un bénéfique.4244 La formation d’un thrombus se produit par déposition locale de fibrine et de collagène dans les premières heures suivant l’angioplastie ou l’implantation d’une endoprothèse. Leur déposition est proportionnelle à l’étendu de la lésion artérielle. La fibrine et le collagène servent également de plateforme à l’agrégation des plaquettes. Les plaquettes ainsi activées induisent avec grande rapidité la principale intégrine responsable de l’agrégation des plaquettes soit l’intégrine Œ11bf33.33 La liaison des plaquettes au collagène stimule la relâche de thromboxane A2 au niveau des complexes de glycoprotéines IIb/IIIa menant ainsi à l’activation et à l’adhésion de plaquettes. Les plaquettes activées libèrent des facteurs de croissance et contribuent à la migration et la prolifération des CML45’ 46 D’autres facteurs tels que le PDGF, le TGF-J3, l’histamine et la production de molécules proinflammatoires sont générés également par les plaquettes et seront décrit dans les prochaines sections.1.2.2.1
Le
ptatetet-derived growth factor
(PDGF)
Le PDGF est un puissant stimulateur de la formation de l’hyperplasie néointimale. Des études animales ont démontré que l’ajout de PDGF favorise la prolifération des CML à la suite d’une lésion endothéliale induite par 1’ACTP.47 De plus, le PDGF agit comme un
puissant chimioattractant pour les CML en contribuant à leur migration. La thrombine, générée par les plaquettes activées, favorise la libération du PDGF. Des études in vivo ont clairement démontré l’importance du PDGF dans la resténose puisque son inhibition par thérapie génique prévient de façon significative l’hyperplasie néointimale dans un modèle de dénudation de la carotide de rat.48
1.2.2.2
Le
transform ing growth factor-beta
(TGF-)
Le TGF-f3 joue un rôle important dans les processus de réparation tissulaire en réponse à un traumatisme. Il exerce de nombreux effets inhibiteurs sur les mécanismes inflammatoires, la croissance et la différentiation des cellules précurseures hématopoïétiques et sur la prolifération et l’apoptose des cellules immunocompétentes. Le TGF-f3, dérivé des plaquettes, stimule la synthèse d’acide désoxyribonucléique (ADN) chez les CML et des protéines de la MEC confirmant son rôle dans I’hyperplasie néointimale. Son implication a également été démontrée par l’augmentation de l’expression des intégrines v et
f33
contribuant ainsi à la migration des CML, monocytes et neutrophiles.49 L’utilisation d’anticorps ciblant spécifiquement la protéine TGF-f3 réduit la formation de la resténose dans les modèles d’animaux de blessures vasculaires.501.2.2.3 Le
ptasm inogen activator inhibitor-1
(PAl-1)
Le PAl-l est présent dans les granulocytes a des plaquettes et peut être activépar diverses cytokines et facteurs de croissance. Sa principale fonction est de s’opposer à l’activation du plasminogène et au système fibrinolytique jouant un important rôle dans l’homéostasie vasculaire.5’ La fibrinolyse implique donc la dissolution des dépôts fibrineux formés en permanence dans la lumière vasculaire. Le précurseur du plasminogène, activé par le t-PA ou le urokinase-type plasminogen activator, est synthétisé et sécrété par les CE qui produisent également le PAT-1. La thrombine et la bradykinine stimulent l’expression du t-PA alors que les médiateurs inflammatoires élèvent le taux de PAT-1. Ce dernier, par liaison au t-PA, forme un complexe inactif diminuant l’activité fibrinolytique.52 Toutefois, le PAT-1 ne se limite pas à la simple régulation des processus thrombotiques. En effet, il
17
module également la dégradation de la MEC, stimule la migration et la prolifération des CML et régule positivement ou négativement l’angiogénèse puisqu’une réduction des tumeur et de l’angiogénèse a été observée chez les souris déficientes pour le gène du PAT-1 alors que sa surexpression favorise la formation de tumeur.53’54 Le PAT-1 est donc impliqué dans les mécanismes thombogéniques, le remodelage vasculaire et au développement de l’athérosclérose.5’
1.2.2.4 Le facteur tissulaire
Le facteur tissulaire est une glycoprotéine intramembranaire présente sur la paroi des vaisseaux et des monocytes circulants. Il se lie au facteur VITIVIIa et ainsi initie la cascade de la coagulation. L’activation de cette cascade contribue à la migration et la prolifération des CML.33 L’expression du facteur tissulaire est induite par divers facteurs de croissance, des cytokines inflammatoires telles que l’TL-1f3 et TNF-a et par des endotoxines.56 Les concentrations plasmatiques du facteur tissulaire augmentent considérablement 4 heures après l’angioplastie.
