Université de Sherbrooke
Association entre le profil de force musculaire et les capacités fonctionnelles aux membres inférieurs chez les personnes atteintes des phénotypes adulte classique et
adulte tardif de dystrophie myotonique de type 1
Par Émilie Petitclerc
Programme des sciences cliniques
Mémoire présenté à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de maître ès sciences (M. Sc.)
en sciences cliniques
Sherbrooke, Québec, Canada Septembre 2015
Membres du jury d’évaluation
Cynthia Gagnon, erg., Ph. D., co-directrice, École de réadaptation Johanne Desrosiers, erg., Ph. D., co-directrice, École de réadaptation Nathaly Gaudreault, pht, Ph. D., évaluatrice interne, École de réadaptation
Jean-Sébastien Roy, pht, Ph. D., évaluateur externe, département de réadaptation, Faculté de médecine, Université Laval
membres inférieurs chez les personnes atteintes des phénotypes adulte classique et adulte tardif de dystrophie myotonique de type 1
Par Émilie Petitclerc
Programme des sciences cliniques
Mémoire présenté à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de maître ès sciences (Ms. S.) en sciences cliniques, Faculté de médecine et des
sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4 But : Les objectifs étaient de 1) décrire les profils de force musculaires aux membres inférieurs (MIs) et les capacités aux déplacements des personnes présentant les phénotypes adulte classique (DM1-AC) et adulte tardif (DM1-AT) de la dystrophie myotonique de type 1 (DM1), et 2) d’explorer l’influence de la faiblesse des MIs sur les capacités aux déplacements dans cette population. Méthode : Cette étude consiste en une analyse secondaire de données issues d’une plus large recherche qui visait à identifier les déterminants de la participation sociale et de la qualité de vie de personnes atteintes de DM1 (n = 158 DM1-AC et n = 42 DM1-AT). La force de quatre groupes musculaires des MIs a été mesurée à l’aide du bilan musculaire manuel (BMM) et du bilan musculaire quantitatif (BMQ) par dynamométrie manuelle. Les capacités aux déplacements ont été évaluées à l’aide de tests standardisés (échelle d’équilibre de Berg, vitesse de marche et
Timed Up & Go). Résultats : Le phénotype DM1-AT présente moins de faiblesse et
d’incapacités que le phénotype DM1-AC (p < 0,001 – 0,020). Le BMM ne détecte pas de faiblesse chez le phénotype DM1-AT mais des pertes de force au BMQ de 12 % à 20 % ont été identifiées chez ce phénotype, excepté pour les fléchisseurs du genou, entrainant des limitations aux déplacements chez 22 % à 48 % de ces individus. Dans le phénotype DM1-AC, l’atteinte musculaire était légèrement plus importante en distal qu’en proximal. Selon ces résultats, les phénotypes DM1-AC et DM1-AT présentent des portraits distincts et les données relatives à chacun devraient être analysées séparément. Une progression générale de la faiblesse au BMQ et des scores aux tests fonctionnels a été observée en fonction des cotes de l’échelle Muscular Impairment Rating Scale (MIRS). Un déficit de force au BMQ (excepté pour les fléchisseurs du genou) et des incapacités fonctionnelles ont aussi été observés dès les premières cotes de la MIRS. Finalement, les dorsifléchisseurs de la cheville et les extenseurs du genou semblent être de bons indicateurs de la fonction des membres inférieurs en DM1. Conclusion : Cette étude a permis de dresser un portrait des atteintes de la force musculaire aux MIs et des capacités fonctionnelles liées aux déplacements pour chacun des phénotypes DM1-AC et DM1-AT de la DM1, ainsi que d’explorer la contribution de la faiblesse des groupes musculaires évaluées sur les capacités aux déplacements dans cette population. Ces résultats contribueront à mieux déterminer les cibles d’évaluation et d’interventions en réadaptation et à mieux définir le processus d’évaluation dans le contexte des essais thérapeutiques à venir.
Mots clés : Dystrophie myotonique de type 1, phénotypes, force musculaire, capacités fonctionnelles aux déplacements, membres inférieurs, facteurs explicatifs, maladies neuromusculaires, physiothérapie.
