FACULTE DES SCIENCES
ÛD
3
-sr
UL
/9
9
J
THESE PRESENTEEA L’ECOLE DES GRADUES DE L'UNIVERSITE LAVAL
POUR OBTENIR
LE GRADE DE MAITRE ES SCIENCES PAR
DENIS LEMAIRE. B.Sc. (LAVAL I
97
O)ETUDE SPECTROSCOPIQUE DES PERCARBONATES ALCALINS.
Décembre 1971
ÿ
%
$
TABLE DES MATIERES
Remerciements ... . Liste des figures... .. . . . . Liste des tableaux... INTRODUCTION . ... . . . CHAPITRE I PARTIE EXPERIMENTALE ... .1 . PREPARATION DE KHCO^... .
1.1. A partir de 11éthoxyde ... 1.2. A partir du bicarbonate . ... .
1.3. A partir de 1'hydroxyde . . . . . 1.4. Propriétés de KHCO^ . , ... 1.5. Préparation de nos échantillons . 1.6. Analyse du produit ... 2. PREPARATION DE KgCgO^ ...
2.1. Méthodes chimiques ... 2.1.1. A partir du peroxyde alcalin
octahydraté M0p*8Hp0(M = Na ou K) ... . . ... 2.1.2. A partir de 1 * hydroxyde . . 2.1.3. A partir du superoxyde . . . 2.2. Méthode électrolytique ... 2.3. Propriétés de K2C2^6 ... 2.4. Préparation de nos échantillons .
page v vi vii 1
3
4 4 5 56
7
8
10
10
10
10
11
1212
14 iii3. PREPARATION DE KgCOU'^HgOg.... 16 3.1. Méthodes de préparation et histo
rique des controverses.... 16 3.2. Propriétés de K2C®3* ’^‘■'^2. . 18 3.3. Préparation de nos échantillons . 18 4. ENREGISTREMENT DES SPECTRES ... 19 4.1. Appareils ... 19 4.2. Techniques expérimentales .... 21
4.2.1. Spectres'infrarouges
(4000-200
cm-1) ...21
4.2.2. Spectres Raman (4000-20 cm ^) 22 CHAPITRE II DISCUSSION DES RESULTATS ... 24 1 . LE PERCARB0NATE KHC0,... 2$ 2. LE PERCARBONATE KgCgOU... 34 3. LE PEROXYHYDRATE KpCO^* 3H2
02
.... 4? CHAPITRE III RESUME ET CONCLUSIONS ...53
BIBLIOGRAPHIE ... ...57
RemenciementA
Çe démine, témoigne*, ma ptuA Aincène gnatitude au pnofeAAeun
''Paul- /htoirie Çiguène poun Aon encounagement et aca conAeitô judicieux
agi. w.'ont penmià de. menen a bien cette iheAe. De ptuA, je. te nemencie
poun t'aAAiAtance qu'il a bien voulu m'accon.deK poun t'inienpnétaiion
deA neAuttatA expénimentaux.
(je veux nemencien également teA docteuAA Kagimiena Henman et ÇoAe Luxa /knau, cottabonateunA du pnofeAAeun Çiguène, poun. teun aide
conAtante; teA pnofeAAeunA Rodnigue Savoie et Otto Çubeti poun t'uti- tiAation de teuKA tabonatoineA de ApectnoAcopie 9,aman et d'anatgAe chi mique neApectivement; et fteAAieunA 'René Robent et Çénand Iranien poun teun ciAMAtance technique.
(f' expnime auAAi ma pnofonde neconnaiAAance a Madame hagdeteine
Çiguène qui a dactgtognanhié cette iheAe.
(je inavail a pu étne exécuté gndce a une Aubvention du ÇonAeil h attonat de RechencneA du Çanada au PnofeAAeun Çiguène.
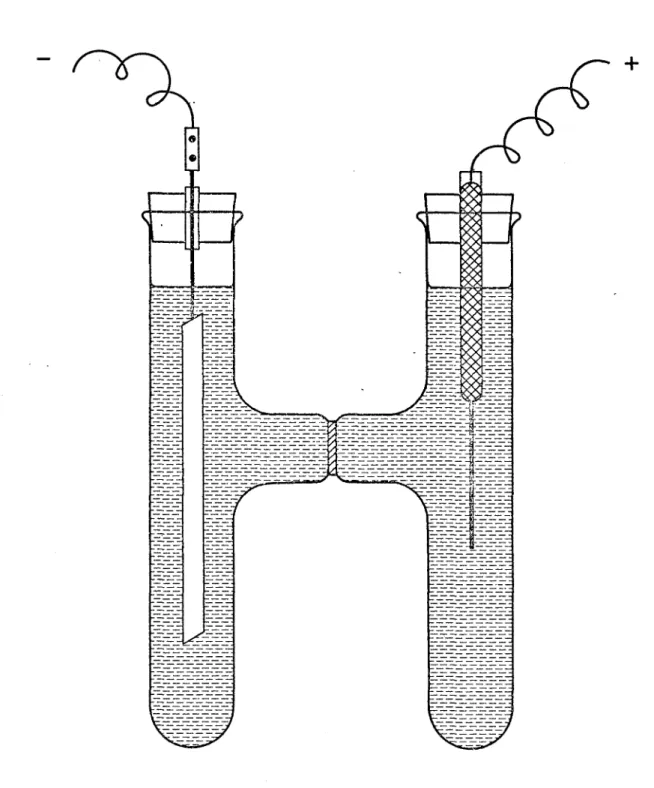
page Figure 1 - Cellule électrolytique... ... 15 Figure 2 - Spectres de vibration de KHCO^...26
Figure 3 - Modèle moléculaire illustrant la configura
tion probable de l’ion permonocarbonate. . 30
2_
Figure 4- - Configuration probable de 11 anion CpO^ dans le perdicarbonate de potassium cristallin... 35 Figure 5 ~ Spectres de vibration de KpCpO^... 38 Figure 6 - Spectre d* absorption de KpCpO,- dans 1 * infra
rouge lointain ... 44
Figure 7 - Spectre infrarouge d'un échantillon de KpCpO^ préparé chimiquement à partir du
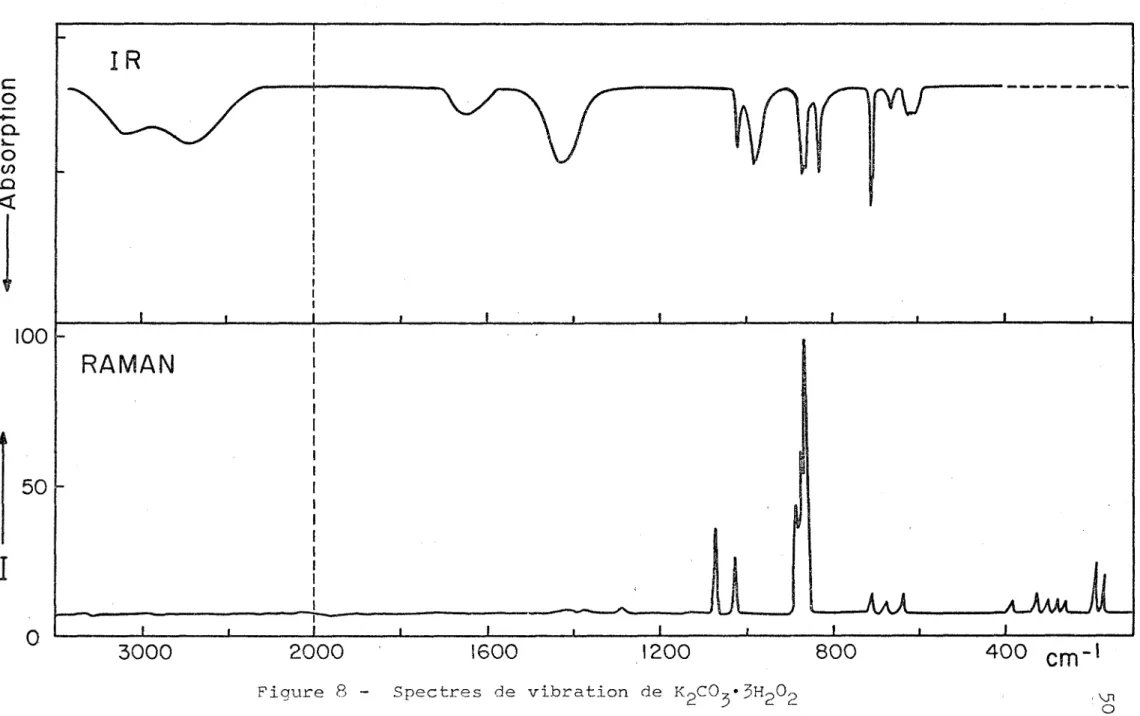
peroxyde de potassium ... 48 Figure 8 - Spectres de vibration de KpCO^^HpOp ... 50
LISTE DES TABLEAUX
page Tableau I - Dosage par émission atomique du potassium
dans le percarbonate KHCO^ . ...