1.2.2.5 La glycoprotéine IIb/IIIa
La glycoprotéine TIb/ITa (CD41/61) est un récepteur pour le fibrinogène, le von Wiltebrand factor, la fibronectine et la vitronectine. Elle participe à l’activation et l’agrégation des plaquettes. Des études ont clairement démontré une augmentation de l’expression du complexe glycoprotéine IIb/ITTa chez les patients ayant développé une resténose 6 mois suivant l’angioplastie ou la procédure interventionnelle.57
1.2.2.6 Le facteur d’activation plaquettaire
Le facteur d’activation plaquettaire est un phospholipide rapidement synthétisé par les plaquettes et les CE stimulées par la thrombine ou par une blessure artérieIle.8 Il est également reconnu pour être un puissant stimulateur d’une multitude voies métaboliques intercellulaires et intracellulaires.59 Il est donc intimement impliqué dans l’hyperplasie néointirnale par sa capacité à induire la migration des CE. Le facteur d’activation
plaquettaire se lie à ses récepteurs au niveau des CE et induit une puissante réponse inflammatoire aigu et chronique produisant l’augmentation du calcium intracellulaire, la contraction du squelette d’actine, le rétrécissement cellulaire et l’hyperperméabiÏité vasculaire influençant ainsi la pression artérielle.60 Le facteur d’activation plaquettaire participe également à l’activation des leucocytes et favorise ainsi leur roulement et adhérence à l’endothélium.6’
1.2.3 La migration et la prolifération des CML et synthèse de
MEC
Il est bien connu que les CML de la paroi vasculaire ont deux phénotypes distincts: un phénotype contractile, que l’on retrouve à l’état normal au niveau de la média, et un phénotype sécrétoire qui apparaît dans les lésions athéromateuses. La fonction contractile permet une vasomotricité régulant le tonus artériel. La vasomotricité est réglée par les messagers agissant sur l’endothélium qui vont ensuite sécréter d’autres second messager (ET-1, NO) aux CML. Les CML assurent des fonctions métaboliques, en particulier la sécrétion de MEC. Le changement de phénotype des CML permettant leur migration de la média vers l’intima et leur prolifération se fait sous l’influence de différents facteurs: le PDGF sécrété par les macrophages et les cellules endothéliales, l’IL-l libéré par les macrophages, l’inactivation du récepteur du TGf-f3, la présence de lysophosphatidylcholine dans les LDL oxydées, la lipoprotéine-A, l’ANG II et la thrombine.6264 Ces facteurs accroissent la capacité de synthèse protéique des CML afin de produire une grande quantité de collagène, de fibres élastiques et de protéoglycanes.
1.2.3.1 Implication du cycle cellulaire dans la prolifération des CML
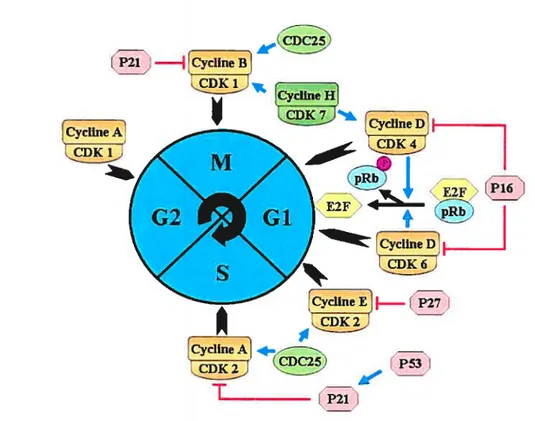
La prolifération cellulaire est un processus biologique fondamental au cours du développement et lors de la réparation tissulaire qui demande donc un contrôle rigoureux, exempt d’erreur et une coordination temporelle et spatiale précise avec d’autres mécanismes.65 Lors du processus de réparation vasculaire, les CML sortent de leur phase quiescente G0 pour entrer dans le cycle cellulaire. Le cycle cellulaire peut se schématiser
19
par une séquence de quatre étapes. L’ensemble des périodes G, S et G2 séparant deux mitoses est nommé interphase.66 En général, les cellules ne progressent pas dans la phase suivante du cycle cellulaire avant d’avoir achevé tous les événements associés à la phase précédente. Pour cela, des mécanismes de surveillance permettent à la cellule de s’assurer de l’exécution complète des événements d’une phase avant d’initier les événements associés à la phase suivante. Ces dernières assurent ainsi une duplication et une transmission fidèle du génome aux cellules filles résultant de la division cellulaire. La machinerie moléculaire du cycle cellulaire est composée de facteurs de régulation qui contrôlent la progression dans le cycle cellulaire. La progression de la cellule au cours des différentes phases du cycle et point de contrôle est régulée par l’activation transitoire de complexes cyclines/kinases dépendantes des cyclines (CDK), de l’état hypo- ou hyperphosphorylé de la protéine rétinoblastome, des phosphatases (Cdc25, cdc2) et de la protéine 53 (Figure 5)6772
Implication des différentes cyclines, CDK et des inhibiteurs des cyclines/CDK au cours des différentes phases du cycle cellulaire.