Relationships between lower limb muscle strength and mobility capacities in myotonic dystrophy type 1 adult and late onset phenotype
By Émilie Petitclerc Sciences cliniques Program
Thesis presented at the Faculty of Medicine and Health Sciences in order to obtain the Master degree diploma maître ès sciences (M. Sc.) in sciences cliniques,
Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada J1H 5N4
Purpose: The purposes of this study were 1) to describe lower limbs muscle strength and mobility capacities, and 2) to explore the respective contribution of lower limb muscle weaknesses on mobility in the adult and late-onset phenotypes of myotonic dystrophy type 1 (DM1). Methods: This study is a secondary analysis of part of the results of a larger study, whose purpose was to identify social participation and quality-of-life determinants in 200 DM1 patients (158 adult and 42 late-onset). The strength of four lower limb muscle groups was assessed using manual muscle testing (MMT) and handheld dynamometry quantitative muscle testing (QMT). Mobility capacities were assessed using standardized tests (Berg balance scale, 10 Meter Walk Test and Timed Up & Go). Results: Although the late-onset phenotype showed less weaknesses and mobility limitations than the adult phenotype (p <0.001-0.020), and although MMT showed no weakness in the late-onset phenotype, quantitative strength losses of 12-20% were measured in this phenotype, with the exception of the knee flexors. These weaknesses led to mobility limitations in 22-48% of participants with the late-onset phenotype. In the adult phenotype, muscle strength impairment was slightly more important distally than proximally (2-2.5/10 and 5.8-8.2% for MMT and QMT, respectively) (p <0.001-0.002). According to those results, the adult and late-onset phenotypes show different profiles of lower limb impairment, and should not be pooled for data analysis. A general progression of quantitative muscle weakness and of mobility scores was observed according to the Muscular Impairment Rating Scale (MIRS) classification. Quantitative weaknesses, with the exception of the knee flexors, and mobility limitations were observed from the first MIRS grades. QMT is therefore definitely a more effective tool for measuring weakness in DM1. Finally, ankle dorsiflexors and knee extensors seem to be good indicators of lower limb function in DM1. Conclusion: This study allowed a better characterization of lower limb weaknesses and mobility limitations in the adult and late-onset phenotypes of DM1, and explored the contribution of lower limb weaknesses on mobility capacities in this population. These results will be useful for developing more specific rehabilitation programs and for optimizing the evaluation of these impairments in the context of the upcoming therapeutic trials.
Keywords: Myotonic dystrophy type 1, phenotypes, muscle strength, mobility capacities, lower limbs, explanatory variables, physiotherapy.
TABLE DES MATIÈRES
RÉSUMÉ ... ii
SUMMARY ... iii
TABLE DES MATIÈRES ... v
LISTE DES TABLEAUX ... vii
LISTE DES ABRÉVIATIONS ... viii
INTRODUCTION ... 1
RECENSION DES ÉCRITS ... 4
2.1 Description générale de la dystrophie myotonique ... 4
2.1.1 Particularités génétiques ... 5
2.1.2 Pathogénèse ... 6
2.1.3 Facteurs explicatifs de la myopathie ... 8
2.2 Atteinte de la force musculaire aux membres inférieurs ... 10
Article 1 ... 11
2.3 Atteinte des capacités fonctionnelles liées aux déplacements ... 36
2.3.1 Capacités fonctionnelles liées aux déplacements en DM1 ... 36
2.3.2 Relation entre la force et les capacités fonctionnelles liées aux déplacements en DM1 ... 37
OBJECTIFS ... 39
MATÉRIEL ET MÉTHODES ... 40
4.1 Dispositif de recherche ... 40
4.2 Population cible et échantillon ... 40
4.2.1 Population cible et critères d’admissibilité ... 40
4.2.2 Plan d’échantillonnage et recrutement des participants ... 41
4.3 Procédure de collecte de données ... 41
4.4 Variables à l’étude... 42
4.4.1 Force musculaire ... 42
4.4.2 Capacités fonctionnelles liées aux déplacements ... 48