9
Tableau II - Fréquences de vibration de KHCO^ ....29
Tableau III - Classification des vibrations fondamentales de
1
' anion CgO^ """ ...36
Tableau IV - Spectres de vibration de l'ion CgO^ . .2
_39
Tableau V - Fréquences de la vibration de valence 0-0dans divers peroxydes organiques .... 41
Le peroxyde d'hydrogène forme deux sortes de composés avec les carbonates alcalins:
a) les peroxyhydrates comme ^
2
^3
" j$^2^2
qui sont, effectivement, des composés d’addition où HgOg joue un rôle analogue à l'eau de cristallisation des hydrates;b) les vrais percarbonates où il y a une réaction chimique entre le peroxyde et les carbonates. On connaît deux sortes de ces percarbonates: i) les peroxymonocarbonates comme KHCO^ qui sont les analogues des peroxymonosulfates, ou caroates KHSCy, et ii) les peroxydicarbonates comme , qui correspondent aux peroxydisulfates K
2
S2^8
au^res*On connaît peu de chose sur la structure de ces compo sés. Sur le percarbonate KHCO^_, aucune étude spectroscopique ni en infrarouge, ni en Raman n'a été faite jusqu'à ce jour. Par contre, le peroxydicarbonate K2<"2®6 étudié Par M.F. Sorokina (1) en "196$ . Il prit un spectre infrarouge et proposa une attribution des bandes observées en assumant pour ce composé la structure suivante:
K K
0
0
0
I0
12
Cette structure entièrement covalente ne saurait être cor recte d’après la théorie de valence qui prévoit la nature ionique pour les composés de ce genre.
Les carbonates alcalins peroxyhydratés furent étudiés au moyen de la spectrographie d’absorption infrarouge par Mlle C. Rocchiccioli (2) en 1965. Elle fit 1’attribution des
bandes d'absorption et confirma
1
'impossibilité de préparer un véritable persel du genre de KgCO^. x HgO (x variant entre1
et 3) .Dans le cadre d'une étude systématique des dérivés de H
2^2
(5
_6
), nous avons pensé qu'il serait intéressant de fai re l'étude de KHCO^, d'approfondir la question de la struc ture de k2
C2®6
compléter les "recherches sur le peroxy-hydrate KgCO^.^2
^2
' nous servant, dans chacun des cas, des spectres i.r. et Raman.PARTIE EXPERIMENTALE
Nous donnerons d’abord pour chacun des composés, un résumé des méthodes de préparation et des propriétés tel les que nous les trouvons dans la littérature, suivi de la description de la méthode employée pour ce travail. Ensuite nous traiterons de la préparation des échantil lons. Enfin, nous donnerons un bref aperçu des conditions d’enregistrement des spectres.
1 . PREPARATION DE KHCO,
I. 1. A partir de l’éthoxyde
J. R. Partington (7) prépara MHCO^ (M =Na ou K) en ap pliquant en partie la méthode de Leblanc et Zellmann (
8
), i.e. par11
action du gaz carbonique sur une suspension al coolique de m00H.TiHgOg , préparée selon d’Ans et Friederich(9). Un mélange de peroxyde d* hydrogène à 30% en poids et d'alcool fut ajouté à une solution d'éthoxyde alcalin dans l'alcool refroidi en-dessous de -10°C. Le rapport K:
est de 1:1. Le produit se solidifie après refroidissement. On fait barboter le COg dans la suspension alcoolique, la
température étant maintenue en-dessous de -10°C. Quand la phénolphtaléine n’est plus colorée le précipité est filtré rapidement et lavé avec de l'éther à froid.
T.P. Firsova (10), en 1969, prépara le peroxybicar- bonate de potassium à partir du bicarbonate de potassium et du peroxyde d'hydrogène. Il prépara une solution sa turée de bicarbonate de potassium dans du peroxyde d'hydro gène à 60%, à la température ambiante. Il filtra la solu tion pour enlever le sel non dissous, la mit dans un dessi- cateur en présence d'acide sulfurique concentré, à 0°C, pendant plusieurs jours pour laisser cristalliser le pro duit. Une autre méthode (10) consiste à ajouter lentement du KHCO? à du peroxyde d* hydrogène de 60 à 90% maintenu à 0°C, jusqu'à ce qu'il y ait un excès, à laisser reposer le tout à 0°C pendant 24- heures, puis à filtrer et à laver le solide avec de l'alcool absolu et de l'éther anhydre.
1.3. A partir de 1'hydroxyde
T.P. Firsova (11) prépara KHCO^ de la façon suivante: il mélangea une solution de KOH à $0% avec du HgOp dans un rapport K0H:HgOg de 1 : 1 . Il fit passer un courant de C0g pendant environ une heure. La réaction fut réalisée à des températures variant entre 0° et -20°C. Par ce procédé, il essaya, mais en vain, de préparer KpCO^. Par une méthode analogue (
12
), il prépara NaHCO^.HpO, mais il fut incapa ble de le rendre anhydre, car les agents de déshydratation causent une perte d'oxygène actif et de C0g.1.4. Propriétés de KHCO;|
L’analyse thermique (11) a révélé que KHCO^ est sta ble jusqu’à 60°C, après quoi il montre deux effets:
a) un effet exothermique à 60°-80°C dû à la décom position en bicarbonate avec dégagement de tout
1
’oxygène actif et une partie considérable du CC>2
et de l’eau,b) un effet endothermique à 185°~200°C dû à la décom position du bicarbonate.
La décomposition thermique se fait selon le schéma: 2khC04 --1 2khc0^ + C>2 —» K2CO^ '+ C02 + HgO + 02.
A la température ambiante, KHCO^_ se décompose avec perte d’oxygène actif, sans perte de COg. A 0°C, il se dé compose lentement.- En présence de ^SO^, il se décompose fortement. Dans l’eau il s’hydrolyse libérant 1’oxygène ac tif et une partie du COg. A l’aide des spectres infrarouges (voir plus loin), nous avons pu confirmer que KHCO^ se dé compose en KHCO? plus ou moins rapidement selon les conditions
La diffraction des rayons X (10) a permis de confirmer
1
’identité des produits obtenus par différentes méthodes de préparation. V.I. Sokol (13) étudia ce composé par les deux méthodes de diffraction des rayons X et de réfraction de lalumière. Cette dernière conduit à des valeurs de 2.080 et 1.467 pour d^° et n^ = (n x n^ x n%)/^ .
La méthode qui semble la meilleure est celle de T.P. Firsova (11), soit à partir de 1'hydroxyde alcalin. Avec huit grammes de KOH, nous préparons une solution de concen
tration d'environ 50%» nous refroidissons à -20°C et nous ajoutons lentement du peroxyde d’hydrogène concentré de façon à avoir un rapport KOH : HgOg = 1:1. Nous faisons passer un courant de CO^ dans le mélange maintenu à -20°C pendant environ une heure. Ensuite, nous filtrons sur un filtre de verre fritté et nous lavons le précipité blanc avec de
1
'alcool absolu froid et de l'éther anhydre froid. Enfin, nous séchons le composé au moyen d'un courant d'air sec.La réaction est la suivante :
KOH + HpOg + COp --1 KHCO, + HgO
En fait, il serait plus exact de dire que le mélange hydroxyde-peroxyde conduit à la réaction d'équilibre sui vante :
KOH + HpOg K00H + HpO
Ensuite, le COg favorise la formation du peroxybi- carbonate selon la réaction qui suit:
K00H + COg -- ► KHCO^
échantillon à partir du bicarbonate de potassium selon une des méthodes décrites par T.P. Firsova (10). Nous avons mélangé très lentement du KHCO^ à du peroxyde d'hydrogène à 91% en poids maintenu à une température de 0°C. Nous
avons traité la solution avec un excès de KHCO^. Nous avons maintenu le mélange solide-liquide à une température de 0°C pendant vingt-quatre heures. Ensuite nous avons filtré la solution et lavé la partie solide à l'alcool absolu et à l'éther anhydre.
Le spectre IR du produit ainsi préparé était identi que à celui de la première préparation.
1.6. Analyse du produit
Une titration avec de l'acide chlorhydrique et une titration du potassium par la méthode, d*émission atomique nous ont permis de confirmer que le produit obtenu avait bien la composition KHCO^.
Pour titrer avec l'acide chlorhydrique, nous avons d'abord dissous
0,98
g. de KHCO^ dans l'eau, en présence de MnOg pour favoriser le dégagement de1
'oxygène actif. La réaction suivante se produit:2khco4 —fr k2co^ +
co
2
+ HgO + o
2
Ensuite nous avons titré le carbonate avec HCl 0.1 N. Lors du titrage, les deux réactions subséquentes se produisent:
a) KgCCy + HCl ► KHCO^ + KC1 b) KHCOz + HCl > HgCO, + KC1
Le volume calculé de solution 0.1 N d'acide nécessaire était de 84.4- ml; la valeur trouvée fut de 84- ml.
Nous avons aussi dosé le potassium par émission ato mique. Des solutions étalons de 5 p.p.m. et de 10 p.p.m. ont été préparées avec du KC1 et de l'eau bidistillée.
Nous avons fait de même avec le percarbonate en supposant, pour le calcul des quantités en poids de potassium, qu'il s ' agissait de KHCO^ pur. Les résultats donnés au Tableau I confirment bien la formule KHCO^ du produit obtenu.
TABLEAU I
Dosage par émission atomique du potassium dans le percarbonate KHCÔ^
Solution Concentration
1
er dosage2
e dosage 5 p.p.m. 21.5#21
.0
# Etalon10
p.p.m. 48.0# 48.0# KHCO^ 5 p.p.m.22
.0
#21
.8
#10
p.p.m. 48.5# 48.0#2
.
PREPARATION DE ^2C2°62.1. Méthodes chimiques
2.1.1. A partir du peroxyde alcalin octahydraté MOq.SHqOCm = Na ou K)
T.P. Firsova (14) prépara du peroxydicarbonate de so dium en faisant passer du gaz carbonique sur du peroxyde de sodium octahydraté. Il prépara ce dernier à partir soit du peroxyde NagOg, soit du superoxyde de sodium NaOg avec le peroxyde d'hydrogène (15). Pour la préparation du peroxy dicarbonate, la pression de COg était maintenue à
5
mm de Hg pendant deux heures, puis à 50 mm de Hg pendant une demi- heure. Enfin, le récipient était rempli, puis évacué àplusieurs reprises. Le rendement maximum, qui était d'en viron 95%, a été obtenu à 10°C. Le produit final contenait
aussi du bicarbonate.
