Evaluation of alkaline electro-activated water and
eggshell as acid mine drainage neutralization and
mine tailing remediation agents
Thèse
Alexey Kastyuchik
Doctorat en sols et environnement
Philosophiae Doctor (Ph.D.)
Québec, Canada
Résumé
Cette étude visait à étudier la capacité d’un biodéchet calcitique seul ou en mélange avec de matériaux chimiques alcalins ainsi que l’efficacité du procédé d’électro-activation dans la neutralisation de l’acidité et le maintien de conditions alcalines dans un résidu minier sulfuré (RMS).
Dans une première série d'expériences, l’évolution du pH du RMS traité avec divers amendements a été suivie en fonction de doses croissantes de coquilles d'œufs de poule (COP) ajoutées seules (2, 4, 6, 8 et 10%) ou en mélange avec 1 et 2% de ciment Portland, 1 et 2% d’oxyde de magnésium (MgO), 1 et 2% de chaux calcique et 1 et 2% de chaux dolomitique. La plus forte dose de COP (10 %) a augmenté la valeur de pH de 2,61 (sans ajout d’amendement) à 7,24. Cependant, les échantillons de RMS mélangés avec COP + ciment (1 – 2%) ou COP + MgO (1 – 2%) avaient un pH très élevé (≥ 8). Les résultats suggèrent que les composés de magnésium ou les produits calcaires riches en oxydes, en hydroxydes et en carbonates, présents dans les RMS chaulés, fournissent une protection à longue terme contre l’acidification anthropique des RMS chaulés.
Dans une deuxième série d'expériences, plusieurs essais ont été effectués pour évaluer l’efficacité du procédé d’électro-activation utilisant deux compartiments, l’anode et la cathode, et certains paramètres géométriques, électriques, qualitatifs et quantitatifs, dans la neutralisation de l’acidité des suspensions de RMS introduites dans le compartiment cathodique. Tous les traitements ont influencé de façon significative les valeurs de pHcatholyte.
Les résultats ont démontré que l’électro-activation permettait de neutraliser efficacement l’acidité du RMS seul ou en mélange avec COP et également d’obtenir des valeurs de pH fortement alcalines (pHcatholyte 8,0 – 10,0). En outre, l’électro-activation utilisant trois
Abstract
This study aimed to investigate the capacity of a calcite biowaste alone or mixed with alkaline chemical materials and the efficiency of the electro-activation process in neutralizing acidity and maintaining alkaline conditions in a sulfide mine tailing (SMT).
In a first set of experiments, chicken eggshell residue (CES) alone (2, 4, 6, 8 and 10%) or mixed with cement concrete (1 – 2%), MgO (1 – 2%), calcitic limestone (1 – 2%) or dolomitic limestone (1 – 2%) was used to neutralize sulfide mine tailing (SMT) acidity and to precipitate trace metallic elements. The highest rate of CES (10%) increased the initial tailing pH value from 2.61 (without amendment) to 7.24, indicating that CES had sufficient lime value to increase the pH of acid SMT. However, the SMT samples mixed with either CES + cement (1 – 2%) or CES + MgO (1 – 2%) had a high pH (≥ 8). The results suggested that magnesium compounds and calcareous products rich in hydroxides, oxides and carbonates present in limed SMT would provide long-term protection against acid deposition or re-acidification of limed SMT.
In a second set of experiments, several trials were carried out to assess the effectiveness of electro-activation process composed by two compartments, anode and cathode, under different electric, geometrical, quantitative and qualitative parameters, in neutralizing acidity and maintaining alkaline conditions in a SMT alone or mixed with CES introduced into the cathode compartment. All treatments significantly influenced the pHcatholyte. The results
demonstrated that electro-activation process is capable of neutralizing the acidity of RMS alone or mixed with COP and also to achieve alkaline pH conditions (pHcatholyte 8.0 – 10.0).
In addition, the electro- activation process using three compartments can remove up to 80% of ferrous iron from an aqueous FeSO4·7H2O solution.
Acknowledgments
It is done. The three years doctoral research is finished and thesis is written. My scientific development could be clearly observed through the pages of my dissertation. But it is just one side of the medal. Another side is the people that I met and that have supported me on this long and sometimes difficult life period.
The first person, that I want to thank, is my beloved wife Christiana. It was necessary cross the ocean (one half of the world) to find her here in Canada. In the most difficult moment of my residence here (like and all time) she gave me moral support. And finally, she made the best present of my life - my son Serge.
Very impressive event in my life, of course, was arriving in Canada. It was a dream of my childhood. Because of that I want to express special gratitude to my professor Mohammed Aider. He accepted my candidacy to work in his research team and proposed very interesting project throughout of my last experience in the electrochemistry. I also want to thank him for his guidance, patience, flexibility, accessibility and criticism.
I do gratefully acknowledge to my co-advisor professor Antoine Karam. For this short three years he has done too much for me. Maybe without his contribution to my fortune it was impossible arrive at the final point of my doctorate program. In the complicated moment he breathed life into the project. His wisdom, altruism, presence, useful accurate hints were always in time. His laboratory, that was accessible for me, had almost all needed equipment and materials. He considerably enriched my mental outlook as a researcher.
I would like to thank Dr Lotfi Khiari for agreement to pre-read this thesis and for his valuable comments.
My appreciation also goes to the members of my thesis committee: Dr Alfred Jaouich and Dr Mathieu Quenum. They have given their time and offered valuable advice and suggestions.
Thanks very much to Daniel Marcotte, Alain Brousseau and Marie-Hélène Lamontagne for their assistance in the special analysis and to Normand Massicotte for his help in the technical questions.
I cannot forget the role of my friends and colleagues Shyam Suwal, Sergey Mikhailin, Luca Lo Verso, Marina Bergoli, Amara Aït Aissa, Alina Gerzhova, Valerie Carnovale, Julien Chamberland, Catherine Couturier, Nassim Naderi, Élizabeth Parent, Sabrine Naimi, Stéphanie Dudonné, Cédric Leterme, Sébastien Goumon, Cheslav Liato et al. The separation with homeland was not so heavy because of you.
My deep appreciation must also be expressed to the faculty direction and especially to vice-doyen Pierre-Mathieu Charest for the foreign student assistance.
And finally I want to thank the most important persons in the life of each man - parents. Galina Alekseevna and Sergey Aleksandrovich, I am deeply grateful to you for all.
