يملعلا ثحبلاو يلاعلا ميلعتلا ةرازو
BADJI MOKHTAR UNIVERSITY-ANNABA-UNIVERSITE BADJI
MOKHTAR-ANNABA-راتخم يجاب ةعماج
ةبانع
Faculté : Faculté des Sciences de l'Ingéniorat
Année 2015 Département : Génie des procédés
M
EMOIRE
Présentation en vue de l'obtention du diplôme de magister
SIMPLIFICATION DU MODELE NRTL-PR
EN VUE DE L’AMELIORATION DE LA REPRESENTATION
DES EQUILIBRES DE PHASES
Option
Génie de l'environnement Par
ZARZOUR Majedi
DIRECTEUR DE MEMOIRE: BOUCHAMI Tidjani, Prof. Univ. Badji Mokhtar, Annaba
DEVANT LE JURY
PRESIDENT: ISMAIL Fadhel, Prof. Univ. Badji Mokhtar, Annaba EXAMINATEURS: NADIA Fertikh, Prof. Univ. Badji Mokhtar, Annaba TOUBAL Abdelaziz, MCA. Univ. Badji Mokhtar, Annaba
RESUME
L’objectif initial de ce travail était de disposer d’un modèle susceptible de représenter les équilibres liquide-vapeur. A l’heure actuelle, seul le modèle proposé par Trassy autorise ce type de calcul d’équilibres de phases. Or ce modèle n’est pas utilisable par les industriels et ceci pour deux raisons. La première est qu’afin d’obtenir des résultats d’une bonne qualité, les expressions utilisées comprennent un grande nombre de termes nécessaire aux calculs, ce qui a pour conséquence des temps de calcul trop longs pour des applications industrielles. La deuxième est que ce modèle a été développé autour d’équations prenant en compte une translation volumique. Cette translation était jusqu’à présent sans conséquences sur les différents modèles utilisés.
Nous avons donc repris ce modèle afin de le simplifier et de l’améliorer. Nous avons tout d’abord modifiée la valeur du facteur de répartition non-aléatoire. Nous avons ensuite proposées une nouvelle décomposition en groupe et sous groupe, ce qui permettre de diminuer le nombre de paramètre ajusté de modèle
Mots clés
: Thermodynamique, Equation d’état, Contributions de groupes, Modèle NRTL-PR, Enthalpie libre d’excès, Corps purs, Mélange pétroliers, Hautes pressions, Zone critique.ABSTRACT
The initial objective of this work was to have a model to represent the vapor-liquid equilibria. At the moment, only the model proposed by TRASSY allows this type of calculation of phase equilibria. However, this model is not used by industry and for two reasons. The first is that to obtain results of good quality, the expressions used include a large number of terms necessary for calculations, and thus the calculation time too long for industrial applications. The second is that this model has been developed about equations taking into account a volume translation. This translation was so far without effect on the
different models used.
We have therefore taken this model to simplify it and improve it. First we changed the value of the factor of non-random distribution. We then proposed a new decomposition group and sub group, which reduce the number of parameters adjusted model.
Key-words: Thermodynamics, Equation of state, Contributions from groups , PR-NRTL model, Excess free energy, Pure substance, Oil mixture, High pressure, Critical zone.
LISTES DES FIGURES
Figure I.1. Diagramme de phases d’un corps pur... 13 Figure II.1. Méthode du « point de bulle » ... 20 Figure II.2. Méthode du « flash » ... 19
LISTE DE TABLEAUX
Tableau II.1. Paramètre critique des corps purs de faible poids moléculaire ... 22 Tableau II.2. Contribution des groupes pour le calcul du paramètre de forme m et du volume de van der Waals V*. ... 24 Tableau II.3. Valeurs des paramètres UNIFAC de volume et de surface considérés dans ce travail. ... 27 Tableau III.1. Base des données expérimentales nécessaires à l’estimation des paramètres d’interaction A0
Groupe principal (1) / Groupe principal ( 2) et B0Groupe principal (1) / Groupe principal ( 2). ... 32 Tableau II.2. Table des paramètres d’interaction A0
Groupe principal (1) / Groupe principal ( 2). ... 34 Tableau II.3. Table des paramètres d’interaction B0
Groupe principal (1) / Groupe principal ( 2). ... 34 Tableau III.1. Résultats globaux obtenus sur les équilibres liquide-vapeur avec la nouvelle méthode et la version originale ... 38 Tableau III.2. Résultats globaux obtenus sur les enthalpies de mélange avec la nouvelle
méthode et la version originale. ... 38 Tableau A1. Systèmes alcane-alcane écarts moyens sur la pression de bulle ΔPbulle% et écarts moyens sur la composition de la phase vapeur Δy1 ... 44 Tableau A2 Systèmes cyclane avec alcane ou cyclane : écarts moyens sur la pression de bulle ΔPbulle%, et écarts moyens sur la composition de la phase vapeur Δy1 ... 46 Tableau A3. Systèmes aromatique avec alcane ou aromatique Ecarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 47 Tableau 4. Systèmes cyclanes-aromatiques écarts relatifs moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 49 Tableau A5. Systèmes méthane-alcane écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 50 Tableau A6. Systèmes méthane-cyclane écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1. ... 51 Tableau A7. Systèmes méthane-aromatique écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1... 52 Tableau A8. Systèmes éthane-alcane écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 52 Tableau A9. Systèmes éthane-cyclanee écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 53
Tableau A10. Systèmes éthane-aromatique écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1... 53 Tableau A11. Systèmes azote-alcane écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 53 Tableau A12. Systèmes azote-cyclane écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 54 Tableau A13. Systèmes azote-aromatique écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1... 54 Tableau A14. Systèmes sulfure d’hydrogène-alcane écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 54 Tableau A15. Systèmes sulfure d’hydrogène-cyclane écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 55 Tableau A15. Systèmes sulfure d’hydrogène-aromatique écarts moyens sur la pression de bulle ΔPbulle % et écarts moyens sur la composition de la phase vapeur Δy1 ... 55 Tableau B1. Système alcane-alcane écarts moyens d’enthalpie de mélange ΔHM% ... 56 Tableau B2. Systèmes cyclane avec alcane ou cyclane : écarts moyens d’enthalpie de mélange ΔHM% ... 58 Tableau B3. Systèmes aromatique avec alcane ou aromatique : écarts moyens d’enthalpie de mélange ΔHM% ... 60 Tableau B4. Systèmes cyclane avec aromatique: écarts moyens d’enthalpie de mélange
TABLE DES MATIERES
RESUME ... I DEDICACES ... III REMERCIMENTS ... IV LISTES DES FIGURES ... V LISTE DE TABLEAUX ... VI TABLE DES MATIERES ... VIII
INTRODUCTION GENERALE ... 10
- CHAPITRE I –EQUATION D’ETAT ... 12
I-1 INTRODUCTION ... 12
I-2 PRINCIPE DES « ETATS CORRESPONDANTS » ... 13
I-3 LES MODELES D’EQUATION D’ETAT ... 15
I-3-2 Les équations non cubiques ... 16
I-3-2-1 L’équation Peng-Robinson augmentée Pra (Solimande et al.[8])... 16
I-3-2-2 l’équation Benedict-Webb-Rubin modifiée par soave[10] ... 16
I-4 CONCLUSION ... 17
- CHAPITRE II -MODELE NRTL-PR ... 18
II-1 INTRODUCTION ... 18
II-2 CONDITION D’EQUILIBRE ... 18
II-3 ENTHALPIE MELANGE ... 20
II-4 CORPS PURS ... 20
II-4-1 COMPOSES LEGERS ... 21
II-4-2 COMPOSES LOURDS ... 22
II-5 MELANGE ... 25
II-5-1 EXPRESSION DU MODELE D’ENTHALPIE LIBRE D’EXCES ... 25
- CHAPITRE III -SIMPLIFICATION DU MODELE PREDICTIF NRTL-PR ... 28
III-1. MODIFICATIONS ... 28
III-1-1. Facteur de répartition non-aléatoire ... 28
III-1-2. Nouvelle décomposition en groupes et sous-groupes ... 28
III-2. AJUSTEMENTS ... 31
III-2-1. Base de données ... 31
III-2-2. Méthode d’ajustement des paramètres d’interaction ... 34
- CHAPITRE IV -RESULTATS ET INTERPRETATION ... 37
IV-1.INTRODUCTION ... 37
IV-2. MELANGES D’HYDROCARBURES ... 37
IV-3. MELANGES CONTENANT DES « LEGERS »... 39
CONCLUSION GENERALE ... 40
BIBLIOGRAPHIE ... 42
ANNEXE A : Equilibre liquide – vapeur ... 44
Introduction Générale
INTRODUCTION GENERALE
Ce travail porte sur la représentation des propriétés thermodynamiques des systèmes complexes à l’aide de modèles thermodynamique basés sur l’utilisation d’une équation d’état.