2.1.2. A partir de 1'hydroxyde
Des solutions concentrées de peroxyde d'hydrogène et d'hydroxyde de potassium sont mélangées à basse température
(11). Ensuite, on fait passer un courant de COg pendant trente minutes. Les rapports KOH : H^Og : HgO sont 1,0 : 0,5 : 6.0 . Entre 0° et -15°C, le produit obtenu a la composition
* x H2^’ °ù x varie entre 0.15 et 0.66 . Le sel de sodium peut être préparé de la même façon (12).
2,1.3. A partir du superoxyde
Riesenfeld et Mau (16) préparèrent le peroxydicar- bonate en saturant une suspension alcoolique de superoxyde
avec CC
>2
à -10°C ou à -5°C.Makarov et Vol'nov (17) 1'obtinrent par 1'action de COg sur du superoxyde finement pulvérisé; MgCO^ serait un produit intermédiaire de cette réaction.
Wolffenstein et Peltner (18), Partington et Fathallah (7), Celis et Masaguer (19) 1
1
obtinrent en faisant passer du COg dans une suspension alcoolique de . x HgOg • Y HgO; (x = 1,2,3; y = '/%, 1 'A).T.P. Firsova (20) prépara KgCgOg en faisant passer sur le superoxyde un mélange de gaz carbonique saturé d’eau à 0°C. Il faut augmenter graduellement la teneur de COp dans le mélange gazeux de 1% à 33% en volume. La réaction à 10°C prend deux heures ; ensuite le produit est séché
par l'air pendant trois heures. Il est brun pâle et contient du bicarbonate de potassium. Sa pureté est d'environ 83%. Selon les auteurs il y aurait formation intermédiaire de
k
2
co4.T.P. Firsova prépara aussi le percarbonate de sodium (
21
) mais en mélangeant le superoxyde avec de la glace et en faisant passer le COg. Le produit obtenu contenait en viron 75% de NagCgO^. A noter que dans ce dernier cas on peut partir du peroxyde de sodium au lieu du superoxyde (21
).12
2.2. Méthode électrolytique
Cette méthode a été d'abord utilisée et bien décrite par T.P. Firsova en 1962 (20) qui électrolysait une solu tion de carbonate de potassium. Il obtint comme produit avec une pureté de 99.9%. Sa couleur était bleue pâle et non pas brunâtre ni orangée comme pour les pro duits des méthodes chimiques. il s'agit, en fait, d'une électrolyse de l'eau avec oxydation du carbonate à l'anode, et dégagement d'hydrogène à la cathode:
2K2C05 + 2H20 -- )
k2C2°6 + 2K0H + 2h2 •
Nous donnerons plus loin les détails techniques.
2.3. Propriétés de
Le perdicarbonate de potassium est un solide pulvé rulent de couleur variable selon la méthode de préparation. Ainsi le composé préparé par voie chimique est généralement coloré en brun ou orange. On a démontré par résonance ma gnétique nucléaire (
22
) que cette couleur est due à la pré sence d'ozonide, K0%. Quant au produit obtenu par électro lyse, il est blanc légèrement teinté de bleu. Cette cou leur s'atténue par lavage à l'éther, mais ne disparaît ja mais complètement. Comme les sels alcalins sont généralement blancs ou incolores, la teinte bleue doit provenir de quel que impureté, encore non identifiée. Il ne saurait s'agird’ozone car la couleur semble persister indéfiniment à tem pérature ordinaire. D’ailleurs aucun des spectres que nous avons obtenus ne montrait d1indication des bandes de
1
'ozo ne à 1035 et 705 cm ^.Par ailleurs les autres propriétés physiques sont les mêmes pour les percarbonates obtenus par les deux mé thodes (
20
,21
)j ainsi la densité,2,02
et1
’indice de ré fraction, nOD= 1.443 (22
) d'où l'on tire la valeur de 23.65 cm^ pour la réfraction moléculaire. La diffraction des rayons X n'a pas permis de déterminer le système cris tallin ni les dimensions de la maille parce que les cris taux sont trop petits et très imparfaits. Cependant il semble que la symétrie du cristal soit assez basse. De plus on a vérifié que les deux.percarbonates K2
C2®6
Na^CgO^ sont isomorphes.Quoique beaucoup plus stable que le permonocarbonate KHCO^, le perbicarbonate K2C2^6 es^ hygroscopique et faci
lement hydrolysé. Cette réaction conduit au bicarbonate, avec dégagement d'oxygène dû à la décomposition du peroxyde d'hydrogène en milieu alcalin*
K2C2°6 +
2
h2° --- * 2KHC0, + H2
02
H2°2
--- & h2
° + %02 .Gardé à sec à température ambiante le percarbonate bien purifié ne se décompose pratiquement pas. Ainsi on
a constaté une perte d’oxygène actif de
1
% seulement après un an (22). Par chauffage vers 130°C il se décompose selon la réaction
k
2C
2°6 ---*k
2c03 + c02 + 'A
°2 •2.4-. Préparation de nos échantillons 2.4-. 4 . Méthode chimique
Quoique la méthode de préparation électrolytique s'a véra pour nous la plus utile à cause de la plus grande pu reté du produit obtenu, nous avons quand même expérimenté un certain nombre de méthodes chimiques. Nous en avons retenu une des plus simples, due à Firsova (21).
Nous avons mélangé du peroxyde de potassium à de la glace dans le rapport KgOg : HgO de 1 : 3. Sur ce mélange maintenu à
0
°C, nous avons fait passer un courant gazeux qui contenait un pourcentage en volume de gaz carbonique augmentant graduellement de10
% à100
%, jusqu'à ce que le composé ne diminue plus de poids. Le produit fut lavé avec de1
'alcool absolu et de l'éther anhydre, puis placé sous vide pendant une heure.2,4.2. Méthode électrolytique
Pour 1'électrolyse, nous avons utilisé un récipient de Pyrex en forme de H dont les deux branches sont séparées par un diaphragme en verre fritté (Figure 1). Un fil de
/ ' +
platine de 40 mm de long servait d'anode et la cathode était faite d'une plaque de platine de 0,3 mm et d1 environ I x 10 cm. Une solution de carbonate de potassium de den sité 1.49 fut soumise à un courant électrique continu de 0.1 à 0.2 ampère de façon à maintenir la tension à
15
ou 16 volts. La cellule était placée dans un bain à -20°C. II fallait de temps en temps agiter la solution à l'anode avec une tige de verre. Après six heures d'électrolyse, nous avons obtenu 1.8 g. de solide bleu pâle. Le pro duit lavé à l'alcool absolu, puis à l'éther était séché dans un courant d'air sec pendant.quinze minutes. Gardé dans un endroit sec à 0°C, il semblait très stable.3. PREPARATION DE K^CO^^HgOg
3.1. Méthodes de préparation et historique des controverses
Les peroxyhydrates furent préparés d'abord par Tana- tar (26) qui pensait avoir obtenu de vrais percarbonates de formule MgCO^.xHgO . Il traitait à froid un carbonate al calin par du peroxyde d'hydrogène à 3%, puis précipitait le produit en y ajoutant de 1'alcool absolu. Au contraire, Riesenfeld (16,27) affirmait que le produit était un per- oxyhydrate. Selon lui, une solution saturée d'iodure de potassium ajoutée à une solution neutre de composé entrai
ne la libération immédiate d’iode s'il s'agit d'un véri table persel. Or, le percarbonate de Tanatar ne libérait pas d'iode. Par ailleurs, Kasanetzky (28) utilisant le bicarbonate au lieu du carbonate obtint des composés qu'il considérait comme étant KgCOr
.2
'A-HgO, K2
C®6
* ^H2
^’CO^.
2
H2
O. Plus tard (29), il crut préparer le peroxyhydra- te CsgCO^.2
H2
O2
. Vers la même époque, Peltner (30) prétendit obtenir de vrais percarbonate s tels que RbgCO^^ j/^^O, Rb
2
C0
^.H202
.2
h20
et RbgCO^.2
h202
.H20
.Il y a" une vingtaine d'années, Partington (?) publiait une étude détaillée de plusieurs percarbonates alcalins. Il contestait la validité du test de Riesenfeld pour établir la distinction entre les vrais percarbonates et les peroxy- hydrates. Il décrivait, entre autres, la préparation de KgCO, .2 Xz/HgO, KgCO^.HgOg.l'/zHgO et KgÇO, ^HgOg.HgO. Sa mé
thode consistait à dissoudre du carbonate de potassium dans du peroxyde d'hydrogène à
30
% selon les rapports de1
:1
, 1 : 2 et 1 : 3. Il maintenait le mélange à ~5°C, puisajoutait un excès d'alcool absolu. Après avoir lavé et filtré le produit, il le séchait en présence de CaClg sous vide.
Par après Celis (19) répéta la même expérience et infirma les conclusions de Partington. Selon lui, il n'existait aucune justification pour conclure à la formation d'un vé ritable persel. Rocchiccioli (2) confirma le point de vue de Celis dans une étude par thermogravimétrie, analyse
18
thermique et spectrographie infrarouge de carbonates alca lins peroxyhydratés. Enfin, Franchuk (31) prétend avoir démontré par les spectres E.P.R. qu'il s'agit vraiment de peroxyhydrates.