Table of contents
Résumé ... iii
Abstract ... v
Acknowledgments ... vii
Table of contents ... ix
Index of tables ... xiii
Index of figures ... xv
List of abbreviations ... xix
Introduction ... 1
Chapter 1 : Review of literature ... 5
1.1 Importance of Canadian mine activity ... 5
1.2 Impacts of mining on environment ... 5
1.3 Acid mine drainage generation ... 5
1.3.1 General consideration ... 5
1.3.2 Chemistry of pyrite oxidation ... 7
1.3.2.1 Geochemistry of iron ... 7
1.3.2.2 Formation of pyrite ... 11
1.3.2.3 Weathering and oxidation of pyritic tailings under natural conditions ... 14
1.3.2.3.1 Reaction 1: Oxidation of pyrite ... 16
1.3.2.3.2 Reaction 2: Oxidation of the ferrous sulfate ... 17
1.3.2.3.3 Reaction 3: precipitation of Fe(III) ... 18
1.3.2.4 Pyrite oxidation in neutral and alkaline medium ... 20
1.3.2.5 Role of carbonate in the reaction of pyrite oxidation ... 21
1.3.2.6 Biological oxidation of metallic sulfides ... 23
1.3.2.7 Factors affecting the oxidation of pyrite ... 23
1.4 Prevention and mitigation of acid mine drainage generation ... 24
1.5 Treatment of acid mine waters and acid mine tailings for environmental control ... 28
1.5.1 Passive treatments ... 30
1.5.2 Active systems ... 33
1.5.2.1 Aeration ... 33
1.5.2.2 Neutralization technologies ... 34
1.5.2.2.1 Alkaline neutralization of mine water and acid mine drainage ... 34
1.5.2.2.2 Lime neutralization processes and high density sludge method ... 37
1.5.2.2.3 Neutralization of acid mine tailing ... 39
1.5.2.2.4 Metal precipitation ... 40
1.5.2.2.5 Alkaline industrial waste as neutralizing agents ... 43
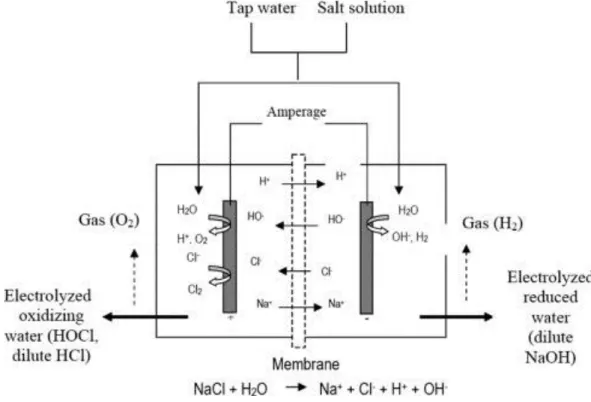
1.5.3 Electro-activated aqueous solutions ... 46
1.5.3.1 Base of electro-activated aqueous solutions ... 46
1.5.3.2 Applications of electro-activated solutions ... 49
1.5.3.3 Application of electro-activated water to acid mine neutralization ... 50
Chapter 2 : Research hypotheses, objectives and significance of the study ... 53
2.1 Hypothesis ... 53
2.2 Objectives ... 53
2.3 Expected results ... 54
2.3.2 Hypothesis 2 ... 54
2.4 Communications ... 55
2.5 Significance of the study ... 56
2.6 Research motivation ... 56
Chapter 3 : Effectiveness of chicken eggshell residue mixed with alkaline amendments in acid mine drainage remediation ... 57
3.1 Résumé ... 57
3.2 Abstract ... 59
3.3 Introduction ... 61
3.4 Materials and methods ... 65
3.4.1 Sulfide mine tailing ... 65
3.4.2 Alkaline amendments ... 68
3.4.3 Methodology ... 68
3.4.3.1 Treatments ... 68
3.4.3.2 Use of chicken eggshell residues mixed with alkaline amendments as acid-neutralising agents ... 69
3.4.3.3 Buffering capacity of sulfide mine tailing amended with alkaline materials70 3.5 Results and discussion ... 71
3.5.1 Use of chicken eggshell residue mixed with alkaline amendments as acid-neutralizing agent ... 71
3.5.2 Buffering capacity of sulfide mine tailing amended with alkaline materials ... 79
3.6 Conclusions ... 83
Chapter 4 : Electro-activation technology in AMD treatment ... 85
4.1 Résumé ... 85
4.2 Abstract ... 87
4.3 Introduction ... 89
4.4 Materials and methods ... 92
4.4.1 Mining tailing and eggshell ... 92
4.4.2 Separator membranes ... 92
4.5 Methodology ... 94
4.5.1 Removal of Fe(II) from aqueous FeSO4 solution by electro-activation process 94 4.5.2 Neutralization of acid aqueous sulfide mine tailing and precipitation of toxic metals using electro-activation process ... 99
4.5.2.1 Experiment 1: Electroneutralization of acid sulfide mine tailing under variable electro-activation conditions including the amount of solid tailing to be treated ... 99
4.5.2.2 Experiment 2: Electroneutralization of acid aqueous sulfide mine tailing previously treated with 0, 4 and 10% chicken eggshell (CES) under variable electro-activation conditions ... 101
4.5.2.3 Experiment 3: Electroneutralization of acid aqueous sulfide mine tailing previously treated with 4% chicken eggshell residue under variable cathodes and membrane type conditions ... 103
4.6 Results and discussion ... 106
4.6.1 Removal of Fe(II) from aqueous FeSO4 solution by electro-activation process 106 4.6.1.1 Evolution of pH of electrolyte solutions ... 107
4.6.1.2 Concentration of Fe in anolyte and central solution ... 110 4.6.2 Neutralization of acid aqueous sulfide mine tailing and precipitation of toxic metals using electro-activation process ... 118
4.6.2.1 Experiment 1: Electroneutralization of acid sulfide mine tailing under variable electro-activation conditions including the amount of solid tailing to be treated ... 118 4.6.2.2 Experiment 2: Electroneutralization of acid aqueous sulfide mine tailing previously treated with 0, 4 and 10% chicken eggshell (CES) under variable electro-activation conditions ... 129 4.6.2.3 Experiment 3: Electroneutralization of acid aqueous sulfide mine tailing previously treated with 4% chicken eggshell residue under variable cathodes and membrane type conditions ... 143
4.6.2.3.1 General results ... 143 4.6.2.3.2 Electro-activation process using cation-exchange membrane as a
separator ... 144 4.6.2.3.3 Electro-activation process using nanofiltration membrane as a separator ... 145 4.6.2.3.4 Electro-activation process using anion-exchange membrane as a
separator ... 149 General conclusions ... 153 References ... 157
Index of tables
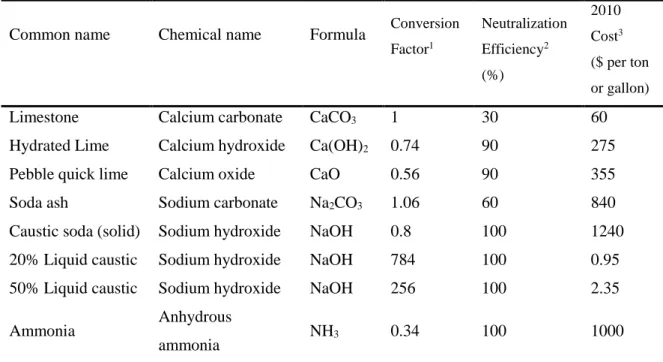
Table 1.1. Chemicals for AMD neutralization (Bise, 2013) ... 34
Table 1.2. Characteristics of chemical compounds used in AMD treatment (Jacobs et al., 2014). ... 35
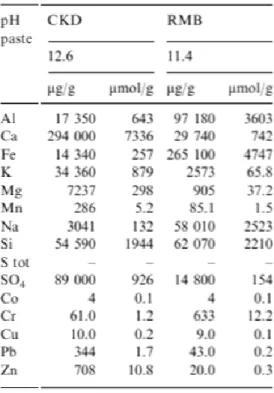
Table 1.3. Element composition of cement kiln dust and red mud bauxite (Doye and Duchesne, 2003). ... 44
Table 3.1. Chemical composition of the Solbec-Cupra tailing (Karam and Guay, 1994) .... 67
Table 3.2. Chemical composition (%) of alkaline amendments ... 68
Table 3.3. Experimental treatments ... 69
Table 3.4. Effect of CES mixed with four alkaline amendments on pHTS of aqueous tailing suspensions ... 76
Table 3.5. Values of pHAA of aqueous treated tailing suspensions before and after addition of 0.025 mmol of acid ... 80
Table 4.1. Composites of ion-exchange membranes MK-40 and MA-40 (Saldadze et al., 1960; Dyomina et al., 2002; Berezina et al., 2008). ... 93
Table 4.2. Manufacturer characteristics of ion-exchange membranes (Pokonova, 2007; Shekino-AZOT, 2015) ... 94
Table 4.3. Experimental parameters studied in experiment 1. ... 101
Table 4.4. Experimental parameters studied in experiment 2. ... 103
Table 4.5. Experimental parameters studied in experiment 3. ... 106
Table 4.6. Analysis of variance on the influence of current intensity and anode-CEM distance on the pH values of anolyte and electrolyte solution in central compartment at t = 120 minutes. ... 107
Table 4.7. ANOVA results (F-value) on concentration of total Fe in central and anolyte compartments at two electro-activation times. ... 110
Table 4.8. ANOVA results (F-value) on the influence of current intensity, solid SMT:water ratio, and rotation speed of stirrer on the pH values of catholyte at six electro-activation times. ... 119
Table 4.9. A partial activity series of elements (Lide, 2000). ... 123
Table 4.10. ANOVA results (F-value) on the influence of CES content of SMT, active membrane surface, cathode-AEM distance on the pH values of catholyte at six electro-activation times. ... 130
Table 4.11. ANOVA results (F-value) on the influence of membrane type, voltage and electrode material, on the pH values of catholyte at six electro-activation times. ... 144
Table 4.12. Values of initial pH and pHcatholyte at the end of electro-activation process .... 147
Table 4.13. Values of initial pH and pHcatholyte at the end of electro-activation process using AEM and variable voltage values and electrode materials. ... 151
Index of figures
Figure 1.1. Acid mine drainage process (MWC, 2006) ... 7
Figure 1.2. Sulfur transformations. 1) Chemolitotrophic oxididation, 2) Phototrophic and chemotrophic oxidation. Adapted from Muyzer and Stams (2008) ... 12
Figure 1.3. Schematic representation of the mechanism of pyrite oxidation in carbonate solutions (Caldeira et al., 2010) ... 22
Figure 1.4. Scheme of dry cover in the abandoned Kettara mine, Marocco (Bosse et al., 2013) ... 27
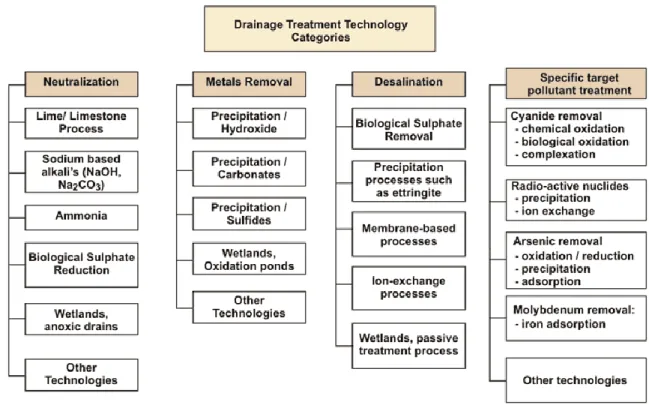
Figure 1.5. Classification of mine drainage treatment technologies (INAP, 2014) ... 29