________ ,CDC25) tP21 —I ______ [yctïii P21
1.2.3.2 Les mitogen-activatedprotein kinases (MAPKs)
Les cellules possèdent de multiples cascades de MAPKs régulant plusieurs activités cellulaires telles que l’expression des gènes, la mitose, le métabolisme de survie et d’apoptose et la différentiation. La voie de signalisation des MAPKs a été largement étudiée pour expliquer les effets de divers stimuli.73 La famille des MAPKs est séparée en cinq groupes distincts: les extracellular signal-regulated kinase (ERK-1, -2 ou p42144 MAPK), les c-Jun N-terminal kinase (JNK-l, -2 et -3), les isoformes de p38 a,
f3, y
et ê, ERK-3 et ERK-4 et finalement ERK-5.74 Les ERK-1 et -2 sont préférablement stimulées par les facteurs de croissance et les esters. Les JNK et p38 MAPK répondent principalement au stress, aux cytokines et aux chocs osmotiques.75 Même si chaque membre de la famille des MAPKs possède des caractéristiques uniques, il partage certaines similitudes. En effet, chaque famille est composée d’une cascade de kinases présentée dans la Figure 6.Cascade des MAPKs
Mitogines Stress, cytokines Stimulus
MEKK3/ MEKK/2/4
Raf/MEKK3 TAK MLK/ASK1 MAPKKK
PD98059
—I
Ir
MEK1/2 MKK3/6 MKK4/7 MAPKK
1
Ir
ERK-1/-2 SB2035$O p38 JNK/SAPK MAPK
‘I
Elk-1
1
ATF-2 c-Jun Facteurs deATF-2 transcription
MAPKAPK-1 MAPKAPK-2
MAPKAP
p7O MAPKAPK-3
MAPKAPK-5 kinases
21
Iniplication des différentes protéines impliquées dans la cascade de signalisation des MAPKs (adapté d’après Pearson G. 75)
1.2.3.2.1 ERK-1 et ERK-2
Les protéines ERK-1 et ERK-2 possèdent 83% d’homologie et sont exprimées par une variété de cellules. Leur activation passe généralement par la stimulation d’un récepteur tyrosine kinase couplé à une protéine G présent à la surface des cellules. Lorsque ce récepteur est stimulé, il induit l’activation de la cascade de Raf/MEKIERK via la petite protéine Ras liant une GTP qui recrute ensuite la protéine SOS. L’activation de Ras, par le changement d’une GDP pour une GTP, permet son interaction avec la kinase Raf Cette dernière phosphoryle deux autres kinases MEK- 1 et MEK-2 qui, à leur tour, activent ERK 1 et -2. Lorsqu’elles sont phosphorylées, ERK-1 et -2 s’accumulent dans le noyau pour induire divers processus dont l’activation de protéines membranaires (CD13Oa, Syk et calnexin), des substrats nucléaires (SRC-1, Pax-6, NA-AT, ELK-1, MEF-2, c-Fos, c-Myc et STAT3), des protéines cytoplasmiques (paxilline et neurofilaments) et autres protéines kinases appelées protéines kinases activées par les MAPK (MK5). Il existe également un inhibiteur de ERK-1 et -2, le PD98059, qui empêche la phosphorylation de ERK-1 et 2.76
77
1.2.3.2.2 p38 MAPK etJNK
L’activation des différents isoformes de p38 est induite par le stress environnant et par des cytokines inflammatoires. La majorité de ces stimuli active également la voie de JNK. La MEK-6 possède la capacité d’induire la phosphorylation de toutes les isoformes de p38 alors que la MEK-3 est sélective pour la p38Œ et p3$3. Suite à son activation, la p38 phosphorylée peut se retrouver dans le cytoplasme ou dans le noyau et ainsi agir sur divers substrats tels que la phospholipase cytosolique A2, la protéine Tau associée aux microtubules et aux facteurs de transcription (ATF-1, ATF-2, MEF2A, Sap-1, Elk-1, nuclear Jactor kappa B (NF-icB), Ets-1 et p53).78 L’inhibiteur de la voie de p38 est le SD203580. Tout comme les p38, les JNK sont activées par des cytokines, du stress, des radiations, des agents dommageables pour l’ADN tels que les peroxynitrites et certains facteurs de croissance. La stimulation des JNK passe par la phosphorylation de la MEK-4
et MEK-7. Lorsqu’elles sont phosphorylées, les INK se localisent dans le noyau pour ainsi induire des facteurs de croissance (ATF-2, NF-ATc1, HSF-l et STAT3).74’78
1.2.4 Le remodelage vasculaire