3.2. Propriétés de KgCO? . 3^2^2
K
2
CO3
. es^ un composé cristallin blanc. Il est très sensible à 1'humidité. A l'air même très sec il finit par se décomposer lentement sans toutefois devenir jaune comme c'est le cas pour KHCO^_. En présence de CaClg il de meure inchangé pendant 48 heures." Toutefois, dans un des- sicateur au-dessus d'acide sulfurique concentré et sous vi de il se décompose totalement en quelques heures. Le test de Riesenfeld (24) est négatif-; mais ceci ne permet pas de conclure à 1'existence d'un peroxyhydrate. En effet il est maintenant admis qu'un test positif, i.e. la libéra tion de l'iode, caractérise un vrai persel, mais que le contraire n'est pas nécessairement vrai (7
,25
).3.3. Préparation de nos échantillons
On a suivi la méthode utilisée par Partington et aussi par Celis. On dissolvait très lentement 4.88 g. de carbonate de potassium dans
12
ml. de peroxyde d'hydrogène à 30% maintenu à -10°C. Ces quantités sont dans le rap port 1:3. Dès que tout était dissous, on ajoutait lente ment 24 ml. d'alcool absolu. On laissait le précipité seformer pendant une demi-heure à -10°C. Après quoi il était filtré et lavé plusieurs fois à l'alcool absolu, puis à l'éther anhydre. Enfin, on le plaçait pendant une heure dans un dessicateur en présence de CaClg (ou HgSO^) sous vide.
Pour fin de comparaison on a aussi préparé I^CO^ . HgOg . 1 'AhgO . La seule différence dans le procédé con siste à prendre trois fois moins de peroxyde d'hydrogène de façon à avoir le rapport K2C<^3 : HgOg de 1:1. Comme le carbonate ne se dissout pas totalement, on peut préci piter le produit après avoir filtré ou non l'excès de
carbonate; dans les deux cas, les résultats sont les mêmes.
4. ENREGISTREMENT DES SPECTRES 4.1. Appareils
Un spectrophotomètre de Perkin-Elmer, modèle 621, à double faisceau a été utilisé pour l'enregistrement des
w zj
spectres infrarouges entre 4000 et 200 cm . L'appareil était calibré périodiquement à l'aide d'un mince film de polystyrène. La précision des fréquences était de + 2 cm dans le cas des bandes fines. Pour l'infrarouge lointaih on disposait d'un interféromètre du type Michelson, de.
-1
f ASCiÜV:-"Research and Industrial Instrument Co.", modèle FS-720.\n \ uvrv- . r.^ Une calculatrice (Fourier Transform Computer, modèle FTC x^o/ir \ -100/7 de Beckman) couplée à un enregistreur X-Y (modèle HR-80),
20
transformait les interférogrammes en spectres d'absorption pour enregistrement direct. Dans le domaine de fréquence utilisé, soit de 4-0 à 4-00 cm la résolution théorique maximum de 11 appareil est de 2.5 cm et la précision sur
+ _y]
les fréquences est d'environ -
2
cmPour les spectres Raman (4-000-20 cm ) nous avons utilisé d'abord un spectromètre Cary (modèle 81) à lampe de mercure (Toronto arc). La raie excitatrice (4-358
X)
était isolée par une solution filtre constituée d'un mélange d'orthonitrotoluène et d'éthyl-violet dans 1'alcool iso propyl ique . La largeur des fentes du monochrometeur
pouvait varier entre 2.0 et 4-.0 cm ^ suivant 1 ’ intensité des bandes étudiées. La précision des fréquences était d'environ - 1 cm \
Nous avons aussi utilisé un spectromètre à source
laser (Coherent Radiation, modèle 52), décrit ailleurs (32), d'une puissance moyenne de
500
mw pour chacune des raies principales à 514-5.25X
et 4-879-9X.
Le faisceau Raman était analysé avec un monochromateur double (spex, modèle 14-00). Le détecteur était une photomultiplicatrice (star Tracker FW-130) refroidie à -20°C par un courant d'azote. Le signal était amplifié au moyen d'un picoampèremètre à courant continu (victoreen, modèle 1001) et les spectres étaient tracés sur un enregistreur-Yew (modèle LER 12a).entre 20 et 200 microns. La résolution spectrale pouvait varier entre
0.5
et3.5
cm ^.4.2. Techniques expérimentales
4.2.1. Spectres infrarouges (4000-200 cm ^) Pour l'étude des trois composés nous avons utilisé la technique de la suspension dans le nujol, dans le fluo- rolube et aussi parfois dans
1
1hexachloro1-3
butadiene. Le premier était surtout utilisé dans la région de 1350 à 200 cm , le second de 4000 à 1350 cm . Les solides étaient d’abord finement pulvérisés au mortier d'agate en atmosphère sèche, puis bien dispersés dans le liquide de manière à obtenir une suspension uniforme. On pressait une goutte de cette dernière entre deux fenêtres de sel.Pour KgCgOc nous avons pu employer des fenêtres de NaCl et de Csl; ces dernières sont transparentes jusqu'à environ 200 cm . Nous avons cependant obtenu des résultats aussi bons avec des fenêtres de AgCl. Par contre pour KHCO^ et k2C^3 * ^H2^2 nous
n1
avons pu utiliser ni NaCl ni Csl, qui semblaient provoquer la décomposition du percarbonate.Seules des fenêtres de AgCl nous ont permis d'obtenir de bons spectres
22
Mentionnons ici que Karetnikov et Sorokina (1) avaient employé la méthode de la pastille de KBr pour leur étude de K2<“2^6 en infrarouge. Nous avons aussi expérimenté cette technique en mélangeant
2
mg de per- carbonate dans 100 mg de KBr finement broyés pour en faire une pastille. Les spectres étaient cependant de qualité très inférieure à ceux obtenus avec une suspen sion dans un liquide. Le percarbonate paraissait réagir lentement avec le bromure à en juger par les fluctuations des bandes d.' absorption; particulièrement celle de la—
'1
vibration 0-0 vers 880 cm . Des tentatives pour appliquer cette même méthode aux deux autres percarbonates n’ont don né aucun résultat valable. Les pastilles devenaient vite blanchâtres puis complètement opaques à cause de
11
oxydation du bromure. Kyrki (42) a noté une réaction semblable, bien que moins forte, avec le persulfate KHSO^.Dans 1'infrarouge lointain, au-delà de 200 cm , nous n'avons étudié que le percarbonate KgCgO^ au moyen d'une suspension dans le nujol entre des fenêtres de silicium et à température ambiante.
4.2.2. Spectres Raman (4000-20 cm
Pour les études avec le spectromètre à arc de mer cure (Cary, modèle 81) le percarbonate finement pulvérisé était pressé dans un tube de Pyrex à extrémité conique
rentrante tel que précédemment décrit (3)• Avec le spectromètre à excitation laser
1
'échantillon, égale ment dans un tube de Pyrex, était éclairé à incidence normale. Le percarbonate KgCgO^ a pu être ainsi étudié à température ambiante. Les deux autres, KHCO^ etK
2
<“<^3
# ’ on^ dû être refroidis avec l'azote liquide pour prévenir toute décomposition thermique sous1
'in fluence du faisceau laserCHAPITRE II
DISCUSSION DES RESULTATS
1. LE PERCARBONATE KHCO^
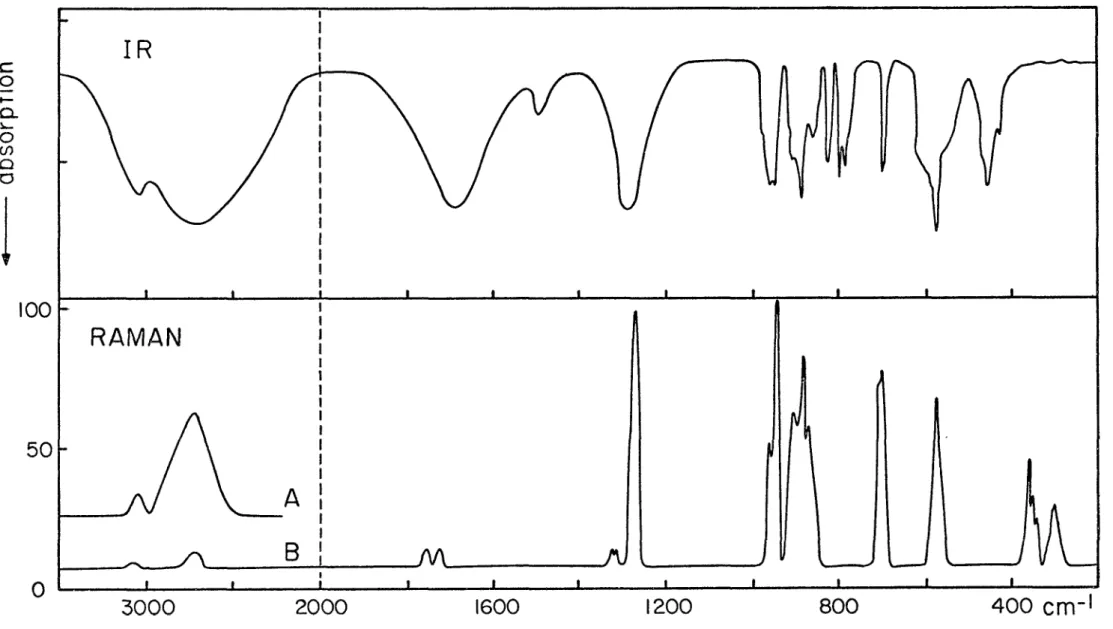
Après de nombreux essais souvent infructueux à cause du caractère instable et hygroscopique de ce com posé , nous avons réussi à obtenir des spectres vraiment représentatifs et reproductibles, tant en infrarouge qu'en Raman. Une illustration, à échelle réduite, en est donnée sur la figure 2. D'après le grand nombre de bandes, et la concordance des fréquences dans les deux
spectres, il est évident que
1
'anion HCO^ n'a pasune structure de symétrie très élevée. On pouvait natu rellement prévoir ceci en se basant sur le modèle le plus plausible d'après les structures connues des molé cules parentes, HCO^ et HgOg. En effet, par analogie avec l'acide de Caro, l^SO^et ses sels (
6
) on peut écri re les formules de résonance suivantes pour1
'anion HCO^_ :0—0
Z
Ho—o
Z
H ©(Z'Z
cZ
\ 0r
zc\
0 o' o (=)
i iiin
RAMAN
2000
3000
Le seul élément de symétrie possible dans ce cas est naturellement le plan (groupe ponctuel C ). De toute façon les spectres ne sauraient nous renseigner là-dessus puisque toutes les vibrations doivent être actives, tant en infrarouge qu'en Raman.