4.5 Taille de l’échantillon ... 50
4.6 Analyses statistiques... 50
RÉSULTATS ... 52
Article 2 ... 53
6. DISCUSSION ... 110
6.1 Retombées de l’étude ... 110
6.2 Limites de l’étude ... 112
6.3 Recommandations pour des études futures ... 113
7. CONCLUSION ... 116
8. REMERCIEMENTS ... 117
LISTE DES TABLEAUX
Tableau 1 Cotation du bilan musculaire manuel selon l’échelle modifiée du
Medical Research Council……….42
Tableau 2 Procédures pour le bilan musculaire quantitatif par dynamométrie
Manuelle………44 Tableau 3 Échelle Muscular Impairment Rating Scale………..46
LISTE DES ABRÉVIATIONS
DM1 : Dystrophie myotonique de type 1 CTG : cytosine-thymine-guanine
DM1-AC : phénotype adulte classique de la DM1 DM1-AT : phénotype adulte tardif de la DM1 MIs : membres inférieurs
MIRS : Muscular Impairment Rating Scale BMM : Bilan musculaire manuel
BMQ : Bilan musculaire quantitatif
COSMIN : COnsensus-based Standards for the selection of health Measurements
INtruments
INTRODUCTION
La dystrophie myotonique de type 1 (DM1) est la forme la plus fréquente de dystrophie musculaire chez l’adulte (Mathieu et al., 1992). Il s’agit d’une pathologie neuromusculaire multisystémique causée par une anomalie génétique sur le locus 19q13.3 du chromosome 19 entrainant une répétition instable des triplets CTG (cytosine-thymine-guanine) (Fu et al., 1993). La prévalence mondiale varie entre 1 et 10 individus par 100 000 habitants (Harper, 2001), et jusqu’à 189 individus par 100 000 habitants dans la région du Saguenay-Lac-Saint-Jean, Québec, Canada (Mathieu et al., 1990). Cette maladie peut affecter les systèmes nerveux, cardiovasculaire, endocrinien, oculaire, respiratoire, digestif et musculaire (Harper, 2001). La DM1 présente une variabilité d’expressions phénotypiques basées sur le nombre de répétitions CTG, la gravité de l’atteinte et l’âge d’apparition des premiers symptômes (Harley et al., 1993). La nomenclature pour chacun des phénotypes n’a pas encore fait l’objet d’un consensus international. Dans le présent mémoire, les termes suivants, ou leurs acronymes, seront utilisés pour désigner les différents phénotypes : congénital, infantile, adulte classique (DM1-AC) et adulte tardif (DM1-AT), en anglais respectivement congenital, infantile, adult et late-onset. Les phénotypes congénital et infantile présentent un tableau clinique ainsi qu’une évolution différents des deux autres (Harper, 2001) et ne seront pas le sujet du présent mémoire.
Dans le phénotype DM1-AC, le plus courant, les symptômes débutent généralement entre l’âge de 10 et 30 ans (International Myotonic Dystrophy Consortium, 2000). Les personnes atteintes développent, entres autres symptômes, de la faiblesse musculaire et de la myotonie (difficulté à relaxer un muscle ou un groupe de muscles une fois qu'ils ont été contractés), notamment aux membres inférieurs (MIs) (Harper, 2001). Dans le phénotype DM1-AT, de moindre gravité, les symptômes apparaissent à partir de 40 ans et ne consistent souvent qu’en de la myotonie aux mains et la présence de cataractes (Arsenault et al., 2006). Les études s’étant intéressé à la description des atteintes musculaires des phénotypes DM1-AC et DM1-AT confondus, aucune étude à ce jour n’ayant exploré ces atteintes séparément pour chacun de ces deux phénotypes, rapportent que la faiblesse musculaire progresserait à un rythme relativement lent (1-3%/année) suivant un patron de
distal à proximal, et généralement symétrique (Mathieu et al., 2003; Hébert et al., 2010). Par ailleurs, l’évolution des symptômes est très variable, la maladie pouvant progresser vers une faiblesse incapacitante en quelques années ou être stable et bénigne pour plus de 20 ans (Mathieu et al., 1992). Le pronostic est donc difficile à établir, même si des moyennes de durée de la maladie sont disponibles pour chacune des cotes de la Muscular Impairment
Rating Scale (MIRS), une échelle de catégorisation de la gravité de l’atteinte musculaire
développé spécifiquement pour la DM1 et largement utilisé en clinique par les neurologues (Mathieu et al., 1992).