Il est reconnu que les artères coronaires humaines peuvent changer de taille en réponse à la croissance des plaques d’athérosclérose. Glagov et ses collaborateurs ont montré, par des études morphométriques, que la coronaire gauche augmente son diamètre interne pour compenser la perte de lumière vasculaire par la sténose tant que la plaque n’occupe pas plus de 40% de la surface de la lumière, phénomène correspondant à un remodelage positif De manière similaire, Losordo et al. ont montré, en utilisant l’échographie endovasculaire intracoronarienne in vivo, que la surface tota’e du vaisseau (tout le contenu à l’intérieur de la lame élastique externe) est plus grande dans les segments d’artères coronaires sténosées que dans les segments d’artères coronaires normales.8° En général, les plaques hémorragiques inflammatoires caractérisées par un large centre lipidique, une infiltration accentuée de macrophages et des dépôts calciques importants ont moins tendance à se remodeler positivement en comparaison aux plaques ne possédant pas toutes ces caractéristiques. Les mécanismes biologiques impliqués dans le remodelage vasculaire ne sont pas très bien connus. Toutefois, la relâche des MMP, particulièrement les MMP-l et MMP-13, par les macrophages est en partie responsable de la dégradation de MEC permettant le remodelage vasculaire.81’82
1.2.5 Les nouveaux traitements pour prévenir la resténose
1.2.5.1 La brachythérapïe
Au cours des dernières années, la brachythérapie endocoronaire a soulevé un réel enthousiasme pour la prévention de la resténose. L’utilisation des radiations ionisantes à faible dose a été proposée dans le but d’inhiber la prolifération néointimale et de prévenir la resténose. De nombreuses études ont démontré une réelle efficacité dans la prévention de la resténose intra-endoprothèse. Toutefois, les premières études cliniques utilisant la brachythérapie ont fait apparaître deux complications majeures: la thrombose tardive et le
23
phénomène de resténose en bordure de la zone irradiée.83’ 84
La thrombose tardive se définit lorsqu’elle se produit au-delà de 30 jours voire même au-delà de 6 mois après ce traitement. Ce type de complication est lié à une réendothélialisation retardée et/ou à un mauvais déploiement de l’endoprothèse coronarienne irradiée sur la paroi. La resténose aux extrémités de l’endoprothèse est due à une hyperpiasie néointimale plutôt qu’à un remodelage de la paroi artérielle.83
1.2.5.2 La thérapie génique
La thérapie génique vasculaire peut potentiellement contrôler la croissance cellulaire impliquée dans la formation de la resténose en modifiant l’expression de certains gènes.85 Les séquences d’antisens d’oligodésoxynucléotides (ODN) se lient par complémentarité au brin d’acide ribonucléique (ARN) messager de la protéine cible, inhibent la synthèse protéique et ainsi bloquent l’expression des gènes impliqués dans la prolifération des CML (Figure 7)86 La livraison intramurale des antisens d’ODN contre le gène c-myc a permis de réduire drastiquernent la prolifération néointimale dans un modèle porcin.87 Toutefois, cette technique ne s’est pas avérée efficace chez l’humain.88 La thérapie génique est également utilisée pour le transfert de gènes ayant des propriétés antiprolifératives et/ou favorisant la réendothélialisation telles que le NO et le vascular endotheliat growth factor (VEGF).899 Cependant, l’induction de ces gènes chez l’humain demeure controversée en autre par le potentiel pro-inflammatoire du VEGF à court terme et par l’absence d’effet vasculaire à long terme.
Figure 7: Blocage de la traduction par les antisens
L’introduction d’un antisens ODN engendre sa liaison par complémentarité à l’ARN messager du gène cible et ainsi bloque sa traduction par les ribosomes (adapté d’après Askari F.K. 86)
1.2.5.3 Les endoprothèses enduits de médicament
(dr;tg etuting stent)
Les endoprothèses pharmacologiques sont des endoprothèses vasculaires classiques enrobées d’un médicament anti-prolifératif intégré dans certains cas dans une couche de polymère. Ce polymère permet de fixer le médicament, d’en contrôler la diffusion, d’obtenir un revêtement uniforme et stable mais qui peut poser des difficultés de fabrication et des risques inflammatoires ou allergiques. Contrairement aux endoprothèses à l’héparine destinées à réduire les risques de thrombose, les endoprothèses pharmacoactives utilisent un médicament destiné à réduire le risque de resténose. Les deux principales molécules utilisées à ce jour sont la raparnycine et ses dérivés (sirolimus, tacrolimus, everolimus, rapalog) et le paclitaxel.92’ Certaines molécules plus toxiques ou inefficaces ont déjà étéAutisensd’ODN — 3, 3, 3, 5, 5, 5, Protéine