En l’absence de toute donnée expérimentale sur la structure de ce percarbonate nous pouvons imaginer un mo dèle basé sur les structures bien connues des composés parents; soit les bicarbonates alcalins et le peroxyde d'hydrogène. Effectivement nous avons pris comme terme de comparaison le cristal de NaHCO^ plutôt que celui de KHCO^. Dans ce dernier, en effet, les anions HCO^ forment des dimères fortement associés par ponts d'hydrogène (33) tandis que dans NaHCO^, les anions forment des chaînes in finies (34). Il en résulte des différences importantes dans les spectres de vibration de ces deux cristaux (35). Or il est à peu près certain que 1'anion HCO^ ne forme pas de dimères. Tout d'abord sa configuration géométrique s'y prête mal. De plus, il n'existe aucune possibilité de structure en résonance qui conduirait à placer une charge formelle positive sur l'oxygène donneur du proton de façon à accroître le caractère ionique de la liaison 0-H, et partant, la charge positive sur l'atome d'hydrogène. Au contraire, dans le cas des bicarbonates une telle possibili té existe, que l'on peut représenter ainsi:
28 Z 0—H © © ,0 - H .... 0^ 0
Les fréquences des diverses bandes présentes dans nos spectres sont données au tableau II. On remarque d'abord que la vibration de valence 0 - H est à fréquence relativement basse, quoique moins basse que dans les bi carbonates alcalins (35~38). Ces derniers sont bien con nus pour avoir des liaisons hydrogène particulièrement fortes (distances 0 - H .... 0 = 2.61.
X)
. D' après des rela tions empiriques comme celles de Nakamoto (39) de Lippin cott (40) et de Pimentel (41) on peut évaluer les distances 0 - H ....0 à. environ 2.64
X,
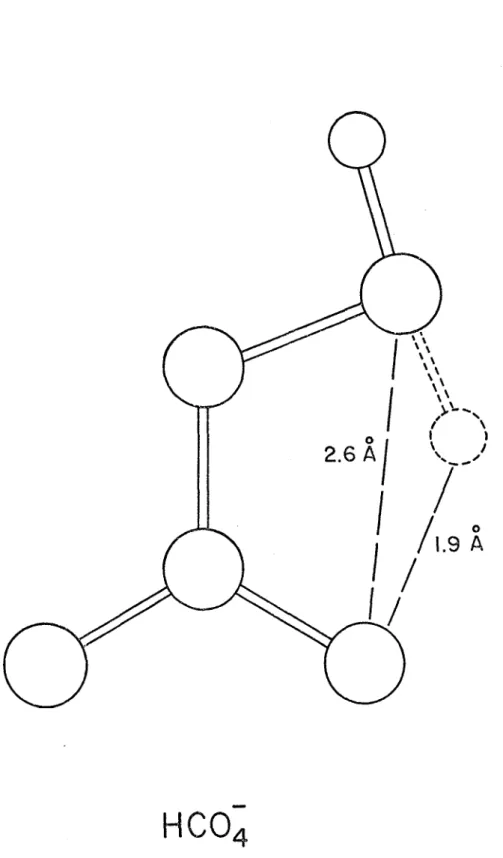
et 0 - H, à environ 1.04 Â dans KHCO^. Ceci correspond à une énergie de quelque 7 kcal/mole par comparaison avec une valeur de 8 kcal/mole pour les bicarbonates (39)•On peut également, à partir de ces données, écarter comme très improbable une liaison intramoléculaire comme celle que représente hypothétiquement la figure 3. En effet, une telle liaison serait beaucoup plus faible, vu la distance 0 .... H de 1.9 X environ comparée à celle pré vue (quelque 1.6
X)
pour une liaison hydrogène entre anions présumément linéaire. Cette dernière condition n'est peut- être pas réalisée, cependant, dans le cristal de KHCO^.Fréquences de vibration de KHCO^
Raman i.r.
NaHCOz
(35) Attributions V 3030m 3030m Combinaison 2910m lî 2730 2700F 2560/2410F Valence 0 - H 1 1960/1943F - 1680F 1698m Valence C = 0 asym. 2 1661/1618F 1492m 1458/1405FF Déformation 00H 3 1273FF 1286F 1300FF Valence C = 0 sym 4 961/942F 955/945F 1052/1038m " C - 0 5 910m 1017F Torsion 0H 10 906 884 869; 882F Valence 0-06
- 855/825 795/782,m 837F Déformation C0p 7 701F 692m 703/695FF C0^ 11 577m 605/575F 650m"
OCOg
8 450m"
coo
9 417f 360m Torsion C-0 (?) 12 300m30
2.6 A
hco
4
Figure 3. Modèle moléculaire illustrant la configuration probable de l'ion permonocarbonate. (Les lignes pointillées se rapportent au cas hypothétique d'une liaison hydrogène intramoléculaire.)
provenir d'une liaison hydrogène plus ou moins coudée. La cause de ces liaisons hydrogène relativement fortes dans KHCO^ aussi bien que dans les bicarbonates doit être d'origine électrostatique. Tout d'abord la
charge négative sur ces anions les rend meilleurs donneurs d'électrons, donc meilleurs accepteurs de protons. De plus, la résonance entre structures canoniques, telles que décri tes plus haut (p.
25
) accroît encore d'avantage la charge négative formelle sur les deux atomes d'oxygène du groupe COg. On a la même situation dans le cas de 1'anion persul fate HSO^ . Aussi devrait-on s'attendre d'y trouver des liaisons hydrogène assez semblables. Or il semble qu'au contraire l'acide de Caro (6) et son anion (4-2) forment plutôt des liaisons intramoléculaires assez faibles: dis tance H .... 0 de l'ordre de 2,1?X,
fréquence 0 - H d'environ 3270 cm . L'explication de cette différence pourrait se trouver dans la présence d'orbitales d sur l'atome de sou fre (contrairement au cas de l'atome de carbone) ce qui contribuerait à délocaliser les électrons autour de l'atome central et à réduire l'électronégativité des atomes d'oxy gène du groupe S0?.Les vibrations fondamentales du squelette C0^ de 1'anion seront interprétées selon une configuration plane
32
(figure 3). Si 11 on fait abstraction de 11 hydrogène, dont 11 orientation doit dépendre surtout des conditions d* empi lement dans le cristal, une configuration plane semble en. effet la plus probable étant donné le léger caractère
double de la liaison C-Og dû à la contribution non négli geable de la structure de résonance III (page 25). En termes d'orbitales moléculaires on peut dire que
11hybri-3
dation de 1 * oxygène, de caractère spv qu'elle a
générale-2
ment dans les peroxydes, prend un certain caractère sp dans ce cas-ci sous 1'influence d'une hybridation sembla ble des orbitales du carbone.
Dans 1'hypothèse d'une symétrie plane (Cs) les douze vibrations fondamentales de HCO^ se répartissent en neuf de classe A* (dans le plan) et 3 A" (hors du plan). Leur numération selon la notation de Mecke.est indiquée dans le tableau II. Les vibrations C = 0 sont à des fréquences légèrement moins élevées que dans 1'anion HCO? , peut être à cause d'un caractère double moins marqué. Par ailleurs la vibration C - 0 subit une assez forte diminution de
fré-— '1
quence, soit quelque 100 cm . Il doit s'agir d'un effet de masse, et peut être aussi de couplage mécanique avec le mode de torsion OH. On a une situation analogue dans H2S05 par rapport à HgSO^ (6). Enfin la vibration de va lence 0-0 se retrouve ici à la même fréquence pratique ment que dans la molécule de H202 (
23
) tout comme dans1'acide de Caro et ses sels. Nous discuterons plus loin de cette question à propos du percarbonate K2C2^6*
Notons en passant que cette vibration 0 - 0 se tra- duit par un triplet en Raman. (Le pic à 906 cm semble trop aigu pour correspondre exclusivement au mode de tor sion OH à 910 cm ^ en infrarouge.) On peut interpréter ceci comme une indication que la maille cristalline con
tient plus de deux anions, ce qui conduit à des vibrations en phase et hors de phase. La même explication vaut na turellement ‘pour la multiplicité des composantes de la bande de déformation Vy en infrarouge. En conclusion on peut dire que la substitution d’un groupe OH pour l’atome d’hydrogène dans 1'anion bicarbonate n'affecte pas telle ment le caractère des liaisons du groupe C0^ ni ses fré quences fondamentales, n serait intéressant de connaître les paramètres de structure de l'ion HCOJpour fin de com paraison. Une étude par diffraction des rayons X serait difficilement réalisable vu la nature instable et pulvé rulente de ce composé. En attendant on peut supposer que les distances interatomiques y sont à peu près égales à celles de NaHCO? (34) et HgOg (23) à savoir: 1,26 Â pour C = 0, 1,34- X pour G - 0 et 1,4-8 Â pour 0-0, et les angles, de 110° à 120°.