La participation sociale de cette clientèle est diminuée dans plusieurs sphères, incluant les habitudes de vie reliées aux déplacements (Gagnon et al., 2008). La force musculaire aux MIs constitue un facteur explicatif majeur pour la réduction de ces habitudes de vie (ratio de risque entre 15.04 et 5.54) (Gagnon et al., 2008). La force des MIs en DM1 a par contre été peu décrite dans les écrits scientifiques et sa relation avec les aptitudes fonctionnelles n’a été que très peu explorée. De plus, à ce jour, l’atteinte de la force musculaire et des capacités fonctionnelles aux MIs n’a jamais été décrite séparément en fonction des phénotypes DM1-AC et DM1-AT. Pourtant, cette différentiation est essentielle pour cibler les besoins en réadaptation qui sont propres à chacun des phénotypes. De plus, la MIRS, qui est couramment utilisée en clinique pour catégoriser le niveau d’atteinte musculaire, n’est pas associé à un profil fonctionnel clair, ce qui pourrait permettre d’avoir une idée rapide des atteintes potentielles de la personne, et ainsi d’orienter les interventions en réadaptation.
En effet, comme il n’existe aucun traitement curatif, les personnes atteintes nécessitent un suivi par une équipe de réadaptation interdisciplinaire en clinique de maladies neuromusculaires, incluant de la physiothérapie. Malheureusement, les services de réadaptation offerts à cette population ont été décrits comme étant peu développés et souvent utilisés de façon inadéquate (Hilton-Jones, 1997; Harper et al., 2004; Cup et al., 2007). Cette situation pourrait s’expliquer en partie par la méconnaissance de l’atteinte musculaire et fonctionnelle propre à chacun des phénotypes, et du processus d’évaluation souvent incomplet qui en découle, ce qui limite l’identification d’objectifs précis d’intervention. De plus, cette méconnaissance des atteintes aux MIs pour chacun des
phénotypes DM1-AC et DM1-AT représente une limite à l’interprétation des résultats des essais thérapeutiques en DM1 (Gagnon et al., 2013) qui sont déjà en phase 1 (essai numéro NCT02312011, www.clinicaltrials.gov). L’absence de consensus international désignant la méthode d’évaluation de la force musculaire (bilan musculaire manuel ou dynamométrie quantifiée) et les tests d’évaluation de la fonction à préconiser auprès de la clientèle DM1 en raison des enjeux méthodologiques entourant les différentes méthodes et tests utilisés (Gagnon et al., 2013) constitue un défi supplémentaire.
Cette étude vise donc à explorer les atteintes de la force musculaires aux MIs et des capacités fonctionnelles liées aux déplacements des personnes atteintes des phénotypes DM1-AC et DM1-AT.
RECENSION DES ÉCRITS
Cette recension des écrits présente d’abord une description générale de la dystrophie myotonique de type 1. Suivra la synthèse des connaissances actuelles sur l’atteinte de la force musculaire aux MIs et des capacités fonctionnelles liées aux déplacements dans la DM1.
2.1 Description générale de la dystrophie myotonique
La dystrophie myotonique de type 1, aussi connue sous le nom de maladie de Steinert, est une maladie neuromusculaire autosomique dominante (Harper, 2001). Cette maladie neuromusculaire multisystémique présente des atteintes variables, notamment au niveau des systèmes musculaire (p.ex. faiblesse progressive, myotonie, atrophie des muscles faciaux et bulbaires), oculaire (p.ex. cataractes), cardio-vasculaire (p.ex. défaut de conduction), endocrinien (p.ex. diabète, hypogonadisme), nerveux central (p.ex. troubles cognitifs, somnolence, fatigue, apathie) et digestif (p.ex. troubles de la motilité intestinale) (Harper, 2001).
La DM1 se transmet par un processus autosomique dominant. La personne responsable de la transmission du gène à sa descendance est nécessairement atteinte par la maladie, à un degré variable (Klein, 1958; Harper, 2001), et présente 50 % de risque de transmettre la maladie si un seul des parents est atteint. La prévalence mondiale varie entre 1 et 10 individus par 100 000 (Emery, 1991; Harper, 2001). Toutefois, dans la région du Saguenay-Lac-St-Jean (Québec, Canada), elle atteint jusqu’à 189 individus par 100 000, en partie en raison d’un effet fondateur (Mathieu et al., 1990). L’effet fondateur réfère à la création d’une nouvelle population à partir d'un nombre relativement restreint d'individus provenant d'une population mère. En général, le hasard fait qu’une partie seulement de la variété génétique de la population d’origine est retenue, ce qui a pour conséquence une plus grande homogénéité dans la nouvelle population formée que dans la population mère (CORAHM, 2002).