Le but de notre travail est d’étudier les équilibres de phases de mélanges pétroliers afin d’obtenir des prédictions sur l’évolution d’un puits pétrolier au cours de son exploitation. Les fluides se trouvant sous hautes pressions il est nécessaire d’avoir recours à une équation d’état, qui fait l’objet de premier chapitre
Nous présentons, dans le deuxième chapitre, le modèle NRTL-PR proposé par Trassy[20], qui est basé sur l’équation d’état de Peng-Robinson associée à l’expression de l’enthalpie libre d’excès gexcès
dérivée de l’équation NRTL de Renon et Prausnitz [18]. L’estimation des paramètres des corps purs est réalisée à l’aide du modèle de Coniglio[22], modifié par Crampon et al.[25]; il utilise la température d’ébullition pour les molécules dites « lourdes » et les propriétés critiques pour les molécules dites « légères ». Pour le calcul des propriétés du mélange, ce modèle fait intervenir une forme généralisée de la fonction d’excès gexcès du modèle NRTL ; elle est composée de deux termes : un terme combinatoire, dépendant des paramètres de taille ri des composés i du mélange, et un terme résiduel faisant
intervenir des paramètres de forme qi ainsi que des énergies d’interaction Akl entre les groupes
fonctionnels k et l représentatifs des composés du mélange. Une fois établies, à partir d’une base d’ajustement de données soigneusement sélectionnées, les valeurs caractéristiques de tous les groupes fonctionnels, le modèle ainsi proposé est totalement prédictif. Il est ainsi possible, sans aucune information préalable, de prédire les propriétés thermodynamiques de n’importe quel mélange.
Cependant, les contraintes économiques imposées à l’exploitation des champs pétroliers en mer, à savoir la suppression des plateformes sur lesquelles était réalisée jusqu’alors l’élimination de l’eau contenue dans le fluide de gisement, imposent que l’on soit en mesure de traiter également les mélanges hydrocarbures - eau. Ces systèmes présentent généralement, dans les conditions de température et de pression au fond de la mer, des équilibres liquide – liquide. Le modèle NRTL est connu pour être le mieux adapté, sous basse pression, à leur représentation. Toutefois, ce modèle qui dérive du concept de la « composition locale » impose que les énergies d’interaction Aeau/hydrocarbure et Ahydrocarbure/eau
soient dissymétriques. Afin de diminuer le nombre total de paramètres d’interaction nécessaires pour représenter tous les mélanges hydrocarbures – eau, nous avons décidé de simplifier la représentation en groupes fonctionnels en faisant intervenir, tout comme dans le modèle UNIFAC, la notion de groupe principal et de sous-groupes. Dans ce travail, nous
Introduction Générale
que les interactions entre groupes d’un même groupe principal soient nulles. De plus, nous avons pris en compte dans la base des données d’ajustement, non seulement des équilibres liquide-vapeur, mais aussi des enthalpies de mélange, dont la connaissance est tout à fait fondamentale pour la modélisation des procédés d’exploitation des gisements. Cette étude fait l’objet du chapitre III.
Dans le chapitre IV, les résultats obtenus dans ce travail sont comparés à ceux présentés par Escandell (2004) avec le même modèle, mais : d’une part, sans tenir compte de la notion de groupe principal et de sous-groupe, donc avec un nombre très élevé de paramètres d’interaction Akl, et, d’autre part, sans prendre en compte les données d’enthalpie
de mélange pour la détermination de ces paramètres. L’objectif de cette comparaison est donc de vérifier si, avec un nombre très restreint de paramètres, il est possible d’obtenir une représentation comparable des équilibres liquide-vapeur et, si possible, de s’assurer que l’on obtient en plus une meilleure restitution des enthalpies de mélange.
Chapitre I Equation d’état
- CHAPITRE I –
EQUATION D’ETAT
I-1 INTRODUCTION
Si pour la représentation des équilibres liquide-vapeur sous « basse pression » il n’est pas déraisonnable de considérer le comportement du gaz comme parfait, la modélisation des équilibres de phase sous hautes n’est pas envisageable sans l’intervention d’une équation d’état. Cette dernière permet de définir de manière cohérente les relations qui existent entre la pression P, le volume v et la température T. Les modèles courants peuvent être classe en deux grandes catégories selon l’origine des bases théoriques auxquelles ils font appel.
La majorité des équations d’état de la littérature se fondent sur la thermodynamique phénoménologique. C’est la voie la plus ancienne puisqu’elle utilise les connaissances acquises a la fin du 19ème siècle. Ses théories découlent d’une observation macroscopique de la matière et bien qu’inexacte, elle reste la plus évident à mettre en ouvre. Son but n’est pas, en effet, de représenter rigoureusement le comportement physique des molécules, mais d’offrir une bonne estimation des variables globales du système. Historiquement, deux modélisations concurrentes se sont opposées. D’un côté, les équations du 3ème
degré en v, dites cubique, dont la plus fameuse représentante reste l’équation d’état de Van der Waals [1]; la pression prendre ici la forme d’une somme d’un terme répulsif et d’un terme attractif. De l’autre, les équations extraites de l’approche dite du viriel (Thiesen[2], Onnes[3]), telle que l’équation de Benedict, Webb et Rubin[4]. La pression, dans leur cas, est exprimée grâce à un développement polynomial de la variable 1/v.
La dynamique moléculaire est une beaucoup plus récente, puisqu’il a fallu attendre l’apparition de la mécanique statistique pour qu’elle prenne son essor. La thermodynamique statistique procède d’une volonté de mieux comprendre le fonctionnement intime des interactions entre les molécules. Ses théories permettent de relier l’étude, à l’échelle moléculaire, des propriétés de la matière aux grandeurs observables. Pour décrire de façon précise les forces entrant en jeu, il est communément admis de faire appel à la mécanique quantique. Les équations mathématiques qui en résultent sont aussi complexes que rigoureuses. Leur résolution nécessite cependant, soit d’effectuer un certain nombre d’approximations nuisant à la qualité des modèles, soit de réaliser des simulations numériques qui ont pour effet d’accroitre considérablement le temps de calcul. En effet, malgré leurs qualités incontestables, ces équations exigent encore pour le moment une capacité de calcul qui ne laisse pas espérer de modélisation des mélanges simples dans des délais raisonnables
Chapitre I Equation d’état
(de quelques minutes à quelques heures). Qu’en serait-il pour des mélanges contenant plusieurs dizaines de composés ?