34
2. LE PERCARBONATE
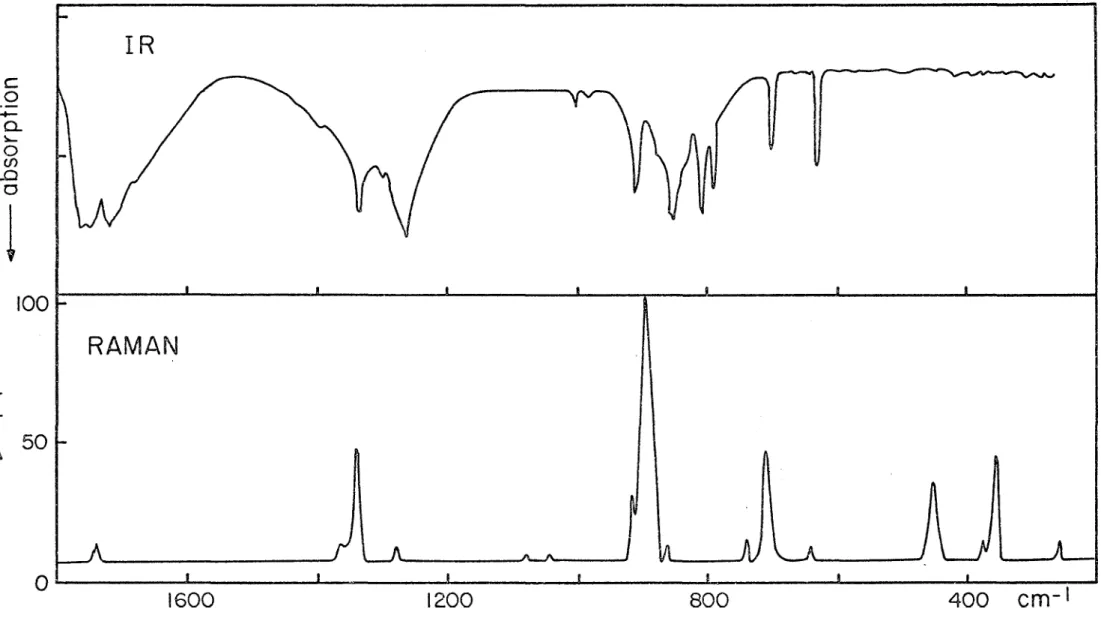
Tout comme dans le cas de KHCO^ on ne connaît pas la structure moléculaire du perdicarbonate KgCgO^. La seule étude par diffraction des rayons X a été faite selon la méthode des poudres (1) pour fins analytiques. Elle n'a fourni, en fait, aucune indication sur la structure, ni même sur la symétrie du cristal ou les dimensions de la maille fondamentale. Nous devons donc encore ici inter préter les spectres d'après un modèle hypothétique basé sur les structures connues des carbonates et des persul fates. A priori deux configurations sont possibles pour
2
-1'anion selon que les deux groupes C0^ reliés par le pont peroxyde sont dans le même plan ou non. Nous avons choisi la première, d'abord par analogie avec le cas de 1'anion (43) et ensuite, comme nous allons le mon trer, d'après les indications de nos spectres.
La figure 4 représente cette configuration plane et centrosymétrique (groupe ponctuel Cg^) suivant laquelle les dix-huit fondamentales doivent se subdiviser en quatre
classes (Tableau III). Les modes symétriques (gerade) seront actifs en Raman seulement, tandis que les modes non symétriques (ungerade) le seront en infrarouge seule ment. Au contraire, dans 1'hypothèse d'une configuration
C20
2
-6
Figure 4„ Configuration probable de 1'anion dans le perdicarbonate de potassium cristallin.
TABLEAU III
2 Classification des vibrations fondamentales de 1'anion CgO^
Au Bg Bu ■Mode s V1 - Valence C = 0 asym. v2 - — v14- ' It C = 0 sym. V3 - - V15 II C - 0 v4- - - - II 0-0 v5 - - v16 Déformation COg v6 - - v1? II OCOg v7 - - ^18 il ooc - v8 V11 ■- h COj (hors-plan) - v9 ^12 - Torsion 0 - C Vyl0 II 0-0 VJ en
gauche (de groupe Cg) comme dans la molécule HgOg, les dix-huit fondamentales (soit 10A et 8b) seraient toutes
actives tant en infrarouge qu’en Raman. Or un examen som maire de nos spectres confirme immédiatement, à la fois le nombre restreint des bandes, une dizaine chacun (Fig.5), et leur non-coincidence en infrarouge et en Raman, à quel
ques exceptions près. D'ailleurs ces dernières (en par- ticulier celle à 702 cm en infrarouge) pourraient aussi s'expliquer par la présence dans nos échantillons de tra ces de bicarbonate KHC0%, résultant de 11 hydrolyse, quasi inévitable du percarbonate. En résumé nos résultats peu vent s1 interpréter de façon satisfaisante, mais non exclu sive, selon une configuration plane et centrosymétrique de
2_
11 anion CgO^ . Pour trancher la question il faudrait une analyse approfondie de la structure cristalline par les méthodes de diffraction. Mais celles-ci exigent la pré paration d’un monocristal, tâche difficilement réalisable vu les propriétés des percarbonates.
L'attribution des bandes que nous proposons au ta bleau IV ne saurait être prise à la lettre vu qu’elle ne
tient pas compte du couplage possible entre vibrations assez voisines. Ce phénomène doit être particulièrement important dans la région de 700 à
900
cm ; soit celle des vibrations de valence C - 0 et 0-0, ainsi que desa b s o r p t i o n
RAMAN
1600
400 cnrr
Spectres de vibration de l’ion CgO^
Raman * * *Infrarouge Attributions V
1742 Valence 11
0
u 11750/1715
1750/1720
If II 131337/1275
11 II 21337/1265 1556/1290
II II 14913
Valence c - 0 3910
920
11 11 15893
II0
10
4861
DéformationCOg
5865/855
878
II II 16810'
820
IIOCOg
17793
IIcOj
8731
II 0C02 6710
702
IIco^
11630
637
II00c
18
450
II00c
7282
Torsion 0 - C02? 9353
II " ? 12162
II0
-0
? 10 * = Ce travail.= Résultats des auteurs russes (1).
4-0
déformations COg. A priori nous croyons qu'il faut at tribuer à cet effet la fréquence remarquablement élevée
_Zj
de la vibration 0-0 à 893 cm . Intuitivement on s'at tendrait à une fréquence sensiblement inférieure à celle de 880 cm dans la molécule de HgOg en se basant unique ment sur l'effet de masse. Ainsi dans le cas quelque peu
2—
analogue de 1'anion SgOo la vibration 0-0 se situe vers 830 cm (4-4-). Mais alors que dans SgOg la vibration la
—4 2—
plus proche est à
1090
cm , dans CgO^ au contraire les vibrations C- 0 sont beaucoup plus rapprochées: soit à-1
910 cm . La même explication vaut évidemment pour l'a- nion HCO^ . Récemment Christe (43) l'a invoquée pour ren dre compte des différences considérables de fréquence 0-0 observées dans les peroxydes de diméthyle. Par contre, elle ne semble pas- applicable aux percarbonates organiques
(46) dont les fréquences fondamentales entre 800 et 1000
-1
cm sont données au tableau V pour comparaison. Seule une analyse des constantes de forces et de la distribution d'énergie potentielle entre les vibrations fondamentales de ces molécules permettrait de tirer au clair la situa tion. Quoiqu'il en soit, il est bien évident que la fré quence de vibration 0-0 n'est pas aussi caractéristique qu'on le croyait jusqu'ici de la liaison peroxyde ; en tout cas, pas de sa longueur. Viz: 1.46 Â dans KgSgOg
Fréquences de la vibration de valence 0-0 dans divers peroxydes organiques
Percarbonates*
vc-c
^c-o0
10
> (CH^CO) - 0 - 0-(COgCH,) 995 950 840 (CH%C0)-0-0-(C02C^Hr) 1000 925 845 (CHzC0)-0-0-(C02C^H^^) 1000 955 840 (C^HX^-O-O-^OgCH^) 100? 926 845 (ZgH^coO-o-o-C^c^^) 990 955/925 850 (CgH^^C02)-0-0-(C02C^H^^) 990-925
—
Peroxydes ** CH, - 0 - 0 - CH-, 779 CD, -0
-0
- CD, 715 CF, - 0 -0 - CF2
890 (CH^)^ C-OO-C(CH^)^ 769 * = voir (46); **= voir (45).42
Les modes de valence C = 0 ont des fréquences un —'I
peu plus élevées ici que dans KHCO^; de 60 cm pour les mo- des antisymétriques V| et et d'environ
25
cm- pourles modes symétriques Vg et Par contre les fréquences de valence C - 0 sont un peu moins élevées que dans KHCO^;
— y|
de quelque 40 cm . Une explication plausible serait ba sée sur les structures de résonnance suivantes:
0 0
À -,
0II
c
o ©
© 0/
0 ©c
0© 0 0y
0© © 0 0V
© y) ©
c
o © iini
La contribution de la structure III doit être né gligeable en fait à cause des charges formelles adjacen tes sur les oxygènes du pont peroxyde. Comme conséquence les liaisons C = 0 auraient un caractère double un peu plus fort, et les liaisons C - 0, un peu moindre encore que dans l'ion HCO^ (page
25
). Par ailleurs ces déplacements peu vent être, du moins en partie, des effets d'ordre mécani que. Ainsi, les groupes C0g sont dans un entourage plus2
Quant aux vibrations de valence C - 0 on peut songer à des effets de masse ou de couplage, comme nous l'avons mentionné plus haut.