Figure 1.6. Scheme of a mine-waste impoundment with a combined remediation complex (Blowes et al., 2014) ... 32
Figure 1.7. Pond treatment (Aubé and Zinck, 1999) ... 37
Figure 1.8. Conventional treatment plant (Aubé and Zinck, 1999) ... 38
Figure 1.9. Solubility of the common heavy metal hydroxides in dependence of pH (HEI, 2012) ... 42
Figure 1.10. Schematic representation of electro-activation apparatus ... 47
Figure 3.1. The Solbec-Cupra site (position A) ... 65
Figure 3.2. Oxidized sulfide tailings on the surface of the site (1994) ... 66
Figure 3.3. Yellow and grey colored sulfide tailings (Karam and Guay, 1994) ... 66
Figure 3.4. Effect of alkaline amendments on tailing pH ... 72
Figure 3.5. Tailing pH values as a function of time for different rates and sources of alkaline amendments ... 74
Figure 3.6. Potentiometric titration curves of acid sulfide mine tailing ... 83
Figure 4.1. The schematic diagram of electro-activation cell units ... 96
Figure 4.2. Plates that function as the anode and the cathode electrodes ... 96
Figure 4.3. Cation- and anion-exchange membranes ... 97
Figure 4.4. The experimental set-up of the reactor ... 97
Figure 4.5. Schematic representation of the two-compartment cell units ... 100
Figure 4.6. Schematic representation of the two-compartment cell units using CEM (a) and NFM (b) ... 105
Figure 4.7. Evolution of pH of solution in the anode (A) and central (B) compartments as a function of electro-activation time ... 108
Figure 4.8. Evolution of Fetotal concentration in the anolyte as a function of electro-activation time ... 111
Figure 4.9. Evolution of Fe2+ concentration in the central solution as a function of electro-activation time ... 112
Figure 4.10. Precipitation of Fe(OH)3 on the CEM from the anode side. (A): Anode-CEM distance = 3 cm and I = 150 mA; (B): Anode-CEM distance = 5 cm, I=250 mA and t=120 minutes ... 114
Figure 4.11. Scanning electron microscope image of two zones: A and B (Fe(OH)3 – light crystals) ... 115
Figure 4.12. EDS spectrum of the grey colored zone (Figure 4.11) ... 116
Figure 4.13. EDS spectrum of white crystal (Figure 4.11) ... 116
Figure 4.14. Precipitation of Fe on CEM (anode side) surface under the following conditions: Anode-CEM distance = 5 cm, V = 126 volts, t = 6 minutes ... 117
Figure 4.15. Effect of the current intensity on pHcatholyte values as a function of
electro-activation time. R = 0.1:1, 0.2:1 and 0.3:1 ... 120 Figure 4.16. Effect of the amount of SMT in cathode compartment on pHcatholyte as a
function of electro-activation time. I = 50 mA, 100 mA and 150 mA ... 121 Figure 4.17. Effect of current intensity on voltage as a function of electro-activation time. R = 0.1:1, 0.2:1 and 0.3:1... 126 Figure 4.18. Effect of solid SMT/water ratios on voltage as a function of electro-activation time. I = 50mA, 100mA and 150 mA ... 127 Figure 4.19 Evolution of sulfur concentration in the cathode compartment ... 129 Figure 4.20. Effect of the CES rate in SMT in cathode compartment on pHcatholyte values as
a function of electro-activation time. DC = 3, 4 and 5 cm. AS = 25% ... 131
Figure 4.21. Effect of the CES content of SMT in cathode compartment on pHcatholyte values
as a function of electro-activation time. DC = 3, 4 and 5 cm. AS = 50% ... 132
Figure 4.22. Effect of the CES content of SMT in cathode compartment on pHcatholyte values
of catholyte as a function of electro-activation time. DC = 3, 4 and 5 cm. AS = 100% ... 133
Figure 4.23. Effect of DC on pHcatholyte values as a function of electro-activation time. CES
rate = 0, 4 and 10%. AS = 25% ... 134 Figure 4.24. Effect of DC on pHcatholyte values as a function of electro-activation time. CES
rate = 0, 4 and 10%. AS = 50% ... 135 Figure 4.25. Effect of DC on pHcatholyte values as a function of electro-activation time. CES
rate = 0, 4 and 10%. AS = 100% ... 136 Figure 4.26. Evolution of voltage as a function of electro-activation time. DC = 3, 4 and 5
cm. AS = 50% ... 138 Figure 4.27. Evolution of voltage as a function of electro-activation time. CES = 0, 4 and 10%. Dc = 3 cm ... 139
Figure 4.28. Sulfur concentration in the catholyte under variable electro-activation conditions at the end of electro-activation treatment. Initial solution = catholyte before starting electro-activation ... 141 Figure 4.29. Calcium concentration in the catholyte under variable electro-activation conditions at the end of electro-activation treatment. Initial solution = catholyte before starting electro-activation ... 142 Figure 4.30. Evolution of pHcatholyte as a function of electro-activation time under variable
conditions using CEM as a separator and a stainless steel cathode material ... 145 Figure 4.31. Evolution of pH as a function of electro-activation time under variable
conditions using nanofiltration membrane as a separator and a stainless steel cathode material ... 146 Figure 4.32. Evolution of current intensity as a function of electro-activation time when applying three different voltages (15 V, 30 V and 60 V) under variable conditions using nanofiltration membrane as a separator ... 147 Figure 4.33. Sulfur concentration in catholyte at the end of electro-activation treatment, using nanofiltration membrane and stainless steel cathode, as a function of voltage. Initial solution = catholyte before starting electro-activation ... 149 Figure 4.34. Calcium concentration in catholyte at the end of electro-activation treatment, using nanofiltration membrane and stainless steel cathode, as a function of voltage. Initial solution = catholyte before starting electro-activation ... 149
Figure 4.35. Evolution of pH as a function of electro-activation time under variable conditions using anion-exchange membrane as a separator and a stainless steel cathode material ... 150 Figure 4.36. Evolution of current intensity as a function of electro-activation time when applying three different voltages (15, 30 and 60 V) under variable conditions using AEM as a separator ... 151
List of abbreviations
A: identification (electro-activation treatment with AEM) AA: alkaline additions
AC: alkaline carbonate
AEM: anion-exchange membrane ALDs: anoxic limestone drains AMD: acid mine drainage AS: active membrane surface, % BC: buffer capacity
C’: electrolyte concentration, mol/L CAL: calcitic limestone
CCM: calcareous products
CEM: cation-exchange membrane COP: coquilles d’œufs de poule CES: chicken eggshell residue CKD: cement kiln dust
CPC: ciment riche en produits calcaires
D: diffusion constant of electrolytes dissolving in a solution, cm2/s DC: direct current
DA: anode-membrane distance, cm
DC: cathode-membrane distance, cm
DMA: drainage minier acide DOL: dolomitic limestone
ECCE: effective calcium carbonate equivalent EDS: energy-dispersive X-ray spectroscopy F: Faraday constant, 9.64853399(24)x104 C mol−1 I: current intensity, mA
ilim: limiting current density
KU-2: cation-exchange resin LD: limestone drain
LR: lime requirement
MA-40: anion-exchange membrane MGO: magnesium oxide
MK-40: cation-exchange membrane MT: mine tailing
N: identification (electro-activation treatment with nanofiltration membrane) N30F: nanofiltration membrane
NFM: nanofiltration membrane OLCs: open limestone channels OM: organic matter
pHAA: pH of aqueous alkaline amended tailing suspensions
pHTS: pH of tailing suspension
pHS: pH de suspensions
R: SMT/water ratio in the cathode compartment, w/v R’: resistance, ohm
RMB: red mud bauxite
RMA: résidus miniers générateurs d’acide RMS: résidus miniers sulfurés
SAPS: successive alkalinity producing systems SMT: sulfide mine tailing
t: time of treatment, min T: treatment
t-- and t-: the transport number of anions in the anion-exchange membrane and in the
solution
TS: tailing suspension
V: voltage voltage of the electro-activation system, V δ: boundary layer thickness, cm.
Introduction
As it is known, oxidation of sulfide minerals, particularly pyrite, which is the most abundant sulfide mineral on the planet (Johnson and Hallberg, 2005), is the major cause of acid mine drainage (AMD) generation (Ritcey, 1989). Sulfide minerals are unstable when exposed to air (oxygen) and rainfall (water), and begin to react almost immediately. This reaction yields high level of sulfuric acid and causes the leaching of heavy metals, in concentrations which may be toxic to aquatic life (Sheoran and Sheoran, 2006; Sadak, 2008).