Cette maladie est causée par une répétition instable (expansion) d’un trinucléotide contenant une séquence cytosine-thymidine-guanosine [CTG]n, localisé sur la région
3’transcrite non-traduite du chromosome 19q13.3 (Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992). Chez un individu normal, cette répétition du couplet CTG est polymorphique et varie entre 5 et 35 répétitions. Chez une personne atteinte de DM1, on observe un nombre plus élevé, allant de 50 à plusieurs milliers de répétitions (International Myotonic Dystrophy Consortium, 2000). Tel que mentionné en introduction, il existe quatre phénotypes de DM1, basés sur le nombre de répétition CTG, la gravité de l’atteinte et l’âge d’apparition des premiers symptômes : le phénotype adulte tardif (AT- entre 50 et 150 répétitions), le phénotype adulte classique (AC- entre 100 et 1 000 répétitions) et les phénotypes infantile et congénital (1 000 répétitions et plus) (Harley et al., 1993; International Myotonic Dystrophy Consortium, 2000). Dans la présente étude, seuls les deux premiers phénotypes (AC et AT) sont considérés.
2.1.1 Particularités génétiques
La DM1 présente certaines particularités génétiques influençant le suivi clinique, soit l’expressivité variable du gène, l’anticipation et le mosaïcisme somatique.
L’expressivité variable du gène désigne le fait que pour un même génotype à risque, la maladie peut prendre différentes formes (Harper, 2001). Par ailleurs, il y a un certain degré de corrélation entre le nombre de répétitions CTG et l’expression clinique globale de la maladie (Udd et Krahe, 2012). Ainsi, d’une part, les individus ayant entre 50 et 100 répétitions CTG n’auront souvent au cours de leur vie que des cataractes sans autre manifestation musculaire ou systémique de la maladie. À l’opposé, les individus porteurs de 1 500 répétitions et plus présentent souvent des atteintes plus graves. L’âge d’apparition des premiers symptômes est aussi inversement corrélée au nombre de répétitions CTG (Harley et al., 1993; Marchini et al., 2000) et constitue donc un facteur influençant le pronostic. De plus, il existe une relation modérée entre le nombre de répétitions CTG et l’espérance de vie (de Die-Smulders et al., 1998). Toutefois, la corrélation entre le nombre de répétitions CTG et le phénotype ou la gravité des différentes atteintes multisystémiques (troubles du rythme cardiaque, somnolence, fatigue, troubles cognitifs, performance intellectuelle) (Harper, 2001) est beaucoup plus faible lorsque les répétitions sont de l’ordre de 200 à 1 000 (Harper, 2001).
Autre particularité génétique, l’anticipation réfère pour sa part à l’apparition plus précoce de la maladie d’une génération à l’autre, avec augmentation de la gravité des symptômes (Ashizawa et al., 1992; Harley et al., 1993; Harper, 2001; Ashizawa et Sarkar, 2011). Ainsi, le nombre de répétitions CTG augmente généralement lors de la transmission du gène d’un parent à son enfant (Harley et al., 1993).
Finalement, le mosaïcisme somatique désigne le fait que le nombre de répétitions CTG varie dans les différents tissus du corps (Ashizawa et al., 1993). Par exemple, la taille de l’expansion est plus grande dans les muscles squelettiques (Thornton et al., 1994), le cerveau (Wong et Ashizawa, 1997) et le cœur (Joseph et al., 1997). Des études ont aussi démontré que le nombre de répétitions est plus grand dans le tissu musculaire que dans les leucocytes (Thornton et al., 1994). Toutefois, la longueur de l’expansion CTG est similaire dans les muscles proximaux et distaux de la jambe (Hedberg et al., 1999). La variation de l’expansion CTG observée entre les tissus est possiblement influencée par les gènes liés au développement puisqu’elle apparait entre la 13e et 16e semaine de gestation (Martorell et al., 1997). Selon l’étude de Martorell, le nombre de répétitions dans les leucocytes ainsi que l’hétérogénéité de l’expansion dans les différents tissus du corps augmentent dans le temps (Martorell et al., 1998). La nature dynamique de la taille de l’expansion pourrait jouer un rôle important dans la variabilité de la gravité des symptômes observée dans la DM1 (Morales, 2010).