Les diverses fréquences de déformation des groupes CO? ne montrent pas de particularités par rapport à HCO^ , ni même HCO? . Viennent ensuite les modes de torsion au tour des liaisons C - 0 et 0-0 dont 1' identification est plus problématique. Nous attribuons la bande Raman à
__/j
353 cm au mode symétrique Vg étant donné que nous n'a vons pas rencontré d'autres bandes aussi fortes dans la
“1 région des basses fréquences (jusqu'à environ 100 cm de la raie excitatrice du laser).. La fréquence la plus rap- prochée en infrarouge est à 282 cm (Figure 6). Ceci re présente un écart considérable par rapport au mode symétri que. Cependant, le couplage interne entre ces deux modes doit être plutôt faible. Les forces de rappel sont surtout de nature électrostatique dans ce cas-ci: attractions et répulsions de Coulomb entre charges ioniques.
Enfin l'autre forte bande dans 1'infrarouge lointain,
wz| _
avec maximum à 160 cm et un vague épaulement vers
190
cm doit appartenir à la torsion autour de la liaison 0-0.80 cm
Spectre d’absorption de K2C?°6 dans 11 infrarouge lointain.
-Les moments d*inertie sont ici beaucoup plus grands que pour les torsions autour de C - 0. Par ailleurs ce mou vement^ d'amplitude relativement grande, doit créer un mo ment dipolaire assez fort, d'où 1'intensité de cette ban de. Les autres bandes faibles à 140, 110, 88 et 64 cm ^ sont des vibrations de réseau; soit de translation des
+ lo
cations K , soit de libration des anions CgO^ . A ce su jet soulignons le dédoublement de certaines bandes fonda mentales (Tableau IV) en infrarouge et en Raman. L'expli cation courante est qu'il s'agit de vibrations en phase
2
-et hors de phase des anions CgOg . dans la maille fondamen tale du cristal.
Il convient maintenant de comparer nos résultats avec ceux des auteurs russes (1). De façon générale ils sont en assez bon accord (Tableau IV) . Il semble cepen dant que leur spectromètre infrarouge était de qualité in férieure au nôtre, tant du point de vue de la calibration (leurs fréquences sont toutes décalées de 10 cm environ par rapport aux nôtres), que du point de vue de la résolu tion. Ainsi, dans la région de 800 à 1000 cm ^ le spectre qu'ils ont reconstitué pour KgCgOg à 100% (le N°6 de leur figure 1) montre une forte bande à double maximum à
920
et 878 cm . N'ayant pu résoudre ce dernier, et en 1'absen ce de données Raman, ils l'ont naturellement attribué à la vibration de valence 0-0. Il est bien connu cependantque cette vibration est toujours faible, sinon absente, en infrarouge pour les peroxydes symétriques.
Toutefois leur erreur principale vient de ce qu'ils ont interprété leurs spectres selon une structure entière ment covalente, comme nous l'avons déjà signalé au début. Ceci les conduit à représenter l'atome de carbone avec une
seule liaison double et deux liaisons simples, soit K — C — 0 — *
II 0
Une telle structure est évidemment inacceptable d'après la théorie de la liaison chimique. Il ne fait aucun dou te que les percarbonates alcalins sont de nature ionique, tout comme les carbonates et les bicarbonates. Les deux liaisons C = 0 de chaque groupe CO^ sont identiques et d'or dre 1,5*
D'autre part, les Russes ont étudié l'effet des impu retés k2C'®5 et KHCO^ sur les spectres d1 absorption infra rouge et de diffraction des rayons X. Leur produit le plus pur, obtenu par voie électrolytique, avait d'après 1'analyse chimique une teneur en percarbonate de 96%. Son spectre infrarouge toutefois montrait nettement les bandes caractéristiques du bicarbonate provenant de la décomposi tion dans la pastille de KBr (page 22). Nous n'avons pas jugé nécessaire d'analyser nos échantillons. A en juger
par la très faible intensité résiduelle des bandes de KHCCy la pureté de notre produit électrolytique devait être au moins aussi élevée que celui des Russes. Par contre le produit de la préparation chimique (page 14) montrait dans son spectre (Figure 7) les principales bandes de
KHCO%; entre autres à 1660/1620/1440/1400/1305/1005/660 cm""\
3. LE PEROXYHYDRATE KgCO^'^HgOg
Disons d'abord que la formule ci-haut est quelque peu idéalisée. En fait les carbonates alcalins peroxyhy- dratés sont trop instables pour être isolables à l'état
pur. On a prouvé par 1'analyse thermique différentielle (2) qu'ils contiennent toujours plus ou moins d'eau; d'où les diverses formules qu'on trouve dans la littérature, par exemple depuis K2C(^3* jusqu'à K^CO,.» ^HgOg * HpO. On ne sait jamais au juste quelle fraction est de l'eau de cristallisation, et quelle fraction, de l'eau adsorbée provenant de la décomposition du peroxyde d'hydrogène. La situation se complique encore du fait que les carbonates eux-mêmes, qui sont assez hygroscopiques, montrent des dif férences considérables dans leurs spectres lors de 11 hydra tation (47). Ainsi certaines bandes infrarouges apparais sent, d'autres sont dédoublées, etc. par la présence d'eau. Il n'est donc pas surprenant que les spectres des carbonates
_ _ i_ _ _ _ _ _ _ _ I_ _ _ _ _ _ _ _ i_ _ _ _ _ _ _ _ I_ _ _ _ _ _ _ _ i- - - 1—- - - 5- - - - -- - - 1- - - »
-3000
2000
1600
1200
800
Figure 7 - Spectre infrarouge d’un échantillon de K2C2^6 préparé chimiquement à partir du peroxyde de potassium.
Dans la figure 8 nous avons reconstitué les spectres typiques d'échantillons censés de composition
Les fréquences des principales bandes correspondent assez bien à celles d'une étude antérieure (2). Notre interpré tation n'est pas toujours la meme, cependant. Nous analy serons les bandes par ordre de fréquence décroissante. La première vers $100 cm , assez large, et la seconde encore plus large, dont le maximum se situe vers
2700
cm , sont sûrement des bandes de valence 0 - H affectées par de fortes liaisons hydrogènes. Il se peut que la seconde doive une partie de son intensité à une harmonique de la forte bande2- -1
de l'ion C0^ à 1420 cm « Ces fréquences sont voisines de celles qu'on trouve dans les composés d'addition de HgOg avec l'urée (3) et le fluorure de potassium ($). Dans le cristal de composition KgOO^ . HgOp . 3/2 HgO, que nous avons également étudié, cette région montre trois maxima vers 3250/3080/2500 cm \ Ce dernier est particulièrement
étalé. Les mêmes fréquences ont été observées par Rocchic- cioli (2) qui a attribué les deux premières aux molécules d'eau et la dernière, au peroxyde d'hydrogène. Cependant on ne voit pas à priori de raison pour une aussi grande différence entre les liaisons hydrogène formées par ces deux molécules. L'absence de bandes correspondantes en
RAMAN
2000
3000
Figure 8 - Spectres de vibration de ^2^2 vn
Raman n'est pas inusitée. Incidemment, la présence d’eau dans tous les échantillons étudiés était attestée par la bande de déformation Vg à 1650 cm
2—
Les vibrations fondamentales de 1'anion CO-, se trou-5
vent diversement affectées dans les peroxyhydrates. Ainsi le mode de valence antisymétrique, qui donne lieu à un dou blet dans le carbonate anhydre et son hydrate, ne produit
ici qu’une seule bande large vers 1420 cm en infrarouge. Par contre le mode symétrique, qui est absent dans le spec tre infrarouge du carbonate anhydre, se manifeste par une bande fine à 1060 cm dans 1'hydrate. Dans le peroxyhy- drate nous n'avons trouvé qu’une bande fine à
1013
cm en infrarouge et un doublet à 1071 -et 1026 cm en Raman. La présence d’eau dans le composé ternaire KgCO^'HgOg'^yS HgO affecte cette vibration; en infrarouge aussi bien qu'en_zj
Raman on trouve alors une seule bande fine a
1050
cmCette vibration, fondamentalement symétrique, est évidemment très affectée par un entourage plus ou moins dissymétrique dans ces divers cristaux.
Vient ensuite la bande assez large à 980 cm ^ en infrarouge, qui n'a pas son équivalent en Raman. Elle a été attribuée par Rocchiccioli à la rotation gênée ou li bration des molécules d'eau. Selon nous elle appartient plutôt aux molécules de peroxyde ; soit la libration autour de l'axe mineur (v^) soit la torsion interne (v^). En effet,
52
elle semble bien correspondre à la bande à 940 cm dans le peroxyhydrate KF'HgOg, qui ne contient pas d’eau; de cristallisation, ou autre. L’identification de la vibration de valence 0-0 du peroxyde d1 hydrogène est ici difficile puisqu'elle coïncide exactement avec la vibration de dé
formation dans le plan de 1'anion carbonate. Cette dernière est sans doute presque seule à 11 origine du doublet à 865~ 830 cm en infrarouge car la bande de valence 0-0 des peroxydes symétriques n'est jamais très intense. En Ra man, au contraire, la vibration Vg du carbonate est souvent absente ou très faible dans les carbonates, même hydratés.
-1
On peut donc conclure que le pic intense à 865 cm avec pics secondaires à 870 et 876 cm provient surtout de la vibration 0-0 du peroxyde d1 hydrogène.
Enfin la bande de déformation hors du plan de l'ion
2- -1
C0? vers 700 cm est accompagnée de pics satellites du côté des basses fréquences, tant en infrarouge qu'en Raman. Une faible bande, un peu plus large que les autres, aux en- virons de 610-630 cm , a été attribuée par Rocchiccioli
à la torsion interne de HgOg. Elle pourrait tout aus si bien être due aux modes de libration des molécules d'eau comme dans de nombreux autres hydrates (48). On voit qu'u ne interprétation définitive des spectres de carbonates peroxyhydratés demanderait une étude systématique assez com plexe dépassant largement le cadre de notre travail.