For many years, the Canadian mining industry is facing to management of large amounts of sulfidic mine tailings. In Québec, the regulations of the Ministère du Développement Durable, de l’Environnement et de la Lutte contre les Changements Climatiques (MDDELCC) require that acid mine tailings do not contaminate surface water and groundwater. Since the adoption of the policy of rehabilitation of contaminated land by the MDDELCC in 1988, the restoration of degraded sites has become a government priority. Acid mine drainage is of particular concern in Canadian mine areas because its high level of acidity, heavy metals (iron, aluminium, copper, and manganese, and possibly other heavy metals) and metalloids (of which arsenic is generally of greatest concern) has adverse effects in the terrestrial and aquatic environments.
The most widespread method used to treat acidic mine tailing effluents or to mitigate the generation of AMD, is an active treatment process involving addition of a chemical- neutralizing agents. There are a large number of alkaline compounds that have been used in neutralizing acid mine drainage or acid generated from sulfidic wastes. These include agricultural lime, dolomite, fly ash, lime marl, soda ash, red mud, unactivated attapulgite and by-product lime from various industrial processes (Adams et al., 1972; Mays and Bengtson, 1978; Ritcey, 1989; Bellaoui et al., 1996; Skousen et al., 2000; Potgieter-Vermaaka et al., 2006; Karam et al., 2009; Fytas, 2010; Falayi and Ntuli, 2014). However, information on acid neutralization of sulfide mine tailings using eco-friendly agro-waste such as chicken eggshells which are rich in calcium carbonate is quite limited.
In the context of sustainable development, there is an increasing interest in the recycling of agrifood waste with high concentration of calcium carbonate such as chicken eggshells.
Canada produces a significant amount of poultry eggshells that are composed primarily of calcium carbonate, which is highly alkaline and neutralizes acid. Eggshells are waste materials from hatcheries, homes and fast food industries (Amu et al., 2005; King’ori, 2011). In 2007, Canadian production of eggs for consumption reached 521.1 million dozen (EFC, 2008). Canada has approximately 1,100 egg farmers, the majority of farms are located in Quebec and Ontario (Farm Credit Canada, 2012). Poultry eggshells are all at once: i) a problem due to the need to manage and ii) a potential source of neutralizing substances and calcium.
Several studies have shown that eggshells could be employed : (i) as a natural and eco-friendly adsorbent material for the removal of either organics (Arami et al., 2006; Zulfikar and Setiyanto, 2013) or heavy metals from aqueous solutions (Rao et al., 2010; Nduwayezu et al., 2013; Ipeaiyeda and Tesi, 2014; Bolboli et al., 2015), (ii) as an alternative soil stabilizer like lime (King’ori, 2011) or as subgrade material for road construction (Olarewaju et al., 2011). Relatively little research has been devoted to assess short-term effect of chicken eggshell waste on acid neutralization and chemical precipitation of heavy metals. In particular, little is known about the effectiveness of chicken eggshell mixed with by-product alkaline materials such as cement concrete rich in calcareous products (Portland cement) or magnesium oxide for acid sulfide mine tailing remediation.
Although most of the research to date has focused on using conventional active and passive treatments, evidence was presented in few studies suggesting that electrochemically activating water technology using ion-exchange membranes could remove metals from AMD (Bunce et al., 2001; Chartrand and Bunce, 2003). The base of electro-activated aqueous solution is electrolysis and it is possible to produce acidic, neutral or alkaline solution at electrode compartment (pH 2 – 12). Electrochemical applications basically use an electrochemical cell with two electrodes submerged in an electrolyte (Doering et al., 2001). Catholyte is an alkaline solution (pH 7 – 12) with a high reduction potential (Marais and Williams, 2001). Electrolysis remediation is often regarded as environmentally friendly technology for remediating sites polluted by heavy metals. However, little is known about the effectiveness of electro-activation for neutralizing tailing acidity and the influence of electro-activation conditions (e.g. current intensity, active membrane surface, distance
between the electrode and the membrane (ion-exchange or nanofiltration membrane), electrode materials and solid/water ratio) on neutralization of sulfide mine tailings amended with chicken eggshell.
This thesis investigates the application of alkaline amendments (chapter 3) and electro-activation technology (chapter 4) in remediation of acid sulfide mine tailings.
Chapter 1 : Review of literature
1.1 Importance of Canadian mine activity
Mining is an important economic activity in many developed and developing countries. Internationally, Canada is one of the leading mining countries and one of the largest producers of minerals and metals. Quebec is a great place for mining (Marshall, 2013). Mining and exploration activities contribute significant economic and social benefits to Canada's provinces and territories (MiningFacts.org, 2012a; Marshall, 2013). Mining also remains an important source of employment in Canada.
1.2 Impacts of mining on environment
The Canadian mineral industry generates annually a significant amount of waste rock and tailings. After being removed, waste rock and tailings are usually stored above ground in large free-draining piles. According to information of the mining association of Quebec, overburden, waste rock and mine tailings were disposed on the surface which was equal 8000 hectares in 1991 (Doye, 2005). As most waste rock and tailings contain metal sulfide minerals, the production of acidic metal-rich mine drainage waters (AMD) generated from sulfide mine tailing piles, as a result of weathering and oxidation of sulfides, constitute an environmental problem to the immediate environment and receiving waters (Ritcey, 1989). AMD severely degrades water quality, and can kill aquatic life and make water virtually unusable (SDWF, 2015).
1.3 Acid mine drainage generation
1.3.1 General consideration
Metal sulfide minerals, primarily pyrites and other sulfide ores, can be oxidized when they come in contact with oxygen (O2) and water (Ritcey, 1989). The oxidation reaction produces
significant amount of sulfuric acid (H2SO4) reducing tailing pH and generating metal-rich
Acidity in AMD is comprised of hydrogen ion acidity or H2SO4 and mineral acidity (iron,
aluminum, manganese, and other metals depending on the specific geologic setting and metal sulfide (Skousen et al., 1996).
The primary factors directly involved in the formation of AMD are the presence, abundance and reactivity or tendency of sulfide minerals to oxidize. The sulfides oxidation reactions are catalyzed by the iron- and sulfur-oxidizing microorganisms (Jacobs et al., 2014). Oxidation of sulfides can be provided by oxygen from the atmosphere and by oxygen that is solubilized in the seepage waters as well as oxidizing inorganic components present in tailings, such as ferric iron, manganese, nitrates, etc. (Ritcey, 1989). The sulfides responsible for acid generation include (Ritcey, 1989; Udayabhanu and Prasad, 2010): pyrite (FeS2), pyrrhotite
(FexSs), arsenopyrite (FeAsS2), chalcopyrite (CuFeS2), sphalerite (ZnS), galena (PbS),
chalcosite (Cu2S), covelite (CuS), molybdenite (MoS2), millerite (NiS).
There are four processes by which sulfides may be oxidized (Ritcey, 1989): 1) chemical oxidation, 2) biological (bacterial) oxidation, 3) electrochemical oxidation, and 4) a combination of chemical, bacterial and electrochemical oxidation.
If uncontrolled, the AMD may runoff into streams or rivers or leach into groundwater (Coil et al., 2014). The resulting fluids may be highly toxic and, when mixed with groundwater, surface water and soil, may have harmful effects on humans, animals and plants. Acid mine drainage can develop at several points throughout the mining process: in underground workings, open pit mine faces, waste rock dumps, tailings deposits, and ore stockpiles. Acid generation can last for decades, centuries, or longer, and its impacts can travel many miles downstream. Roman mine sites in Great Britain continue to generate acid drainage 2,000 years after mining ceased (Figure 1.1) (MWC, 2006). That is why AMD is a priority ecological problem for many countries (Leopold and Lapakko, 2001).
Figure 1.1. Acid mine drainage process (MWC, 2006)
1.3.2 Chemistry of pyrite oxidation
1.3.2.1 Geochemistry of iron
Fe is one of the most abundant elements in the lithosphere, and its common range in magmatic rocks is 1.4 to 10% (Kabata-Pendias, 2011). Its highest concentrations are usually associated with ultramafic rocks.
In nature, Fe readily undergoes oxidation or reduction, depending upon prevailing environmental physicochemical parameters, such as pH, oxygen concentration, availability of electrons and redox potential (Eh) (Ritcey, 1989; Stumm and Morgan, 1996; Hedrich et al., 2011; Kabata-Pendias, 2011). Its oxidized and reduced forms are referred to as ferric (Fe3+) and ferrous (Fe2+) iron, respectively. Fe readily participates in subsurface redox reactions (Vance, 1994).