Dans le cadre de travail, nous nous sommes donc focalisés sur les équations d’état découlent de la thermodynamique phénoménologique qui s’avèrent être idéale pour les applications à des mélanges complexes tels que les fluides pétrolières.
L’objectif principal de ce premier chapitre est d’évaluer les qualités et les limites de différents modèles d’équation d’état afin de déterminer lequel pourrait apparaître le plus adapté à nos besoins.
I-2 PRINCIPE DES « ETATS CORRESPONDANTS »
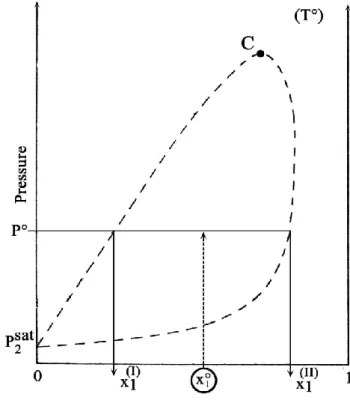
Dans la nature, la matière se présente sous trois états physiques fondamentaux : gaz, liquide et solide. L'état physique d'un composé pur est lié à l'organisation moléculaire de la matière, c'est-à-dire, à la taille, à la forme des molécules, à la distance et aux interactions intermoléculaires. L'état physique d'un composé pur donné dépend essentiellement des deux variables intensives que sont la température T et la pression P. Pour des raisons scientifiques et pratiques, il est intéressant de connaître l'état physique d'un composé à une température et une pression données. Le diagramme de phases P=f(T) d’un corps pur, proposé par la figure I.1, permet de définir les domaines d’existence des trois états physiques, délimités par trois courbes d’équilibre, courbe de fusion, courbe de sublimation et courbe de vaporisation, qui caractérisent le passage d’une phase à une autre.
Chapitre I Equation d’état
Les trois courbes d'équilibre se coupent en un point unique, appelé point triple (t), où coexistent les trois phases. La courbe d’ébullition s’achève dans sa partie supérieure par un point particulier dit point critique (c). Dans le plan (P, T), la zone délimitée par les droites d’équations respectives P Pc et T Tc est connue sous le nom de domaine supercritique. Le fluide supercritique a des propriétés physico-chimiques intermédiaires entre celles du liquide et du gaz. Sa masse volumique élevée, proche de celle des liquides, lui confère un pouvoir solvant plus important que celui des gaz. Il possède, de plus, les caractéristiques d’un gaz en raison de sa faible viscosité et de sa diffusivité élevée.
C’est Van der Waals [1] qui a été le premier à en formuler l’énoncé. L’observation du plan (P, T) montre que le point critique est un lieu vraiment remarquable du domaine. Il est, en effet, le seul point où l’on observe à la fois un point d’inflexion et une tangente horizontale.
Si on considère l’équation de Van der Waals[1], on remarque que celle-ci dépend non seulement de deux paramètres a et b, mais également d’une troisième grandeur notée ici, R (R étant une constante universelle, dénommée « constante des gaz parfait »). La relation de P en fonction des variables T et v, se présente comme la somme d’un terme répulsif et d’un terme attractif telle que,
2 v a b v RT P (I-1) La détermination des paramètres a et b du composé étudié apparaît idéale, au point critique, puisque l’on peut appliquer en ce lieu caractéristique, les conditions suivantes :
0 0 ) , ( , 2 2 , c c c c v T v T c c c v P v P v T P P (I-2)
Les équations qui composent le système (I-2) sont fréquemment appelées « spécifications critiques ». Lorsque l’on écrit l’équation d’état de Van der Waals[1] sous sa forme adimensionnelle à l’aide des expressions de a et b trouvées grâce à la résolution du système (I-2), on revient à une relation qui ne dépend plus que des propriétés réduites, c’est-à-dire des grandeurs Tr = T/Tc, Pr = P/Pc, vr = v/vc :
r
r r r v T v P 32 3 1 8 (I-3)Chapitre I Equation d’état
Suite à l’examen de l’équation (I-3), le principe des « états correspondants » stipule que deux substances placées dans les mêmes conditions de pression et température réduites doivent posséder un volume réduit identique. L’expérience démontre que ce n’est malheureusement que très rarement le cas (le principe est essentiellement valable pour les fluides monoatomique). L’analyse des origines de cet écart au principe des « états correspondants » formulé pare Van der Waals[1], conduit à supposer que de nombreux paramètres, autre que les grandeurs critiques, peuvent influencer la nature des équilibres de phases. Outre la forme géométrique des molécules, des caractéristiques physiques telles que le moment d’inertie, les interactions électrostatiques ou le rayon de giration de la molécule sont susceptibles d’expliquer l’existence de cet écart. Pitzer[5] à suggérer d’introduire une nouvelle grandeur permettant de corriger la loi des états correspondants de Van der Waals[1]. Cette grandeur est appelée facteur acentrique dont la valeur est pratiquement nulle pour les molécules sphériques et augmente régulièrement avec la longueur de la chaîne pour les hydrocarbures paraffiniques. Le facteur acentrique est défini de telle sorte qu’il permette la représentation de la pression de saturation du composé à la température réduite de 0,7. Son expression est la suivante :
1 log 7 , 0 10 r T c sat P P (I-4)
I-3 LES MODELES D’EQUATION D’ETAT I-3-1 Les équations cubiques
A la suite des travaux de Van der Waals[1], de nombreux auteurs, séduits par la simplicité d’emploi des équations cubiques, ont proposé des modèles de plus en plus élaborés. Devant leur grande nombre, la tentation était grande de parvenir à généraliser l’écriture de ces équations. Dans la solution, la plus fréquemment proposée, pour des équations cubiques possédant deux paramètres a et b, la pression s’écrit :
2 2 wb ubv v ) T ( a b v RT P (I-5) avec, a(T), le terme d’interaction ;
v, volume molaire ; b, covolume molaire ;
u et w, deux paramètres caractéristiques de l’équation cubique choisie : ainsi, pour Van der Waals[1] : u = 0 et w = 0 ;
pour Redlich- Kwong[6] : u = 1 et w =0 ; pour Peng-Robinson[7] : u = 2 et w = -1.
Chapitre I Equation d’état
Il faut noter que les paramètres a(T) et b sont spécifiques à chaque équation d’état et qu’ils dépendent des propriétés des corps purs et des mélange. Leur estimation dans le cadre du modèle NRTL-PR sera abordée au chapitre II.
Les deux modèles cubiques les plus couramment utilisés dans la littérature sont l’équation de Redlich et Kwong[6] et l’équation de Peng et Robinson[7]. Ces derniers auteurs ayant eu pour soucis, lors de l’élaboration de leur modèle, l’amélioration de la restitution des densités pour les molécules de taille moyenne, c’est sur l’équation de Peng-Robinson[7] que notre intérêt s’est naturellement, porté.
I-3-2 Les équations non cubiques
On peut noter deux exemples de l’équation non cubique les plus utilisée dans la littérature :
I-3-2-1 L’équation Peng-Robinson augmentée Pra (Solimande et al.[8])
Le trait essentiel de toutes les équations cubiques est qu’elles ne sont pas capables de représenter correctement la courbure des isothermes autour de la zone critique. PRc n’échappe malheureusement pas à cette règle. C’est à partir de cette constatation que Solimando[8] a envisage d’ajouter, à l’équation de Peng-Robinson[7] corrigée, un terme empirique dont le rôle est de modifier la concavité des isothermes voisines du point critique.