RESUME ET CONCLUSIONS
Les percarbonates alcalins sont connus depuis très longtemps. La première description d'une préparation par voie électrolytique remonte à la fin du siècle dernier
(4-9»50)« Cependant, leur nature et leur constitution a fait l'objet d'une longue controverse. Ce n'est que ré cemment que la question a pu être élucidée grâce à la spectroscopie moléculaire. En effet on a longtemps con fondu entre les véritables percarbonates et les carbonate peroxyhydratés, divers auteurs optant pour l'une ou 11 au tre formulation, la plupart du temps sans grande justifi cation expérimentale. On sait maintenant que les deux existent bien distinctement.
Les persels sont de deux types : les permonocarbo- nates, MHCO^ et les perdicarbonates, MgCgO^. On les ob
tient par carbonatation ménagée, à basse température, de solutions aqueuses de peroxydes ou de superoxydes alca lins. Les seconds s'obtiennent aussi par électrolyse: oxydation anodique de solutions saturées de carbonates. Les deux sont difficiles à purifier parfaitement. Ils
sont assez fragiles, surtout les permonocarbonates, et s'hydrolysent facilement avec dégagement d'oxygène actif.
se décomposent spontanément, en sorte que il est quasi impossible d'isoler et de caractériser une espèce cristal line donnée. Les diverses formules qu'on trouve dans la littérature à ce sujet correspondent probablement à des mélanges plutôt qu'à des composés bien définis.
Quant aux percarbonates de formule hypothétique MgCO^ rapportés dans la littérature (7) ils ne semblent pas réels; du moins on ne possède pas de preuve physique de leur existence. En ceci les percarbonates ressemblent aux persulfates car on ne connaît pas de composés de for mule MpSOc;, On pourrait dire que l'acide percarbonique, HgCO^, s'il existait serait monobasique tout comme HpSO^, l'acide de Caro.
Notre contribution au problème des percarbonates peut se résumer ainsi:
- Nous avons observé pour la première fois les spectres de vibration en infrarouge et en Raman de l'espèce ionique HCO^ . Ces spectres correspondent à ceux qu'on attendrait d'une molécule de configuration plane avec des paramètres de structure assez voisins des espèces parentes HCO% et HgOp' Le cristal est probablement constitué de chaînes infinies d'anions reliés par des ponts d'hydrogène assez forts. La cohésion est assurée en grande partie par les
56
forces de Coulomb entre ions.
- Nous avons repris le spectre infrarouge du perdicarbonate K2C2®6’ déjà étudié par des chimistes russes (1), et nous enregistré pour la première fois son spectre Raman. Le petit nombre de bandes observé dans les deux cas, et leur alternance en infrarouge et en Raman sont une forte indi cation d'une structure plane et centrosymétrique de 1’anion
2_
C2^6 * De Plus nous avons confirmé 1'identité des pro duits obtenus par voie électrolytique et chimique ; ces derniers étant cependant moins purs que les premiers.
- Enfin, nous avons étudié de la même façon deux peroxyhy-drates de formule approximative et K^CO^*^2^2*^/2 H
2
O. Leurs spectres assez peu caractéristiques, peuvent né anmoins s*interpréter par analogie avec ceux des peroxyhydra- tes d'urée et de fluorure de potassium, étudiés précédemment dans ce laboratoire (5,5). Une étude spectroscopique plus poussée de ces composés mal définis ne semble pas justifi able pour le moment vu 11 intérêt purement académique de la question.1. Karetnikov, G.S. et Sorokina, M.F., Zh.Fiz.Khim.
59(2), 364 (1965). [Russ.J.Phys.Chem. 39(2), 18? (1965)]. 2. Rocchiccioli, C., C.R. 261, 361 (1965).
3. Arnau, J.L. et Giguère, P.A., J.Mol.Struct. 3, 483 (1969). 4. Arnau, J.L. et Giguère, P.A., Can.J.Chem, 47, 3745 (1969). 5. Roy, Hélène et Giguère, P.A., Rev.Chim.Min, 7, 1053 (1970). 6. Arnau, J.L. et Giguère, P.A., Can.J.Chem. 48, 3903 (1970). 7. Partington, J.R. et Fathallah, A.H., J.Chem.Soc. 1934 (1950) 8. Leblanc, M. et Zellmann, R., Z.Elektrochemie, 29, 179 (1923) 9. D'Ans, J. et Friederich, W. , Z.Anorg.Chem. 73, 325 (1912). 10. Firsova, T.P. et Duganova, V.M., Zh.Neorg.Khim. 14(1), 22
(1969); [Russ.J,Inorg.Chim. 14(1), 10 (1969)].
11. Firsova, T.P., Molodkina, A.N., Morozova, T.G. et Akse nova, I.V., Zh.Neorg.Khim. 9(5), 1066 (1964); [Russ.J. Inorg.Chem. 9(5), 563 (1964)J.
12. Firsova, T.P., Molodkina, A.N., Morozova, T.G. et Akse nova, I.V., Zh.Neorg.Khim. 8(2), 278 (1963); [Russ.J. Inorg.Chem. 8(2), 140 (19637].
13. Sokol, V.I., Bakulina, V.M,, Filatov, E.Ya., Firsova, T.P., Izv.Akad.Nauk SSSR, Otd.Khim.Nauk, 6, 1163 (1968).
14. Mel'nikov, A.Kh. et Firsova, T.P., Zh.Neorg.Khim. 6(11), 2470 (1961); [Russ .J. Inorg.Chem. 6(11 ) , 1251 ( 1961)"] 7 15. Mel'nikov, A.Kh. et Firsova, T.P., Zh.Neorg.Khim. 6(1),
169 (1961;; [Russ.J.Inorg.Chem. 6(1), 83 (1961)].
16. Riesenfeld, E.H. et Mau, W., Ber. 44, 3589-3595 (1911).
58
17. Makarov, S.Z. et Vol'nov, I.I., Izv.Akad.Nauk. SSSR, 0td. Khim. N auk , 4, 370 (195*1).
18. Wolffenstein, R. et Peltner, E., Ber. 41, 280 (1908). 19. Cells, M.G. et Masaguer, J.R., An.R.Soc. Esp.Fis.Quim.
51B, 695 (1955).
20. Mel'nikov, A.Kh., Firsova, T.P. et Molodkina, A.N., Zh.Neorg.Khim. 7(6), 1237 (1962); [Russ.J.Inorg.Chem. 7(6) 637 (1962)TT
21. Mel'nikov, A.Kh., Firsova, R,P., Zh.Neorg.Khim. 6(10), 2230 (1961); [Russ.J.Inorg.Chem. 6(10), 1137,
(T9£yT)
J . 22. Sokol, V.I., Bakulina, V.M., Filatov, E.Ya. et Firsova,T.P. , Zh.Neorg.Khim. 13(9)2347 (1968) ; [Russ. J. Inor g. Chem. 15(9), 1211 (1968)].
23. Schumb, W.C., Satterfield, C.N. et Wentworth, R.I., "Hydrogen Peroxide", Reinhold, New York, 1955, p.460. 24. Riesenfeld, E.H. et Reinhold, B., Ber. 42, 4377 (1909).
25
. Giguère, P.A. et Olmos, A.W., Can.J.Chem. J54, 689 (1956). 26. Tanatar,S.,.
Ber. 32, 1544 (1899); et 43, 2149 (1910). 27. Riesenfeld, E.H., Ber. 43, 566 (1909).28. Kasanetzky, P., J.Russ.Phys.Chem.Soc. _34, 202 et 388 (1902). 29. Kasanetzky, P., J.Russ.Phys.Chem.Soc. 46, 1110 (1914).
30. Peltner, E., Ber. 42, 1777 (1909).
31. Franchuk, I.F., Teor. i Eksperim.Khim., Akad.Nauk Ukr.
SSSR,
1(4), 531 (1965).32. Guay, M., Thèse de doctorat, Université Laval, Québec 1971. 33. Nitta, I. Tommiie, Y. et Hoe Koo, C., Acta cryst. 5,292
(1952).
34. Sass, R.L. et Scheuerman, R.F., Acta cryst. 1_5, 77 (1962). 35. Novak, A., Saumagne, P. et Bok, L.D.C., J.Chim.phys. 60,
1385 (1963). —
36. Nakamoto, K., Sarma, Y.A., et Ogoshi, H., J.Chem.Phys. 43, 1177 (1965).
37. Bernitt, D.L. , Hartman. K.O. , et Iisatsune, I.C., J. ' Chem.Phys. , 42, 3555, ('1965).
38. Couture-Mathieu, L., J.Phys.Rad., 11, 541 (1950) et 15,
551 (1954). —"
59. Nakamoto, K. Margoshes, M. et Bundle, P.E., J.Am.Chem, Soc., 77, 6480 (1956).
40. Lippincott, E.R. et Schroeder, R., J.Chem.Phys. 23,
1099 (1955). ""
41. Pimentel, G.C. et Sederholm, C.H., J.Chem.Phys., 24,
639 (1956). —
42. Kyrki, J.R., Suomen Kemistilehti, B38,51, (1965)• 43. Zachariasen, W.H. et Mooney, R.C.L. , Z.Krist. 88, 63
(1954). —
44. Simon, A. et Richter, H., Z. anorg.allgem.Chem. 315,
196 (1962). —
45. Christe, K.O., Spectrochim.Acta 2?A, 463 (1971). 46. Lapshin, N.M. Lodonov, V.A. et Khorshev, S, Y a., Zh.
Org.Khirn. , 4, 952 (1968). .
47. Schutte, C.J.H. et Buijs, K.. Spectrochim. Acta, 17 j 921 (1961).
48. Brun, G., Rev,.Chim. minérale, 5, 899 (1968).
49. Constam, E.J. et von Hansen, A. , Z.Elektrochem. 3, 137 (1896).