The reactions of Fe in processes of weathering are dependent largely on the availability of electrons, the Eh-pH system of the environment and on the stage of oxidation of Fe compounds involved (Kabata-Pendias, 2011). Redox reactions involving Fe and S can be catalyzed by enzymes (Bohn et al., 1985). Oxygen has a great affinity for electrons and is the primarily and strongest electron acceptor in nature (lithosphere) because it generates the
greatest Gibbs free energy change and produces the most energy. Under aerobic conditions, it accepts electrons according to the following reaction (Ebsworth et al., 2013):
O2 + 4 e- + 4H+ ↔ 2H2O (Eh0 = 1,229V) [1.1]
Oxidizing agents have high electrode potentials and are electron acceptors. In aqueous solutions O2 is thermodynamically a powerful oxidizing agent (Ebsworth et al., 2013). In
chemical industries, atmospheric O2 is by far the cheapest oxidizing agent since other
oxidizing agents such as H2SO4, HNO3, MnO2 and H2O2 need O2 for their preparation,
resulting in higher investment (Nayak and Rao, 2003).
Oxygen is supplied to the surface water layers and soils primarily from the air (Rowell, 1981). However, some biological processes occurring in aerobic soils could oxidize water under dark conditions releasing O2 (Fleischer et al., 2013). It has been suggested that O2 is produced
by dismutation of nitric oxide (NO) in anaerobic methane oxidisers (Ettwig et al., 2010; Ettwig et al., 2012). NO and nitrous oxide (N2O) in soils are among nature's most powerful
electron acceptors (Ettwig et al., 2012). NO dismutase has been involved in O2 production in
soils containing nitrate and nitrite (Fleischer et al., 2013).
Oxic conditions occur when the supply of O2 exceeds the consumption (Dillmann et al.,
2013). Aerobic conditions prevails in aerated soils, well drained soils, coarse-textured soils, areas with a low water table, and any other waterlogged or flooded media.
Ferrous iron (Fe2+) is a soluble form of Fe that is stable at extremely low values of pH or
under anaerobic conditions (Boundless.com, 2014). Anaerobic conditions prevails in aquatic ecosystems and terrestrial ecosystems such as wetland soils, paddy soils, organic soils, poorly drained soils, clayed soils, areas with a high water table, soils amended with heavy rates of organic materials, and any other waterlogged or flooded media (Inglett et al., 2005).
Under aerobic and moderate pH conditions, Fe2+, as Fe(OH)2, is susceptible to spontaneous
chemical oxidation by molecular oxygen (O2), which acts as electron acceptor, to the ferric
form, according to simple rate law following the reaction path (Nayak and Rao, 2003): Fe(OH)2 + ¼O2 + ½H2O → Fe(OH)3 [1.2]
In natural water, the rate at which the spontaneous chemical oxidation occurs depends on the temperature and concentrations of protons (pH), dissolved oxygen and Fe2+ (Stumm and
Morgan, 1996):
[1.3]
where k is a temperature-dependent constant (3×10−12 mol. l−1 min−1 at 20 °C).
Oxidation of Fe2+ is easily carried out by contact with air (Wong, 1984) or by oxygenation. During oxidation of Fe2+ salt aqueous solutions, poor soluble compounds including Fe3+ oxides are formed (Domingo et al., 1994). Alicilar et al. (2008) studied the air oxidation of Fe2+ ions in water. While the maximum yield of 86 % is catalytically achieved by blowing air at a neutral medium, the oxidation was almost completed in an alkaline solution even at stationary atmosphere. The reaction was first order with respect to Fe2+.
In aerobic soils, Fe(II) minerals in parent material slowly oxidize spontaneously (Bohn et al., 1985). In most environments, rates of spontaneous chemical oxidation of Fe2+ are very low at pH <4, though these become appreciably greater at higher pH values (Hedrich et al., 2011). The ferric iron (Fe3+) is the stable oxidative state of Fe in aerobic conditions and
predominates under strongly acidic and oxidizing conditions (Bohn et al., 1985). Under anaerobic conditions, Fe3+ acts as an electron acceptor (oxidant) and is reduced to Fe2+.
The reduction of Fe(OH)3 in anaerobic soils can be considered in two steps. Firstly, Fe(OH)3
dissolves and Fe3+ goes into solution, and then Fe3+ is reduced to Fe2+ (Rowell, 1981): Fe(OH)3(s) ↔Fe3+ + 3OH- [1.4]
Fe3+ + e- ↔ Fe2+ [1.5]
Although ferric iron is thermodynamically stable in aerated waters, its strong tendency to hydrolyse (react with water). At the pH of typical agricultural soils, Fe3+ ion is hydrolyzed to Fe(OH)2+ (Bohn et al., 1985), so the reduction reaction of dissolved Fe(III) is more realistically.
Fe(OH)2+ + e- + 2H+ ↔ Fe2+ + 2H
2O [1.6]
Ferric iron can be reduced to Fe2+ by strong reducing agents in the medium. Iron reducing
agents in soils are humic acid, ferrous iron, and sulfide demonstrated that the reducing agents, particularly sulfides, in soils may affect redox chemistry and soil pH, ultimately affecting the electrokinetic remediation process. Ferrous hydroxide generated from the spontaneous chemical oxidation is susceptible to hydrolyze abiotically to insoluble ferric hydroxide (Fe(OH)3) (Boundless.com, 2014).
In soil system, ferric hydroxide (Fe(OH)3) is the direct result of Fe2+ oxidation and
precipitation. The time required for uncomplexed ferrous iron to undergo oxidation to the ferric state is dependent on many factors, the dominant being: pH; temperature; dissolved oxygen level; and the presence of other soluble ions (Vance, 1994). The oxidation of Fe2+ to Fe3+ is accelerated with increasing pH and temperature (Vance, 1994). With time, ferric hydroxide in soils could be transformed into amorphous hydrous ferric oxide (Fe2O3·XH2O),
maghemite (γ-Fe2O3), lepidocrocite (γ-FeOOH), hematite (α-Fe2O3), and goethite
(α-FeOOH) (Vance, 1994). Jones et al. (2014) investigated the kinetics of Fe(II) oxidation in the presence of the iron oxyhydroxides ferrihydrite, Si-ferrihydrite, schwertmannite, lepidocrocite and goethite over the pH range 4 – 5.5. Despite limited sorption of Fe(II), the rate of Fe(II) oxidation was up to 70-fold faster than in the absence of any Fe oxyhydroxide phase over pH 4.5 – 5.5. Enhanced Fe(II) oxidation was minor or negligible at pH 4 with undetectable amounts of Fe(II) adsorbed to the iron oxyhydroxides at this pH.
The general rule governing the mobilization and fixation of Fe in lithosphere are that acid and reducing conditions promote the dissolution of Fe compounds, whereas oxidizing and alkaline conditions promote the precipitation of Fe (Kabata-Pendias, 2011). Under reducing conditions, the stable oxidation states are Fe2+ and sulfide. Under oxidizing conditions, O2 is
available to take up electrons and reduce electron availability and the stable oxidation states are Fe3+ and SO42- (Kabata-Pendias, 2011). In sulfate soils, pyrite is oxidized chemically at
high pH, but at pH values below 4.5, it is mediated microbially (Rowell, 1981). When sulfate containing soils are waterlogged, anaerobic conditions prevail, and consequently, the reduction of Fe3+ to Fe2+ takes place (Bohn et al., 1985).
Another important aspect of iron chemistry, is its use by prokaryotes as a source of energy. Most species of iron-oxidizing bacteria are affiliated with the Proteobacteria. The latter can be subdivided into four main physiological groups (Hedrich et al., 2011): (i) acidophilic, aerobic Fe oxidizers; (ii) neutrophilic, aerobic Fe oxidizers; (iii) neutrophilic, anaerobic (nitrate-dependent) Fe oxidizers; and (iv) anaerobic photosynthetic Fe oxidizers. Some species (mostly acidophiles) can reduce Fe(III) as well as oxidize Fe(II), depending on prevailing environmental conditions.
The acidophilic iron-oxidizing proteobacteria as well as sulfur-oxidizing bacteria have been the focus of a great deal of research since 1940, primarily because of their role in generating acidic and metal-enriched mine drainage waters (Hedrich et al., 2011). The most widely studied of all iron oxidizers is the acidophile Acidithiobacillus ferrooxidans and Thiobacillus
thiooxidans, a sulfur-oxidizing bacteria. A comprehensive review of the mechanisms of
biotic oxidation of Fe and pyritic tailings by both iron-oxidizing bacteria and sulfur-oxidizing bacteria have been reported by Ritcey (1989) and Hedrich et al. (2011).
1.3.2.2 Formation of pyrite
Iron sulfides, as well as occurring as accessory minerals in many common rocks, are major phases in the polymetallic sulfide ore deposits that are still the main suppliers of many base (Cu, Pb, Zn), ferroalloy (Co, Mn, Ni), and rare or precious (Ag, Au, Ga, Ge, Pt) metals (Lennie and Vaughan, 1996).