Le modèle de Solimando[8] se présente sous la forme suivante : 2 1 exp ) ( ) ( ) ( v b C v b M b v b b v v T a b v RT P x (I-6)
M et C1 des paramètres du terme correctif.
Le calcul des grandeurs b et a(T) s’effectue de la même manière que pour l’équation de Peng-Robinson, c’est-à-dire grâce aux expressions présente dans le chapitre II.
L’écriture de l’équation Pra, dans sa version présentée par Solimando[8] et al, est intéressante car elle permet de retrouver le formalisme de l’équation cubique de Peng-Robinson corrigée proposée par Rauzy[9], lorsque l’on s’éloigne de la zone critique.
I-3-2-2 l’équation Benedict-Webb-Rubin modifiée par soave[10]
Soave[10] a propose récemment une version modifiée de l’équation BWR (Benedict et al[4]). L’originalité de son travail est fondée sur la constatation que la distribution des puissances de 1/v, dans le modèle BWR originel, est irrégulière. Ainsi, l’équation présentée par Soave[10] substitue au terme en (1/v)[5] de l’équation BWR originelle, un terme en (1/v)4. De fait, le facteur de compressibilité s’exprime alors,
Chapitre I Equation d’état 1 2 4 2 1 2 exp 2 v F v F v E v D v C v B RT PV z (I-7)
B, C, D, E et F sont des paramètres déterminés au point critiques.
I-4 CONCLUSION
Ce chapitre a permis de mettre en lumière les difficultés qui peuvent être rencontrées lorsque l’on décide de représenter des mélanges, dont les propriétés thermodynamiques des constituants sont mal connues, à l’aide d’équations d’état.
Deux points importants doivent être soulignés sur l’utilisation de l’équation d’état : - premièrement, L’utilisation d’équation d’état doit toujours garder à l’esprit que le
caractère prédictif des modèles à considérer, dans le cas de mélanges complexes, est un critère fondamental. Sans cette qualité, bon nombre d’excellentes équations d’état se trouvent dans l’incapacité de faire preuve de leurs talents ;
- il n’est pas certain due l’utilisation d’une équation complexe soit en mesure de garantir une bonne restitution des valeurs expérimentales dans le cas de composés pour lesquels peu d’informations sont disponibles. Des équations d’état cubiques, malgré leur rusticité et leurs nombreux défauts, peuvent conduire à des résultats tout à fait honorables, et de meilleure qualité que ceux obtenus avec des équations plus complexes, surtout si l’on prend en considération les incertitudes expérimentales et la nécessité de faire intervenir des contributions de groupes pour estimer les gradeurs critiques.
Chapitre II Modèle NRTL-PR
- CHAPITRE II -
MODELE NRTL-PR
II-1 INTRODUCTION
Dans ce chapitre, nous allons présenter la modèle NRTL-PR, le choix du modèle NRTL-PR permettant de représenter les propriétés des mélanges est basé sur le formalisme «Equation d’état – Fonction d’excès» initialement proposé par Huron et Vidal[11], puis modifié par Mollerup[12], Péneloux et al[13], et Michelsen[14]. Il permet d’exprime le paramètre a(T) du mélange à partir d’un modèle d’enthalpie libre d’excès gExcès(T,xi), tel que
ceux proposés dans la littérature pour la représentation de la phase liquide sous basse pression. Cette méthode permet donc aux modèles moléculaires, valables uniquement dans le domaine des basses pressions (Van Laar[15], Wilson[16], UNIQUAC[17], NRTL,…), d’être appliqués aux hautes pressions grâce à l’intervention de l’équation d’état.
L’originalité du modèle NRTL-PR est d’avoir choisi le modèle NRTL de Renon et Prausnitz[18] pour le calcul des équilibres de phases de mélanges complexes pouvant contenir des hydrocarbures et des composés polaires ou très associés tels que l’eau. Dans ce dernier cas, des phénomènes de démixtion apparaissent et, sous basse pression, il est reconnu que seul le modèle NRTL (ou la version LEMF proposée par Marina et Tassios[19]) basé sur le concept de la « composition locale » permet de modéliser correctement ces équilibres liquide - liquide.
Il s’agit maintenant d’expliciter pour les corps purs, puis pour les mélanges, les différentes méthodes d’estimation des paramètres caractéristiques a et b. En effet, la résolution de l’équation d’état et donc le calcul des coefficients de fugacité qui interviennent dans la condition d’équilibre ne sont possibles qu’une fois ces paramètres déterminés. Une fois choisies l’équation d’état et les méthodes d’estimation des paramètres a(T) et b, on peut aborder le calcul des conditions d’équilibres de phases et des enthalpies de mélange.
II-2 CONDITION D’EQUILIBRE
Quel que soit le modèle thermodynamique utilisé, l’équilibre entre deux phases (I) et (II) s’exprime analytiquement par l’égalité des potentiels chimiques ou bien par l’égalité des fugacités iI et iIIde chaque composé (i) dans chaque phase :
Chapitre II Modèle NRTL-PR
II i II II II i I i I I I i T,v ,x x T,v ,x x (II-1)
I I
II II
x v T P x v T P , , , ,Le coefficient de fugacité φi d’un composé i est calculé à partir de l’énergie résiduelle d’Helmholtz Ares, selon l’expression suivante :
i j n V T i res i n RT A z , , / ln ln (II-2) En effet, l'énergie libre est la fonction caractéristique dont les variables sont le volume et la température. Nous présentons l'expression de l'énergie libre résiduelle molaire ares:
ideal v res V dV z nRT n V T A n V T A n V T A , , , , , , 1 (II-3) et : nRT PV z (II-4)où z est le facteur de compressibilité du fluide, égal à l’unité dans le cas d’un gaz parfait. Ainsi, pour une équation d'état donnée, et donc d'une expression du facteur de compressibilité z(T,v) (relation II-4), on effectuera le calcul de l'intégrale (II-3) afin d'obtenir la forme explicite de l'énergie libre résiduelle molaire ares(T,v). On peut alors calculer les
différentes propriétés thermodynamiques telles que l'énergie interne résiduelle et le coefficient de fugacité.
L’équation (II-2) devient :
v v dv z z z 1) ln ( 1) ( ln (II-5)Il existe deux méthodes pour résoudre les conditions d’équilibre (I-1) :
- une première, spécifique du calcul des équilibres entre le liquide (I) et la vapeur (II),
consiste à effectuer un calcul de « point de bulle », comme il est montré sur la figure II.1, ou un calcul de « point de rosée ». Pour le point de bulle, on fixe la composition de la phase liquide x1° et la température T° et on calcule, à l’aide des équations (II-8), la composition y1
de la phase vapeur et la pression P.