2.1.2 Pathogénèse
L’amplification de l’expansion CTG se trouve sur le gène DMPK (Dystrophy Myotonic Protein Kinase) qui sert à coder une protéine de type kinase, la myotonine, servant à la régularisation d’autres protéines. La fonction de cette protéine est encore obscure, tant chez les sujets sains que chez les personnes atteintes de DM1 (Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992; Wansink et al., 2003; Kaliman et al., 2008; Forner et al., 2010; Pantic et al., 2013). Dans une maladie autosomique dominante, on observe généralement une diminution de la production de la protéine codée par le gène en cause, car elle ne peut être produite que par l’allèle normal. Dans la DM1, la situation est différente puisque la mutation se retrouve sur une région non-codante. Deux théories ont
été avancées pour expliquer la pathogénèse de cette maladie: un gain de fonction de l’ARN messager et une perte de fonction de la protéine DMPK (Udd et Krahe, 2012).
Dans un processus normal de transcription d’un gène, l’ADN contenu dans le noyau de la cellule est transcrit en ARN messager (ARNm) afin de lui permettre de traverser la membrane cytoplasmique. L’ARNm est ensuite traduit en protéines par les ribosomes. Dans la DM1, l’expansion CTG est située dans une région non-codante. Lors de la transcription de l’ADN en ARNm, l’ARNm issu de l’allèle anormal reste piégé dans le noyau de la cellule sous forme d’inclusions ribonucléaires. Cela créerait une accumulation toxique (Furling et al., 2001). Cet ARNm mutant se lie à la protéine CUG-PB qui est impliquée dans le processus de la myogenèse. Chez un modèle animal (souris), l’expansion CTG a été introduite en grande quantité dans les cellules musculaires (Timchenko et al., 2004). Cette souris a développé des inclusions ribosomiques, de la myotonie, des changements histologiques dans les tissus musculaires et de la dystrophie musculaire. Par contre, malgré de larges expansions CTG, une souris étudiée par Mankodi et son équipe (Mankodi et al., 2000) n’a pas développé d’atrophie musculaire, ce qui suggère que le processus de diminution de la force musculaire dans la DM1 pourrait impliquer d’autres mécanismes.
La deuxième hypothèse avancée serait la perte de fonction de la protéine DMPK. Dans la DM1, une diminution de la production de la protéine DMPK a été démontrée et expliquée par l’accumulation toxique d’ARNm dans le noyau (Furling et al., 2001). L’expansion CTG affecterait la structure de la chromatine, produisant une répression des gènes adjacents et une diminution de la production de protéines associée à ces gènes. Par exemple, le gène SIX5/DMAHP est situé en amont du gène DMPK et démontre une diminution de son activité chez les personnes atteintes de DM1 (Thornton et al., 1997; Lopez Castel et al., 2010). La suppression du facteur de transcription SIX5 chez un modèle animal a aussi causé l’apparition de cataractes (Klesert et al., 2000). La réduction de la protéine DMPK chez la souris a entrainé des défauts de conduction cardiaque, ce qui peut suggérer un effet similaire de la réduction de la protéine DMPK chez les humains atteints de DM1 (Berul et al., 2000). Cette théorie semble cependant moins probable depuis que le gène responsable de la DM2 a été identifié. Les deux maladies sont similaires à plusieurs
points de vue, mais ne proviennent pas d’une anomalie sur le même gène. La DM2 présente plusieurs caractéristiques communes à la DM1, dont les cataractes, la myotonie et l’atrophie testiculaire. Par contre, la progression de la faiblesse dans la DM2 implique les muscles proximaux de manière beaucoup plus marquée que dans la DM1 (Ranum et Day, 2004). Le gène responsable de la DM2 est situé sur le chromosome 3 alors que celui impliqué dans la DM1 se situe sur le chromosome 19. Comme les gènes adjacents ne sont pas les mêmes pour les deux maladies, il est difficilement justifiable que la DM1 ou la DM2 soit en partie causée par l’effet de la diminution de la production de protéines sur des gènes adjacents (Ranum et Day, 2004).