Among iron sulfide minerals, pyrite (ferrous disulfide) is the most stable form of iron sulfide mineral and is by far the most common and abundant sulfide mineral in most soils and sediments (Fanning, 2002; Rabenhorst, 2011). Pyrite and other sulfide minerals are found primarily in bituminous coal mines, metal sulfide ore bodies, pyritic coal spoil, lignite, soils of coastal marshes, acid sulfate soils (cat clays) and waterlogged environments, such as mangrove swamps or estuaries and wetlands. Acid sulfate soils are formed during aeration of marsh muds and are widespread around coastal regions, and are also locally associated with freshwater wetlands and saline sulfate-rich groundwater in some agricultural areas (wikipedia.org, 2015). Acid sulfate soils are described by Rowell (1981); Fanning (2002); Rabenhorst and Fanning (2002).
Pyrite is often discarded together with low grade metal-containing rocks and wastes on mine tailings (Van Straaten, 2002). Pyrite that forms in soils and sediments as well as that found in many sulfide mine tailings most commonly occurs as raspberry-like framboids (Caruccio and Geidel, 1978; Fanning, 2002).
Historically, pyrite was formed under severely reducing conditions by the reduction of sulfates derived from the sea to sulfide by sulfate-reducing bacteria (SRB). The reduction of sulfate to sulfide occurs in nature under anaerobic conditions in the presence of sulfate, SRB and a source of organic carbon. SRB have a key role in both the S and C cycles (Muyzer and Stams, 2008). Under severely reducing conditions, several genera of SRB including Desulfovibrio, Desulfotomaculum, Desulfobacter, and Desulfococcus species (Melville and White, 2002; Muyzer and Stams, 2008) obtain oxygen for respiration through the reduction of sulfate ions in sea, groundwater, sediments and sulfate soils, producing hydrogen sulfide (H2S) (Fig. 1.2). Sulfate reducers can also reduce other sulfur compounds like sulfite, sulfur
or thiosulfate, to sulfide (Muyzer and Stams, 2008).
Figure 1.2. Sulfur transformations. 1) Chemolitotrophic oxididation, 2) Phototrophic and chemotrophic oxidation. Adapted from Muyzer and Stams (2008)
1 Oxic conditions 1 SO42- reduction 2 Anoxic conditions 2 S0 S0 H2S SO42
-In absence of O2, SRB obtains energy for growth by both oxidation of organic substances
and reducing sulfate. They are able to remove hydrogen atoms from the organic molecules (CH2O) and to use sulfate as the terminal electron acceptor of their electron transport chain
(Barton, 1995; Harmon et al., 2007; Muyzer and Stams, 2008). Dissimilatory sulfate reduction at low pH is a proton-consuming process, as illustrated in the following equation (Nancucheo and Johnson, 2011):
SO42− + 2CH2O + 2H+ → H2S + 2CO2 + 2H2O [1.7]
Environmentally important activities displayed by SRB are a consequence of the unique electron transport components or the production of high levels of H2S (Barton and Fauque,
2009). Most of them are anaerobic microorganisms, however there are examples of sulfate-reducing bacteria that are tolerant of oxygen. Under oxygenated conditions these bacteria switch to aerobic respiration before reducing sulfate (Muyzer and Stams, 2008).
Telang et al. (1994) used nucleic acid hybridization techniques to characterize the SRB at seven mine waste water and two soil sites in Canada. It was found that most SRB isolated from distinct sites are genomically different and differ also from SRB found in oil field production waters. Desulfotomaculum and Desulfotomaculum-like sequences were the most dominant SRB genes detected in contaminated groundwater associated with uranium mill tailings (Chang et al., 2001).
So while SRB are respiring, they produce sulfide that will sequester a variety of available metal cations (including Fe2+ ion) present in coastal environments into a highly insoluble
precipitate (Harmon et al., 2007; Shao et al., 2012) such as pyrite (Melville and White, 2002) and sometimes orthorhombic marcasite (Bloomfield, 1981). Fe enters coastal environments as iron oxides sorbed to the surface of clay and silt particles (Rabenhorst and Fanning, 2002). Under severely anaerobic conditions, iron oxides or Fe hydroxide minerals are reduced to Fe(II) which react with H2S to form ferrous monosulfide (mackinawite), and then slow
conversion to pyrite in presence of elemental sulfur (Berner, 1963; Rowell, 1981). Pyrite is the thermodynamically favored phase (Fanning, 2002). At temperatures of 20-30°C the conversion of FeS to FeS2 can require a few years to occur, assuming continuously reducing
formed in reducing solutions, would provide facile topotactic sites for pyrite or marcasite nucleation (Lennie and Vaughan, 1996). Changes in the environmental conditions, including pH, temperature and exposure to SRB, however, could alter the iron sulfide minerals to other crystalline structures such as greigite and pyrrhotite (Gander et al., 2013).
Mechanisms for pyrite formation may follow several possible pathway including: 2) partial oxidation of monosulfide, and 3) reaction of monosulfides with H2S 1) reaction of
monosulfide or Fe2+ with polysulfide such as greigite (Fe3S4). The reactions can be written
as follows:
Fe2+ + S2- → FeS (Rabenhorst and Fanning, 2002) [1.8] Fe(OH)2 + H2S → FeS + 2H2O (Rowell, 1981) [1.9]
FeS + (+ Sxy-, loss of ē, or + H2S) → FeS2 + various (Rabenhorst and Fanning, 2002) [1.10]
FeS + S0 + e- → FeS2 (Rowell, 1981) [1.11]
Lennie and Vaughan (1996) studied the transformation of synthetic iron monosulfides mackinawite (Fe1+xS) and "amorphous FeS" (fine-grained "X-ray amorphous" black
precipitate), the first formed iron sulfides in aqueous systems at low temperatures, using X-ray photoelectron and X-X-ray absorption spectroscopies. The predominant sulfidation sequence at low temperatures was as follows: [cubic FeS]/amorphous FeS → mackinawite → greigite → marcasite/pyrite.
Altschuler et al. (1983) studied the pattern of sulfur transformation in peat across the Everglades basin. They found that pyrite formation in organic-rich swamps depends on the use of organic oxysulfur compounds in dissimilatory respiration by sulfur-reducing bacteria. This paragenesis accounts for the occurrence of framboidal pyrite bound in fossil tissue in coal and sediments.
1.3.2.3 Weathering and oxidation of pyritic tailings under natural
conditions
Many base metal ores are sulfidic, and they frequently contain large concentrations of pyrite (FeS2), which, with other sulfide minerals, ends up mostly in the tailing wastes (Nancucheo
Pyrite and other sulfide minerals are potentially highly reactive minerals (Nancucheo and Johnson, 2011). In the presence of both air (oxygen) and water, pyrite oxidizes ultimately to soluble sulfate and ferric iron with the concomitant production of proton acidity, as illustrated in the equations 1.12 to 1.17.
Weathering of rocks is due to exposure to water, O2, and CO2 at concentrations different from
those in which the rock formed, and the major reaction that weathers minerals is the strong tendency of ions in solids to dissolve in water (Bohn et al., 1985). Pyrite is generally stable under reducing conditions (below the water-table), however, when the soil is drained and pyrite exposed to the atmosphere acidity is produced due to pyrite oxidation. In soil system, pyrite is oxidized chemically at high pH, but at pH values below 4.5, it is mediated microbially (Kabata-Pendias, 2011).
Basic of AMD chemistry is reaction of pyrite with oxygen and water producing a solution of ferrous sulfate (FeSO4) and (H2SO4). Further oxidation of ferrous ion (Fe2+) to ferric sulfate
(Fe2(SO4)3) and subsequent hydrolysis to Fe(OH)3 as well as the regeneration of Fe3+ ion
(which is reduced to Fe2+ on reaction with pyrite) produce additional acidity (Ritcey, 1989; Evangelou and Zhang, 1995; Johnson and Hallberg, 2005).
Presence of certain bacteria particularly ion and sulfur oxidizing bacteria catalyzes chemical oxidation of sulfides in the tailings (Silverman, 1967; Arkesteyn, 1979; Guay et al., 1989; Karavaiko et al., 1994; Nowaczyk and Domka, 2002) and facilitates reactions of acidification of mine wastes thereby increasing the rate of oxidation by several orders of magnitude (Nordstrom and Southam, 1997). A comprehensive review of the mechanisms of weathering and oxidation of pyritic tailings as well as the role of bacterially-assisted oxidation of pyritic tailings have been reported by Ritcey (1989).
The general chemical reactions explaining the oxidation of pyrite, the most ubiquitous sulfide mineral responsible for acid mine generation, are a heterogeneous reaction, and are given by the following steps (reactions 1 to 3) (Caruccio and Geidel, 1978; Ritcey, 1989).
1.3.2.3.1 Reaction 1: Oxidation of pyrite
In the presence of atmospheric air, i.e., oxygen (O2), and water, pyrite is oxidized and
breakdown to yield Fe2+ as ferrous sulfate, and sulfuric acid (Singer and Stumm, 1970) according to the following equation (Burns, 1987):
FeS2(s) + 7/2O2 + H2O → Fe2+ + 2H+ + 2SO42- → FeSO4 + H2SO4 [1.12a]
or
FeS2(s) + 8H2O → Fe2+ + 16H+ + 2SO42- + 14e- [1.12b]
This first step illustrates the conversion of solid mineral pyrite to dissolved Fe2+ and dissolved sulfate. It is considered as the primary mechanism by which the acid is released into water drainage (Akcil and Koldas, 2006; Abbassi et al., 2009). The stoichiometry of this reaction shows that 1 mole of FeS2 will produce 2 moles of H+ (acidity).