- La deuxième méthode est tout à fait générale. C’est la méthode de « calcul de Flash »
(figure II.2), dans laquelle les variables T° et P° sont fixées, ainsi que la fraction molaire globale x1° du mélange. Dans ce cas, on calcule les fractions molaires
II 1 I 1 , x x et les volumes molaires 1II I 1 , v
Chapitre II Modèle NRTL-PR
Figure II.1. Méthode du « point de bulle » Figure II.2. Méthode de « flash »
II-3 ENTHALPIE MELANGE
L’enthalpie de mélange est obtenue à partir des enthalpies d’écart du mélange (H-H*) et celles des corps purs (H-H*) i dans les mêmes conditions de température et de pression :
p i i i M H H x H H H 1 *) ( *) ( (II-6)L’enthalpie de mélange dépend, d’une part de l’équation d’état, et d’autre part de l’expression du terme d’énergie libre résiduel Ares
: ) 1 ( ) / 1 ( / , nRT z T T A H n V res M (II-7)
( ) / 1 / / 1 / ) , , ( ) , , ( ) ( ) ( , , i i TPx iTPx i i i M x P T RT x z z T d T b da x T d T da H i (II-8) avec : , ln ( , , ) ln ln( ) ( ) ( ) / ln ( ) η η η i i nj i nj T nb T v x z z b n na bRT n 1 1 1 1 1 1 2 2 2 1 1 2 , v b η = , (II-9)II-4 CORPS PURS
i i i v b η =
Chapitre II Modèle NRTL-PR
Quelles que soient les méthodes employées pour le calcul des propriétés thermodynamiques des fluides pétroliers, il est d’usage de se placer dans les conditions du modèle « à un fluide », c’est à dire que l’expression de l’équation d’état (Eq. I-5) est la même pour la représentation des corps purs et celle des mélanges. Seules les expressions de b et a(T) sont différentes :
- Pour le corps pur : b = bi et a(T) = ai(T) sont estimés uniquement à partir des
propriétés des corps purs (cas des composés « légers » ou « lourds » dans le paragraphe suivant)
- Pour le mélange : b =f(bi, xi ) et a(T) = f(ai(T), xi ), ce qui impose que l’on définisse
des « règles de mélange » dépendant de la composition xi .
Trassy[20] avait considéré différentes méthodes d’estimation des paramètres afin d’obtenir la meilleure représentation possible du corps pur. La méthode distingue ainsi deux ensembles de molécules :
- les composés « légers » tels que l’azote, le dioxyde de carbone, l’hydrogène sulfuré, les
hydrocarbures ayant moins de cinq atomes de carbone.
- les composés « lourds » tels que tous les autres hydrocarbures.
II-4-1 COMPOSES LEGERS
Les propriétés critiques apparaissent comme des éléments incontournables pour qui cherche à employer une équation d’état. Leur obtention doit être, ainsi, la plus précise possible car toute incertitude les concernant, entraîne l’apparition d’erreurs significatives lors du calcul des grandeurs fondamentales de l’équation d’état. L’utilisateur d’équations d’état
peut être confronté à deux types de situation lorsqu’il recherche les propriétés thermodynamique de corps purs:
* soit les compensés qu’il désire étudier possèdent des propriétés physiques accessibles expérimentalement, et dans ce cas les grandeurs critiques sont le plus souvent disponibles dans la littérature. Parmi les nombreuses sources de données de qualité, les Thermodynamic Tables[21] offrent un large éventail de propriétés physico-chimiques à destination des pétroliers.
* soit les propriétés critiques des corps purs étudiés sont totalement inconnues. Cette situation se produit principalement pour les composés lourds possédant de très faibles tensions de vapeur; dans les conditions ambiantes, ces composés lourds se trouvent ainsi placés dans des états très éloignés de leur domaine critique. A très haute température (au-déla de 600 Kelvins), l’énergie apportée au système est suffisante pour permettre la rupture de certaines liaisons moléculaires. Dans ce cas, les grandeurs critiques calculées ou estimées
Chapitre II Modèle NRTL-PR
pour ces composés n’ont aucune réalité physique. Malgré tout, pour aider l’utilisateur d’équation d’état à estimer les grandeurs réelles ou fictives qui lui sont dispensables, quelques hauteurs de la littérature proposent des corrélations ou des méthodes de contributions de groupes.
Les composés considérés comme légers sont (dans ce travail) N2, H2S, CH4 et C2H6 les paramètres critique considérés pour ces composés sont donnés dans le tableau II.1, l’estimation de a(T) et b est basée sur les grandeurs Tc et Pc qui sont connues
expérimentalement : - Covolume b : C C b P RT Ω b = , Ωb = 0,07780 (II-10) - Fonction a(T) : f(T ) P T R ) T ( a r C 2 c 2 a , Ωa = 0,45724 (II-11) avec :
0.5
2 r r) 1 m(1 T ) T ( f , C r T T T (II-12)f(Tr) est la fonction de la température réduite (Tr) originellement proposée par Soave[10] afin
d’améliorer la représentation des pressions de vapeur en fonction de la température ; elle introduit un nouveau paramètre m, le facteur de forme, qui est corrélé au facteur acentrique ω de Pitzer[5]. Dans le modèle NRTL-PR on utilise la forme généralisée du paramètre m proposée par Robinson et Peng 7pour tenir compte de la taille des molécules :
m =0,37464+1,54226ω-0,26992ω2 ω≤0.49
m =0,374642+1,4503ω-0,164423ω2 +0.016666ω3 ω≥0.49
Tableau II.1. Paramètre critique des corps purs de faible poids moléculaire
Paramètres N2 H2S CH4 C2H6
Tc(K) 126,2 373,2 190,4 305,4
Pc (bar) 33,9 89,4 46,0 48,8
Ω 0,039 0,081 0,011 0,099
II-4-2 COMPOSES LOURDS
Chapitre II Modèle NRTL-PR
Les composés « lourds » sont décrits par la méthode de Coniglio[22], qui présente l’intérêt d’être basée sur les températures d’ébullition Teb, et non plus sur les spécifications
critiques difficilement accessibles pour les molécules de haut poids moléculaire. Les températures d’ébullition sont généralement obtenues expérimentalement, sinon elles sont estimées par contribution de groupes d’Avaullée[23], Avaullée et al, permettant d’accéder facilement à une bonne estimation des propriétés thermodynamiques des hydrocarbures lourds.
- Fonction a(T) : elle s’exprime de la manière suivante :
) f m f m exp( ) T ( a ) T ( a eb 1 T0,4 2 T2,5 reb reb , eb T 1 TT f reb - (II-14)
avec : a(Teb) , la valeur de la fonction a(T) à la température normale d’ébullition. Teb m1 et
m2 sont deux paramètres spécifiques de la molécule; ils peuvent s’exprimer en fonction d’un
seul paramètre m :
m1 = 1,80546m + 0.21887 , m2 = 0,11113m – 0.03502 (II-15)
Le paramètre de forme m est calculé par contribution de groupes. Trassy[20] propose l’expression suivante de m : 5 4 ) 5 , 0 1 ( 3S C ln(S) C C m = S0,6+ + + (II-16)
où, C3, C4 et C5 sont des constantes universelles : C3 = 0,322693, C4 = 0,087814 , C5 = 0,591802. S est une variable intermédiaire sur laquelle portent les contributions de groupes :
k k 15 1 k N M S (II-17)
avec, Nk le nombre d’occurrences du groupe k et Mk la contribution du groupe k. Les valeurs
de ces paramètres sont indiquées dans le tableau II.2.
- Covolume b:
Dans la méthode de Coniglio[22], le covolume n’est plus calculé à partir des coordonnées critiques du corps pur, mais à partir de la contribution de groupes de Bondi[24]. Il s’agit d’une contribution de groupes qui permet de calculer les volumes de van der Waals V* pour n’importe quel type de composé.
Le volume de van der Waals est calculé à partir du rayon de Van der Waals et de la longueur de la liaison covalente. Les atomes qui constituent la molécule sont supposés
Chapitre II Modèle NRTL-PR
sphériques et ils s’interpénètrent jusqu'à ce que les forces attractives et répulsives s’équilibrent. La molécule de méthane est prise comme référence dans l’expression du covolume, car il est le premier des hydrocarbures.
avec * 4 * 4 CH CH V V b b (II-19)
Dans ce travail nous avons utilisé les contributions de groupes publiées par Crampon et al[25], qui correspondent à une simplification de la méthode originale de Bondi[24] pour le calcul du volume de Van der Waals.
k k 15 1 k N V * V (I-20)
Les valeurs des paramètres de groupes Vk de la méthode de Crampon et al[25], sont
données dans le tableau II.2.