2.1.3 Facteurs explicatifs de la myopathie
Plusieurs aspects ont été étudiés pour mieux comprendre la physiopathologie de la myopathie présente dans la DM1. D’abord, une grande variabilité d’anomalies histologiques, parfois contradictoires, a été observée dans les muscles squelettiques des personnes atteintes de DM1. Des biopsies musculaires ont démontré un plus grand pourcentage de fibres de type 1 (Grimby et al., 1988), qui seraient moins affectées par l’atrophie, alors que d’autres ont observé une atrophie de ces mêmes fibres (Brooke et Engel, 1969) ou des fibres de type 2 rares ou inactives (Borg et al., 1987). En DM1, le tissu musculaire montre une régénération imparfaite se traduisant par une plus grande présence de noyaux centralisés et la présence d’amas de noyaux (Grimby et al., 1988; Harper et al., 2004), une proportion plus élevée de tissus conjonctifs (Grimby et al., 1988), de la fibrose (Grimby et al., 1988), la présence de dépôts adipeux anormaux (Harper et al., 2004; Grimby et al., 1988), des changements architecturaux (Grimby et al., 1988; Harper et al., 2004). Ces observations suggèrent une homéostasie musculaire altérée et un processus de réparation inadéquat.
La capacité de myogenèse du muscle DM1 est significativement réduite, ce qui se traduit par des myoblastes montrant une senescence prématurée (Bigot et al., 2009), une capacité de prolifération diminuée (Furling et al., 2001) et un retard dans leur différentiation et leur fusion en myotubes (Sabourin et al., 1997; Furling et al., 2001). Une autre étude a rapporté une augmentation du nombre de myoblastes afin de compenser leur dysfonction (Thornell et al., 2009). Lorsque la condition s’aggrave, la compensation par le
nombre n’est plus possible, ce qui entraîne des anomalies dans la régénération musculaire. La prolifération des myoblastes serait diminuée par une sur activation de la cascade p16, qui empêche la synthèse d’ADN et donc la prolifération cellulaire, qui est, entre autres, déclenchée par l’expansion CTG (Rayess et al., 2012; Harashima et al., 2013). La perte de masse musculaire peut être causée par une diminution de la synthèse ou à une augmentation de la dégradation. En DM1, les conclusions de quelques études tendent vers une diminution du processus anabolique (Halliday et al., 1985; Griggs et al., 1986). Finalement, un taux élevé de facteur TNF (facteur pro-inflammatoire) est observé dans le muscle DM1 entrainant une inhibition de la myogenèse et de la régénération protéique ainsi qu’une résistance à l’insuline, ce qui pourrait expliquer en partie l’atrophie musculaire observée (Zhang et al., 2008). L’ensemble des processus énumérés ici semble jouer un rôle dans la myopathie observée dans la DM1 mais leur importance respective dans le développement de la maladie reste à déterminer.
2.2 Atteinte de la force musculaire aux membres inférieurs
Article 1
Lower limb muscle impairment in myotonic dystrophy type 1: the need for better guidelines
Auteurs: Émilie Petitclerc, Luc J. Hébert, Johanne Desrosiers, Cynthia Gagnon.
Statut de l’article: L’article original a été publié (Petitclerc E, Hebert LJ, Desrosiers J, Gagnon C (2015) Lower limb muscle impairment in myotonic dystrophy type 1: The need for better guidelines. Muscle Nerve 51:473-8). La reproduction de cet article dans le présent mémoire est autorisée par Wiley Periodicals, Inc.
Avant-propos: L’article consiste en une revue systématique des écrits portant sur la description de l’atteinte de la force musculaire aux MIs en DM1. J’ai effectué la recension des écrits, l’extraction des informations relatives aux qualités métrologiques à l’aide de la grille de cotation développée par le groupe COSMIN (COnsensus-based Standards for the selection of health Measurements Instruments) pour les instruments de mesure en santé, l’analyse et la rédaction de l’article. Les co-auteurs ont supervisé les étapes de réalisation du projet et de la rédaction de l’article.
Résumé : Dans la DM1, la faiblesse musculaire aux MIs est une atteinte majeure. L’obtention d’un portrait clair de l’atteinte de la force musculaire présente plusieurs défis. Une recension systématique des écrits portant sur l’atteinte de la force aux MIs chez les phénotypes DM1-AC (adult en anglais) et DM1-AT (late-onset en anglais) a été réalisée pour documenter les variables qui influencent la mesure de la force. Trente-deux articles ont été révisés en utilisant les lignes directrices développées par le groupe COSMIN. Seulement le tiers des études ont décrit un protocole reproductible. La fidélité du bilan musculaire quantitatif a été évaluée pour deux groupes musculaires seulement, tandis que celle du bilan musculaire manuel n’a été mesurée que dans une seule étude et uniquement sur un score total. Dans la plupart des études, les variables influençant l’atteinte de la force en DM1 ne sont pas décrites. Cette recension illustre la variabilité de l’évaluation de la force musculaire en relation avec les caractéristiques propres à la DM1 et la validité questionnable des résultats en raison de la faible documentation des qualités méthodologiques des protocoles et méthodes utilisés. La nécessité d’adopter un consensus
sur l’utilisation d’un protocole d’évaluation de la force musculaire standardisé est manifeste.