In a laboratory kinetic study of the oxidation of pyrite in acid aqueous suspension by molecular O2, McKay (1957) demonstrated that the oxidation reaction may be affected by at
least three parallel reactions. The predominant of these was the direct oxidation of S atoms from pyrite by molecular O2 dissolved in acid water as suggested by the following two
simultaneous paths, the first one is a rapid reaction, and the second a slow reaction:
(i) One molecule of O2 is chemisorbed rapidly on the pyrite surface, which is covered by a
mono-layer of O2, comprised of one O2 molecule at each FeS2 site, i.e.:
FeS2(s) + O2(aq) → FeS2·O2 [1.12c]
(ii) A second molecule of O2 is sorbed on an O2 covered site, i.e.
FeS2·O2 + O2(aq) → FeS2·2O2 → FeSO4 + S0 [1.12d]
A separate study of the aqueous oxidation of FeSO4 by molecular O2 revealed a second order
dependence on Fe2+ ion and a first order dependence on molecular O2, i.e., d [Fe2+] /dt = k
[Fe2+]2 [O 2].
In addition, sulfuric acid generated by the reaction of equation 1.12 (a, b) might oxidize pyrite in the absence of O2 to ferrous sulfate.
FeS2(s) + H2SO4 → FeSO4 + H2S + S [1.12e]
1.3.2.3.2 Reaction 2: Oxidation of the ferrous sulfate
If dissolved oxygen is present in the acidic water in sufficient quantity or when the acidic water has a contact with atmospheric oxygen, additional pyrite is oxidized (equation 1.13a) (Burns, 1987) and the dissolved Fe2+ ions generated by the reaction of equation 1.13 (a,b) can readily oxidize either in situ or downstream into Fe3+ (equation 1.13b) unless it is
otherwise immobilized (e.g., as a sulfide mineral) (Nancucheo and Johnson, 2011).
4FeS2 + 15O2 + 2H2O → 4Fe3+ + 8SO42- + 4H+ [1.13a]
Fe2+ + ¼O2 + H+ → Fe 3+ + ½H2O [1.13b]
The oxidation of Fe2+ to Fe3+ (equation 1.13b) consumes protons (i.e., H2SO4 is consumed in
the reaction) which happens at all pH values (Nordstrom, 2007). This step is referred to as rate determining step for the overall sequence (Singer and Stumm, 1970), thus indicating that the oxidation rate is of the first order with respect to both the concentration of Fe(II) and dissolved O2. Stumm and Lee (1961) reported that 50% of the acidity in AMD arises from
the oxygenation of Fe(II) ions, and reminder arises from the oxygenation of sulfide. The rate of oxygenation of Fe2+ ions was found to be proportional to the concentration of Fe2+, strongly influenced by pH and dependent on the partial pressure of O2 (Stumm and Lee,
1961).
The Fe3+ ion produced will precipitate and hydrolyze, with concomitant net production of protons (equation 1.14). It has been shown that the pyrite can also be oxidized in the presence of excess Fe3+ generated by the reaction of equation 1.13b (Ritcey, 1989) further hydrolyze to form additional acidity.
2Fe3+ + FeS2 → 3Fe2+ + 2S0 [1.13c]
Fe2(SO4)3 (equation 1.13a) could also contribute to the oxidation of pyrite (McKay, 1957)
according the following equation (Ritcey, 1989): S2- + Fe
The elemental sulfur produced in reactions 1.13c and 1.13d can be oxidized by Thiobacillus
thiooxidans and T. ferrooxidans as follows (Ritcey, 1989):
S0 + 3O2 + 2H2O → 2SO42- + 4H+ [1.13e]
The oxidation of pyrite in aqueous ferric sulfate was also given by the following chemical reaction (Zheng et al., 1986):
FeS2 + 14Fe3+ + 8H2O → 15Fe2+ + 2SO42- + 16H+ [1.13f]
The reaction 1.13f was found to be kinetically limited and the oxidation rate for the pyrite depended on the total Fe concentration and on ratio of Fe3+/Fe2+ under constant acid concentration (Zheng et al., 1986).
Most researches and authors reported that the Fe2+ ion in this step (equation 1.13b) is only chemically very slowly oxidized, but accelerated in presence of some bacteria.
1.3.2.3.3 Reaction 3: precipitation of Fe(III)
The hydrolysis and precipitation of a Fe(OH)3 phase produces protons (Nordstrom, 2007):
Fe3+ + H2O → Fe(OH)2+ + 3H+ [1.14a]
Fe3+ + 3H2O → Fe(OH)3(s) + 3H+ [1.14b]
When the insoluble form of Fe(III) precipitates as a hydroxide (red orange), three additional hydrogen ions are generated for each dissolved iron ion which precipitates. The hydroxide ions thus are removed from solution and the water becomes more acid through the process of hydrolysis (ASTM, 1969).
It is known that Fe(III) salts including sulfates hydrolyze strongly in aqueous solution to give acid solutions (Discher and Medwick, 2006). The degree of hydrolysis of Fe3+ ions in aqueous solution is a function of pH. As pH increases, protons are lost from waters of hydration in a step-wise fashion (Diz et al., 2006). Nordstrom (2000) used PHREEQCI program to simulate pyrite oxidation by adding increments of O2 to pyrite in water and stated
of hydrolysis, i.e., near or above a pH = pK1 = 2.2 for Fe3+ hydrolysis (Nordstrom, 2007). At
pH 2.2, no hydrolysis occurs and the pH can only increase on oxidation.
Fe(OH)3 also may oxidize additional pyrite to produce more ferrous and hydrogen ions as
illustrated in equation 1.15.
14Fe3+ + FeS2 (s) + 8H2O → 2SO42- + 15Fe2+ + 16H+ [1.15]
The character of the reactions 1.14 and 1.15 is spontaneous or bio-catalytic. The microorganisms play a role of catalytic agents that derive energy from the oxidation reaction (Jennings et al., 2008).
If Fe(II) is produced (equation 1.15) in presence of enough quantity of dissolved oxygen, reactions 1.13 and 1.14 are put into the cycle. The absence of dissolved oxygen promotes the equation 1.15 that will continue to completion and water environment will be characterized by elevated concentration of ferrous iron (Younger et al., 2002). In general, the regeneration of Fe3+ ion (which is reduced to Fe2+ on reaction with pyrite) is the key reaction in promoting the ongoing oxidation of the pyrite.
In the presence of bacteria, particularly Thiobacillus ferrooxidans and Thiobacillus
thiooxidans, the rates of chemical reactions (equations 1.12, 1.13, and 1.14) can be
significantly accelerated. Thiobacillus ferrooxidans is the most important biological catalyst agent responsible for the fast oxidation process of the pyrite (Singh et al., 1997). Its activity has optimum in the pH range varying from 1.5 to 3.5. Therefore, any pH changing from the optimum could considerably reduce its vital activity, and consequently the acidification process (McGuire et al., 2001). Malhotra et al. (2002) developed a chemo-biochemical process using Thiobacillus ferrooxidans for desulphurization of gaseous fuels and emissions containing hydrogen sulfide (H2S). The bio-oxidation of ferrous ions to ferric ions has been
achieved efficiently in the temperature range of 20 – 44 °C. A pH range of 1.8 – 2.2 was optimum for the growth of culture and effective bio-oxidation of ferrous ions to ferric ions. The efficiency of bio-oxidation of ferrous ions to ferric ions was not affected in the presence of ferric ions up to a concentration of 500 mg/L.
The net result of all reactions is to remove pyrite, to form solid Fe(OH)3 and to form dissolved
sulfuric acid, generating 4 hydrogen ions (acid) for each iron atom initially present as pyrite. Most of the acidity generated results from precipitation of Fe(OH)3, which has very low
solubility. Often equations are added giving the following:
FeS2(s) + 15/4O2 + 7/2H2O → Fe(OH)3(s) + 2SO42- + 4H+ (Ritcey, 1989) [1.16a]
or
8FeS2 +30O2+ 18H2O→Fe8O8(SO4)(OH)6 + 15SO42- + 30H+ (Nancucheo and Johnson,
2011) [1.16b]
1.3.2.4 Pyrite oxidation in neutral and alkaline medium
It was shown in the previous works of Bailey and Peters (1976), Reedy et al. (1991), Taylor et al. (1984) and Usher et al. (2004), isotope markers demonstrated that the nature of sulphate oxygen is rather from water molecules than from dissolved molecular oxygen.