Tableau II.2. Contribution des groupes pour le calcul du paramètre de forme m et du volume de van der Waals V*.
Groupes principaux Sous-groupes Mk Vk
« ALC » CH3 0.085492 13.67 CH2 0.082860 10.23 CH 0.047033 6.78 C -0.028020 3.33 « CYC » CH2 0.062716 10.23 CH substitué 0.034236 6.78 CH jonction de cycles 0.01003 6.78 C substitué -0.010213 3.33 C jonction de cycles 0.051147 3.33 « ARO» CH 0.050476 8.06 C substitué 0.071528 5.54 C jonction de cycles 0.013697 4.74 bCH4 = 26,80 cm3.mol-1 V*CH4 = 17,12 cm3.mol-1 (II-18)
Chapitre II Modèle NRTL-PR
II-5 MELANGE
Pour l'étude des mélanges sous des pressions élevées, on utilise généralement la même équation d'état que celle ayant servi à déterminer les propriétés des corps purs. Nous décrivons ici les règles de mélange qui permettent au modèle NRTL-PR d’être totalement prédictif.
Pour un mélange contenant p constituants, les paramètres de l’équation d’état (Eq. I-5) s’expriment de la façon suivante :
) ln( ) ( ) ( 1 1
p i i i Excès p i i i i b b x RT g b T a x b T a (II-21)
p i i ib x b 1 (II-22)Les grandeurs indicées i font références aux corps purs.
II-5-1 EXPRESSION DU MODELE D’ENTHALPIE LIBRE D’EXCES
La méthode de Trassy[20] propose une enthalpie libre d’excès gExcès de type NRTL, composée de deux termes : un terme dit "combinatoire" ou "athermique" qui prend en compte les différences de taille ri des molécules et un terme "résiduel" qui est du aux interactions
entre les molécules et fait donc intervenir, outre les différences de forme qi des molécules, les
énergies d’interaction Γij entre molécules. Tous les paramètres du modèle sont calculés par
contributions de groupes, ce qui conduit à une expression totalement prédictive de l’enthalpie libre d’excès : Excès res Excès Comb Excès
g
g
g
=
+
(II-23) Terme combinatoireLa partie athermique, appelée aussi combinatoire, représente les effets dus aux différences de taille et de forme des molécules présentes dans le mélange. Pour un mélange à p constituants, le terme combinatoire s’exprime de la façon suivante :
p i i i Excès Comb r r ln x RT g 1 ,
p i i ir x r 1 (II-24)ri est le facteur de volume ou de taille de la molécule i défini par :
i i
i b
r (II-25)
Chapitre II Modèle NRTL-PR 2 i 3 i 2 i 1 2,1432 .10 L 2,15430 .10 L δ = + - + - (II-26)
Quant au paramètre de longueur de chaîne, Li de la molécule i, il est estimé à partir des
paramètres de groupes Rk de la méthode UNIFAC (Fredenslund et al.,[27]) Les valeurs des
paramètres Li sont présentées dans le tableau II-3.
N 1 k k ik i v R L (II-27)avec : Rk , le paramètre de volume du sous-groupe k, vik le nombre d’occurrence de ce
sous-groupe et N le nombre total de sous-groupes. Les valeurs des paramètres Rk sont
présentées dans le tableau II.3
.
Terme résiduel
La partie résiduelle est chargée, quant à elle, de prendre en considération les effets des interactions moléculaires. On postule qu’elle peut se calculer en remplacent la solution de molécules par une solution des groupes constitutifs de ces molécules. Pour un mélange de p constituants, l’expression du terme d’excès résiduel proposée par Trassy, s’écrit alors :
p j ordre onde ij ordre premier ij p m mi m m ji j j p i i i Excès resx
q
x
q
x
q
g
1 sec 1 1
(II-28)Nous avons pris la formule de terme résiduel proposée par Escandell, qui est celle de Trassy[18] sans le terme de deuxième ordre, qui complique inutilement l’expression du terme résiduel :
p 1 j ij p 1 m mi m m ji j j p 1 i i i Excès resx
q
x
q
x
q
g
, RT exp ij ji (II-29)qi est le facteur de forme ou de contact extérieur à la molécule i :
∑
= = N 1 k k ik i v Q q (II-30)Où, Qk est le paramètre de surface du sous-groupe k. Les valeurs de ce paramètre sont issues
du modèle UNIFAC, sont reportées dans le tableau II.3.
τij est le facteur de répartition moléculaire ; il dépend du facteur de répartition non-aléatoire
α, dont la valeur a été modifiée dans ce travail (chapitre III). Γij est l’énergie d’interaction entre les molécules i et j. Elle est calculée par la méthode de « contributions de groupes » suivante :
Chapitre II Modèle NRTL-PR
N l kl jl il N k ik ij A 1 1 2 1 (II-31)
N l l il k ik ik Q v Q v 1 1 T T B A Akl kl kl , T° = 298,15k (II-32)où , νik est le nombre d’occurrence sous-groupes fonctionnels k dans la molécule i et Akl et le
paramètre d’interaction entre ces sous-groupes. Son estimation fait l’objet du chapitre III. Tableau II.3. Valeurs des paramètres UNIFAC de volume et de surface considérés dans ce travail.
Il faut remarquer que:
-les molécules légères (N2, H2S, CH4, C2H6) ont un paramètre de longueur de chaîne égale à l’unité
- pour les molécules telles que méthane et l’éthane, le paramètre de longueur de chaîne correspond au nombre d’atomes de carbone.
Groupes principaux Sous-groupes Qk Rk Li
« ALC » CH3 0.848 0.9011 1,0000 CH2 0.540 0.6744 1,0000 CH 0.228 0.4469 0,7025 C 0.000 0.2195 0,3821 « CYC » CH2 0.540 0.6744 0,6666 CH substitué, jonction de cycles 0.228 0.4469 0,6000 C substitué, jonction de cycles 0.000 0.2195 0,0000 « ARO» CH 0.4000 0.5313 0,6666
C substitué, jonction de cycles 0.1200 0.3652 0,5000
« CH4 » CH4 1.1600 1.1290 1,0000
« C2H6 » C2H6 1.6920 1.8022 2,0000
« N2 » N2 1.0400 0.8560 1,0000
Chapitre III Simplification du modèle prédictif NRTL-PR
- CHAPITRE III -
SIMPLIFICATION DU MODELE PREDICTIF NRTL-PR
III-1. MODIFICATIONS
Nous allons aborder maintenant les modifications qui ont été faites dans ce travail sur le modèle NRTL-PR présenté précédemment.
III-1-1. Facteur de répartition non-aléatoire
La valeur du facteur de répartition non-aléatoire α intervenant dans l’expression du terme résiduel (II-29) du modèle NRTL avait initialement été choisie à 0,2, c’est à dire à la valeur originale proposée par Renon et Prausnitz. Cependant des essais ont montré que les résultats sont sensibles à une variation du facteur de répartition non-aléatoire et qu’une valeur de α négative était préférable. Nous avons donc retenu la valeur α = –1. , c’est à dire celle du modèle LEMF, proposé par Marina et Tassios pour représenter les équilibres liquide-liquide se produisant sous basses pressions dans les mélanges hydrocarbures – eau.
