A statistical theory of polymer network degradation
Texte intégral
Figure
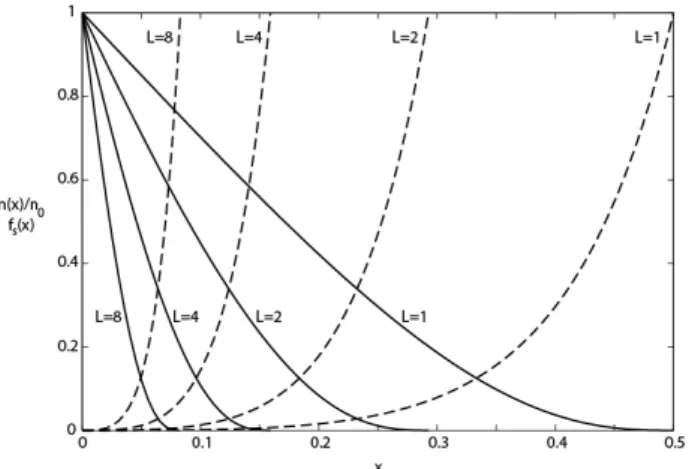
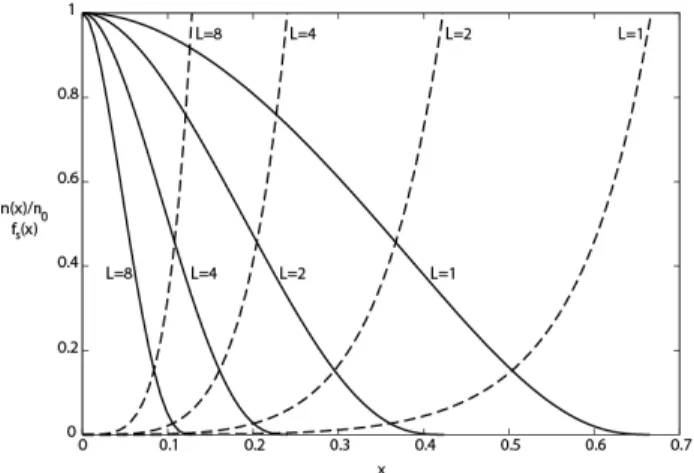
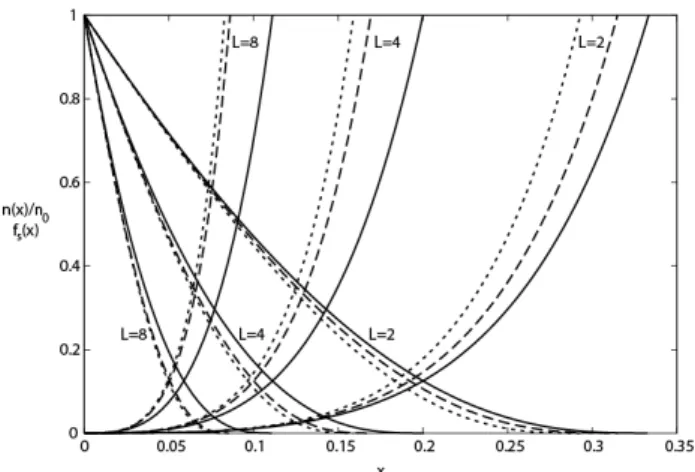

Documents relatifs
sont toutes r´ ealis´ ees ici comme sous-quotient de SHM d´ efinie en haut sur des ouverts de X lisse. Nous assumons par r´ ecurrence la d´ ecomposition sur le compl´ ementaire de
(Int.) Combust. Ceamanos, The influence of the percentage of oxygen in the atmosphere on the thermal decomposition of lignocellulosic materials, J. Martín-Gullón,
Regardless of whether the process starts from abiotically available building blocks and energy sources present in the environment, or directly from simple activated chemical
The intermediate coherent scattering function of entangled polymer melts: a Monte Carlo test of des Cloizeaux’ theory... Classification
Figure 1 shows the thermodynamic analysis results in terms of the dependence of Gibbs energy change of the CuCl 2 decomposition reaction on temperature at different
populations, and housing discrimination and segregation. It describes the evolution of U.S. housing policy over time, explains the federal housing policies that exist in 2010, and
Isolated microspore culture techniques and recent progress for haploid and doubled haploid plant production.. Microspore culture for haploid
According to the literature, the actualization of an affordance may depend on the presence of appropriate enabling, stimulating, and releasing conditions (Volkoff and Strong