FeS2 + 8H2O → Fe2+ + 2SO42- + 16H+ + 14 e- [1.17]
It is based on electrochemical mechanism that is realised in the cathodic and anodic reactions at the pyrite surface (Caldeira et al., 2010). The key role of holes were determined in the pyrite anodic dissolution by Mishra and Osseo-Asare (1988) and Wei and Osseo-Asare (1997). The first step of the anodic reaction consists in the reversible H2O (or OH-) adsorption
by means of interaction with an iron (Fe) hole 3d t2g, nonbonding pyrite orbital and hydroxyl
radical generation (Caldeira et al., 2010).
FeS2 + H2O + H+ → Fe(OH·)S2 + H+ [1.18]
The nonbonding orbital contains the hole that doesn’t weaken the iron-sulfur bond in the pyrite. In the sequel, it was assumed that the hydroxyl radical is relocated to sulfur sites on the pyrite surface (Caldeira et al., 2010).
Fe(OH·)S2 → FeS2(OH·) [1.19]
Sulfur site oxidation follows after complete surface hydroxylation of pyrite (Mishra and Osseo-Asare, 1988).
FeS2(OH·) + 3H2O + H+ → Fe(OH·)2S2(OH·)2 + 3H+ [1.20]
Fe(OH·)2S2(OH·)2 + 2H+ → Fe2+ + S2O32- + 2H+ + H2O [1.21]
Thiosulfate is an intermediate product of pyrite oxidation reaction. The subsequent stage of oxidation process depends of pH. For low pH conditions the thiosulfate ion splits with S (elemental) and HSO3- (bisulfate ion) formation that are oxidized at the end to sulfate (Mishra
and Osseo-Asare, 1988). High pH and presented oxidants (oxygen and ferric iron) result in thiosulfate oxidation to sulfate.
Cathodic reaction in the pyrite oxidation process is an electron receiving by dissolved oxygen and ferric ion (oxidants). In the work of Singer and Stumm (1970) was noted that major pyrite oxidant in the acidic medium is a ferric ion due to difference in the oxidation rate. This regularity was determined also for the circumneutral pH. It was shown also that effective pyrite oxidation could be realized by Fe(III) only in the presence of dissolved oxygen and adsorbed Fe(II) (Moses et al., 1987). An adsorbed ferrous iron is oxidised to ferric iron by dissolved oxygen. At the same time, an adsorbed ferric iron is reduced by pyrite. It forms a Fe(II)-Fe(III) electron transfer between pyrite and dissolved oxygen (Caldeira et al., 2010). As it was discussed above, OH- plays a role in the hydroxyl adsorption on the iron sites, and in the radical formation. Their contribution to this process is increased with pH growing. Thus, high pH promotes to the Fe-S2 bond breaking and liberation of ferrous iron
hydroxocomplexes (Fe(OH)+, Fe(OH)
20 and Fe(OH)3-) (Caldeira et al., 2010).
1.3.2.5 Role of carbonate in the reaction of pyrite oxidation
Smith and Shumate (1970) showed that Fe(II)-Fe(III) oxidation in the carbonate absence is slow in strong acidic conditions and accelerates with pH above 4 due to iron hydrolysis. The pyrite has similar character of oxidation. Thus, the surface mineral oxidation hypothesis is supported by the rate controlling of ferrous iron oxidation and iron hydrolysis (Caldeira et al., 2010).
Investigations of Millero et al. (1987) and King (1998) determined catalytic effect of oxygen containing ligands in the ferrous iron oxidation rate. Their results were revised by Pham and Waite (2008) and were confirmed by King (1998): k1(Fe(OH)20) > k1(Fe(CO3)22-) >
k1(Fe(CO3)(OH)-). The contribution of each compound is 20, 40 and 40% respectively. Thus,
the carbonate contained particles play the main role in the ferrous iron oxidation (80%). It is possible due to the less energy bond between ferrous iron and hydroxide ion or carbonate (bicarbonate) ion that is formed in the high spin complexes (Caldeira et al., 2010).
In addition, the buffering properties of carbonate minimize pH changing in the solution and preserve favorable conditions for oxidation of pyrite. Hydroxide ions produced in the anodic reaction will be bounded in HCO3- (Caldeira et al., 2010). Carbonate complexes of iron also
are more soluble in the water solutions under alkaline conditions. It makes iron more available to the chemical reactions. A schematic model of the pyrite oxidation process proposed by Caldeira et al. (2010) is shown in Figure 1.3.
Figure 1.3. Schematic representation of the mechanism of pyrite oxidation in carbonate solutions (Caldeira et al., 2010)
Fe(III) carbonate complexes (electron acceptors) are adsorbed on the pyrite surface in the presence of carbonate. Electron transfer from anodic to cathodic side of pyrite surface reduces the Fe(III)-CO3 to Fe(II)-CO3. Produced Fe(II) carbonate complex is oxidized by
oxygen. It forms the cycle of electron transfer from the pyrite sulfur to dissolved oxygen. The water or OH- adsorption from the anodic side leads to radical formation on the iron site and further radical transport to sulfur. It results in thiosulfate formation and pyrite dissolution (Caldeira et al., 2010).
1.3.2.6 Biological oxidation of metallic sulfides
Several sulfide minerals can biologically be oxidized and generating acid mine drainage. These include (Ritcey, 1989): arsenopyrite, bornite, bravoite, chalcosite, chalcopyrite, cobaltite, covellite, enargite, marcasite, marmatite, millerite, molybdenite, orpiment, pyrite, phyrrhotite, sphalerite, stannite, tetrahedrite and violarite.
Many common metals may participate in sedimentation and generation of H+ ions during reactions of hydrolysis. These reactions usually occur in the area of blending of acidic waters with 3 major dissolved metals mix with cleaner waters resulting in metal hydroxides precipitation on stream channel substrates.
Al3+ + 3H2O ↔ Al(OH)3(s) + 3H+ [1.22]
Fe2+ + 0.25O2 + 2.5H2O ↔ Fe(OH)3(s) + 2H+ [1.23]
Mn2+ + 0.25O2 + 2.5H2O ↔ Mn(OH)3(s) + 2H+ [1.24]
Metal sulfide minerals like pyrite may interact with valuable mineral deposits and some of these minerals may also fabricate acidity and sulfate ions. Oxidation and hydrolysis of metal sulfide minerals pyrrhotite (Fe1-xS), chalcopyrite (CuFeS2), sphalerite [(Zn, Fe)S] and others
release metals such as zinc, lead, nickel, and copper into solution in addition to acidity and SO42- (Jennings et al., 2000; Younger et al., 2002).
1.3.2.7 Factors affecting the oxidation of pyrite
There are several factors that can influence the kinetics of pyrite oxidation. These include (Ritcey, 1989): oxygen content of the gas phase, if saturation is less than 100 %, oxygen concentration in the water phase, degree of surface saturation with water, pH of the solution in contact with pyrite, temperature, chemical activity of Fe3+, surface area of exposed metal sulfide, chemical activation energy required to initiate acid generation and bacterial activity (Akcil and Koldas, 2006).
There are a number of common microscopic bacteria which oxidize sulfide minerals as a source of energy. These bacteria can accelerate the rate of S oxidation and are an important factor in pyrite oxidation and consequently in AMD formation. The kinetics of microbial oxidation of pyrite are influenced by many factors, including (Ritcey, 1989): oxygen
availability, carbon dioxide requirements, temperature dependence, pyrite reactivity, amount of pyrite and its surface area, bacterial contact with pyrite, inhibitors.
1.4 Prevention and mitigation of acid mine drainage generation
Preventing and controlling AMD is a concern at operating mine sites and after mine closure. Advances continue to be made in research and the development of technology to improve ARD prediction, prevention, and treatment.The prevention and mitigation of AMD generation is based on the methods that minimize the presence of the primary reactants for sulfide oxidation (water and oxygen), and/or maximize the quantity and availability of acid neutralizing reactants. These methods could be classified as shown below (Ritcey, 1989; Lottermoser, 2012):
1) minimizing oxygen penetration by diffusion or advection;
2) minimizing water infiltration and leaching (water as a reactant and a transport agent); 3) minimizing, removing, or isolating sulfide minerals;
4) controlling pore water solution pH by addition of lime, limestone, etc.;
5) maximizing availability of acid neutralizing minerals and pore water alkalinity; 6) controlling bacteria and biogeochemical processes (bactericides using): organic
chemicals designed to kill or to inhibit sulfide-oxidizing bacteria activity have been applied to sulfide wastes in order to slow the rate of AMD generation. However, there is concern that some of these chemicals may kill beneficial microorganisms in the environment, thus becoming pollutants themselves.
Various approaches have been evaluated for inhibition of pyrite oxidation or to prevent or minimise the generation of AMD from pyritic tailings. Such techniques are known collectively as ‘source control’ measures. These include (Ritcey, 1989; Johnson and Hallberg, 2005; Lottermoser, 2012; MiningFacts.org, 2012b):
1) mixing acid-producing materials with acid-buffering materials, combining sulfide wastes with limestone or calcite can result in AMD neutralization on site;
2) total solidification of tailings;