III-1-2. Nouvelle décomposition en groupes et sous-groupes
Comme il a été indiqué dans l’introduction, la principale modification réalisée dans ce travail consiste à reprendre la définition de « groupes fonctionnels » d’une façon beaucoup plus simple. Le tableau II.3 du chapitre précédent montre quels sont les nouveaux « groupes » et « sous-groupes ». Tout comme dans le modèle UNIFAC (Fredenslund et al.,) par exemple ; les interactions entre sous-groupes appartenant à un même groupe sont considérées comme nulles, ce qui conduit à une forte diminution du nombre des paramètres du modèle.
Chapitre III Simplification du modèle prédictif NRTL-PR
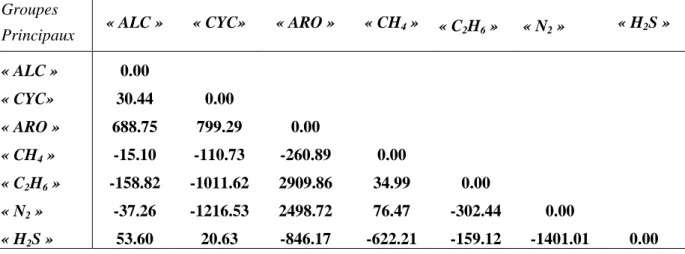
D’après les expressions (II-31,32) développées au chapitre précédent, l’énergie d’interaction Гij entre deux molécules i et j s’exprime en fonction des énergies d’interaction Akl entre groupes fonctionnels k et l ; Ces derniers dépendent des paramètres A°kl et B°kl suivant la relation : -1 T T B A Akl kl kl , T°=298,15K (III-1)
Les paramètres d’interaction entre groupes, A°kl et B°kl , sont les véritables grandeurs qui permettent de rendre la méthode NRTL-PR totalement prédictive, après l’étape préalable d’ajustement décrite dans ce chapitre. Ces paramètres obéissent aux conditions générales suivantes :
Lorsque l’on s’intéresse à des composés non polaires, comme c’est le cas pour les hydrocarbures ou composés « légers » considérés dans ce travail, les valeurs des paramètres A°kl et B°kl sont symétriques, soit :
A°kl = A°lk , B°kl = B°lk (III-2)
Ce n’est que pour la représentation des interactions avec un composé polaire, telles que celles entre l’eau et les hydrocarbures par exemple, que la nécessité de représenter le phénomène de démixtion impose que l’on dissymétrise ces interactions, ce qui conduit alors à la non égalité des interactions entre groupes k et l d’une part, et l et k d’autre part.
Nous considérons dans ce travail que les paramètres d’interaction à l’intérieur d’un même groupe principal sont nuls, contrairement à la dernière version du modèle NRTL-PR proposée par Escandell[24], dans laquelle ces paramètres étaient tous différents. On suppose donc que, entre deux sous-groupes k et l appartenant au même groupe principal :
A°kl = 0 , B°kl = 0 (III-3)
Cette dernière condition conduit donc à déterminer des paramètres d’interactions non nuls, A0 et B0, non plus entre sous-groupes mais entre groupes principaux. On comprend ainsi que cette hypothèse simplificatrice permet considérablement de diminuer le nombre total de
paramètres à ajuster lors de la mise au point de la méthode de contributions de groupes, ainsi
que ceux nécessaires à la représentation des systèmes complexes.
On peut noter également que, avec cette nouvelle version, les paramètres A°lk et B°lk
Chapitre III Simplification du modèle prédictif NRTL-PR
des paramètres A°k et B°lk une prédiction totale des propriétés thermodynamiques des
mélanges contenant un nombre quelconque d’alcanes. Pour les mélanges contenant d’autres composés, la méthode proposée conduit bien évidemment à ajuster préalablement un certain
nombre de paramètres. Toutefois, leur nombre est très restreint, comme on peut le voir sur les
exemples ci-dessous.
Exemples de la nouvelle décomposition comparée à la décomposition originale Nous considérons les mélanges binaires suivants.
Système Propane / Toluène : Notre nouvelle version conduit à :
ACH3/CH2 = 0 , ACHaro/Caro = 0
ACH3/Caro = ACH3/CHaro = ACH2/Caro = ACH2/CHaro ≠ 0
Il faut donc m = 2 paramètres A°k l et B°kl pour représenter ce système. Avec le modèle NRTL-PR original (Escandell), aucun de ces paramètres n’étaient nuls, ce qui conduisait à estimer :
ACH3/CH2, ACH3/CHaro, ACH3/Caro ACH2/CHaro, ACH2/Caro
ACHaro/Caro
Soit un ensemble de m = 12 paramètres A°k l et B°kl.
Système Ethyl-benzène / Methyl-cyclopentane : Notre nouvelle version conduit à :
ACH3/CH2 = 0 , ACHaro/Caro = 0 , ACH2cycl/CHcycl = 0 ACH3/Caro = ACH3/CHaro = ACH2/Caro = ACH2/CHaro ≠ 0
ACH3/CH2cyc = ACH3/CHcyc = ACH2/CH2cyc= ACH2/CHcyc ≠ 0 ACH2cyc/CHaro = ACH2cyc/Caro = ACHcyl/CHaro = ACHcycl/Caro ≠ 0
Il faut donc m = 6 paramètres A°k l et B°kl pour représenter ce système. Avec le modèle NRTL-PR original (Escandell[24]), on devait estimer :
ACH3/CH2, ACH2cycl/CHcycl, ACHaro/Caro
ACH3/Caro , ACH3/CHaro, ACH2/Caro, ACH2/CHaro
ACH3/CH2cyc , ACH3/CHcycl , ACH2/CH2cycl , ACH2/CHcycl
ACH2cyc/CHaro , ACH2cyc/Caro , ACHcyl/CHaro , ACHcyc/Caro
Chapitre III Simplification du modèle prédictif NRTL-PR
III-2. AJUSTEMENTS
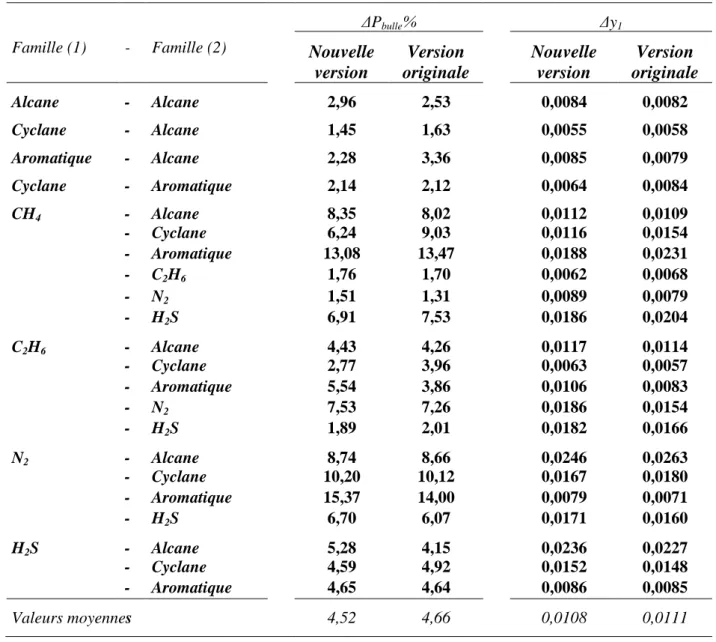
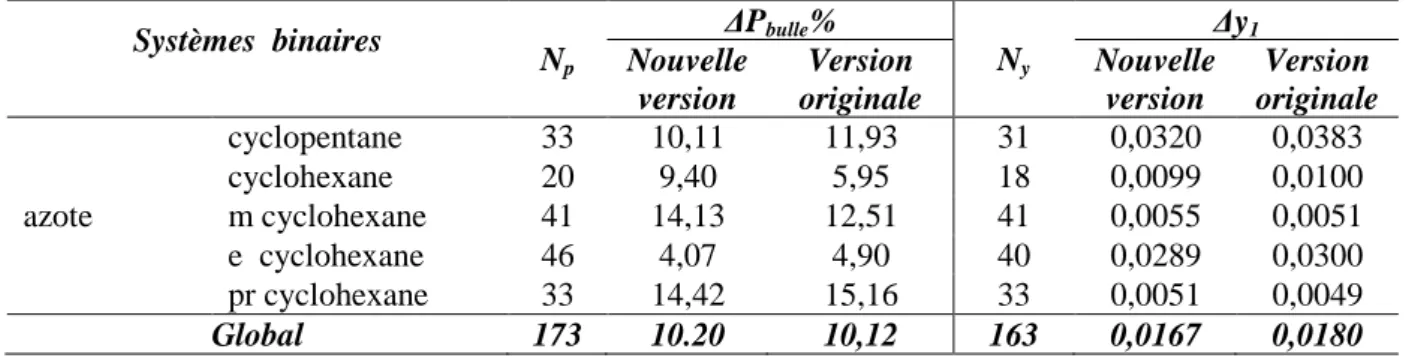
Nous présentons ici la base de données d’équilibres liquide–vapeur et d’enthalpies de mélange considérées dans ce travail, ainsi que la méthode utilisée pour l’ajustement des paramètres d’interaction entre les groupes principaux.
III-2-1. Base de données
La base de données est récapitulée dans le tableau III.1. Nous indiquons, pour chaque type de paramètres d’interaction à déterminer, A0
Groupe principal (1) / Groupe principal ( 2) et B0Groupe principal (1) / Groupe principal ( 2), le nombre de systèmes binaires comportant des équilibres liquide-vapeur (Nsys_LV)et des enthalpies de mélange (Nsys_H), ainsi que le nombre total des points expérimentaux de chaque type : NP, Ny et NH étant, respectivement, le nombre de points expérimentaux de pressions de bulle, de compositions y1 de la phase vapeur et d’enthalpies de mélange.
Chapitre III Simplification du modèle prédictif NRTL-PR
Tableau III.1. Base des données expérimentales nécessaires à l’estimation des paramètres d’interaction A0
Groupe principal (1) / Groupe principal ( 2) et B0Groupe principal (1) / Groupe principal ( 2).
Groupe principal (1) - Groupe principal (2) Données d’équilibres Liquide-vapeur Données d’enthalpies de mélange Nsys_LV NP Ny Nsys_H NH « ALC » - « ALC » 58 3814 1457 97 1963 « CYC» - « ALC » 46 2094 1041 66 3831 « ARO » - « ALC » 99 6771 4074 107 4555 « CYC» - « ARO » 35 3120 2198 35 1712 « CH4 » - « ALC » 22 3901 2370 - « CYC» 8 455 326 - « ARO » 17 900 516 - « C2H6 » 1 325 289 - « N2 » 1 471 170 - « H2S » 1 117 108 « C2H6 » - « ALC » 17 2174 935 - « CYC» 4 220 61 - « ARO » 8 327 236 - « N2 » 1 170 175 - « H2S » 1 82 69 « N2 » - « ALC » 14 1323 1130 - « CYC» 5 173 163 - « ARO » 9 584 184 - « H2S » 1 74 71 « H2S » - « ALC » 13 473 334 - « CYC» 5 134 134 - « ARO » 5 150 334
Chapitre III Simplification du modèle prédictif NRTL-PR
Le calcul des propriétés thermodynamiques, telles que les équilibres liquide-vapeur et enthalpies de mélange, est réalisé à l’aide d’un programme qui utilise le langage FORTRAN et qui a été mis au point par le LCPM (Laboratoire de Chimie Physique de Marseille). Il permet non seulement de calculer ces propriétés à l’aide d’équations d’état, mais aussi d’ajuster les paramètres de groupes A0
kl et B0kl. Plusieurs opérations ont été nécessaires afin de rendre nos données compatibles avec le programme de calcul :
Vérification des données déjà existantes,
Saisie de nouvelles données de la littérature,
Transformation de données existantes afin de pouvoir les utiliser dans les dernières versions du programme de calcul,
Vérification des paramètres des corps purs,
Classification des données de systèmes binaires en fonction des groupes principaux qu’ils comportent (tableau III.1) :
Alcane – Alcane Cyclane – Alcane Aromatique – Alcane Aromatique - Cyclane
Méthane – Alcane, - Cyclane, - Aromatique Ethane - Alcane, - Cyclane,-Aromatique Azote - Alcane, - Cyclane, - Aromatique
Sulfure d’hydrogène - Paraffine, - Cyclane, - Aromatique Composé « léger » - Composé « léger »
Chapitre III Simplification du modèle prédictif NRTL-PR
III-2-2. Méthode d’ajustement des paramètres d’interaction
Les paramètres d’interaction A°kl et B°kl obtenus dans ce travail sont donnés respectivement dans les tableaux III.2 et III.3. Ils ont été obtenus de façon successive, en se référant au classement proposé dans le tableau III.1. Il faut remarquer que:
La représentation des systèmes binaires Alcane - Alcane est totalement prédictive, puisque les paramètres A0 ALC (1) / ALC ( 2) = B0ALC (1) / ALC ( 2) sont nuls.
L’ajustement des paramètres doit être effectué dans l’ordre indiqué dans le tableau III-1. En effet, si l’on veut par exemple déterminer les interactions A0
CH4 (1) / ARO ( 2), il faudra considérer une base de données d’équilibres liquide-vapeur et d’enthalpies de mélange qui comprennent des systèmes binaires tels que : Méthane - Benzène, -Toluène, -Xylènes …., c'est-à-dire des données qui font intervenir, non seulement les interactions CH4 / ARO mais aussi CH4 / ALC. C’est pourquoi l’étude de ces dernières interactions doit être effectuée préalablement.
Tableau II.2. Table des paramètres d’interaction A0Groupe principal (1) / Groupe principal ( 2).
Groupes
Principaux « ALC » « CYC» « ARO » « CH4 » « C2H6 » « N2 » « H2S »
« ALC » 0,00 « CYC» 80,38 0,00 « ARO » 942,92 822,08 0,00 « CH4 » 646,22 1394,19 2117,96 0,00 « C2H6 » 170,12 571,29 1931,06 225,68 0,00 « N2 » 1973,76 2564,29 4359,15 648,49 1600,94 0,00 « H2S » 2480,49 2603,47 638,43 3376,52 2801,12 5105,76 0,00
Tableau II.3. Table des paramètres d’interaction B0
Groupe principal (1) / Groupe principal ( 2).
Groupes
Principaux « ALC » « CYC» « ARO » « CH4 » « C2H6 » « N2 » « H2S »
« ALC » 0.00 « CYC» 30.44 0.00 « ARO » 688.75 799.29 0.00 « CH4 » -15.10 -110.73 -260.89 0.00 « C2H6 » -158.82 -1011.62 2909.86 34.99 0.00 « N2 » -37.26 -1216.53 2498.72 76.47 -302.44 0.00 « H S » 53.60 20.63 -846.17 -622.21 -159.12 -1401.01 0.00