Abstract
In myotonic dystrophy type 1 (DM1), leg muscle weakness is a major impairment. There are challenges to obtaining a clear portrait of muscle strength impairment. A systematic literature review was conduct on lower limb strength impairment in late-onset and adult phenotypes to document variables which affect strength measurement. Thirty-two articles were reviewed using the COSMIN guidelines. Only a third of the studies described a reproducible protocol. Only 2 muscle groups have documented reliability for quantitative muscle testing and only 1 total score for manual muscle testing. Variables affecting muscle strength impairment are not described in most studies. This review illustrates the variability in muscle strength assessment in relation to DM1 characteristics and the questionable validity of the results with regard to undocumented methodological properties. There is therefore a clear need to adopt a consensus on the use of a standardized muscle strength assessment protocol.
Original Title
Lower limb muscle impairment in myotonic dystrophy type 1: the need for better guidelines.
Journal
Muscle & Nerve Authors
Émilie Petitclerc PT, MSc(c)1, Luc J. Hébert PT, PhD2,3, Johanne Desrosiers OT, PhD4, Cynthia Gagnon OT, PhD5
1. Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Canada.
2. Faculty of Medicine, Rehabilitation and
3. Radiology Department, Université Laval, Québec, Canada.
4. School of rehabilitation, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, Canada.
5. Groupe de recherche interdisciplinaire sur les maladies neuromusculaires,
Neuromuscular Clinic, Centre de Santé et de Services Sociaux de Jonquière, Jonquière, Canada.
Acknowledgments
The corresponding author has received bursaries from the Corporation de recherche et d’action sur les maladies héréditaires (CORAMH), the Faculty of medicine and health sciences of the Université de Sherbrooke, the Fondation Go, and the Ordre professionnel de la physiothérapie du Québec (OPPQ), as well as the financial, professional and technical support of the Groupe de recherche interdisciplinaire sur les maladies neuromusculaires (GRIMN) of the Neuromuscular Clinic of the Centre de santé et de services sociaux de Jonquière (Fonds de dotation du CSSS Jonquière).
INTRODUCTION
Myotonic dystrophy type 1 (DM1) is the most common form of muscular dystrophy in adults1. It is a neuromuscular disease2 resulting from a mutation on the 19q13.3 locus of chromosome 19, leading to an unstable repetition of cytosine-thymine-guanine (CTG) base pairs3. It is a multisystem disease with various symptoms including loss of muscle strength, myotonia, dysphagia, and cognitive impairment, among others4. As recently stated in an international workshop report, the promising therapies in DM1 have led researchers and clinicians to believe that this population will soon have access to new therapeutic trials such as gene therapy5. As muscle strength is greatly affected in this population, it will be 1 of the main outcome measures to use to monitor disease progression. Indeed, lower limb (LL) weakness and high level of fatigue are the 2 most important variables for disrupted social participation in DM1 patients6. However, presenting a clear and complete portrait of muscle strength impairment and properly assessing muscle strength in the context of therapeutic trials offers some challenges.
The first challenge is related to the description of muscle strength impairment according to the specific characteristics of the DM1 population. There are 4 DM1 phenotypes based on disease severity and age of onset (congenital, childhood, adult, and late-onset)4. The pattern of muscle strength impairment and the rate of progression of weakness are quite different in the congenital and childhood phenotypes compared with the 2 others4. This paper will focus on the adult and late-onset phenotypes. In the adult phenotype, symptoms generally appear in the second or third decade of life4. Affected patients develop, among other symptoms, myotonia and progressive loss of muscle strength4. The adult phenotype is heterogenous, as some patients have severe impairment affecting several systems early in life, while others are not as severely affected7,8. There is no clear cut-off between the 2 phenotypes, but the late-onset phenotype is characterized by older age of onset (>40years) and usually less severe muscle strength impairment9. In addition, the potential variability in rate of progression is an additional variable to take into consideration while describing muscle strength impairment. Indeed, the disease can progress to debilitating weakness in a few years, or it can be stable and benign for more than 20 years1. The prognosis is thus difficult to establish even though Mathieu et al.1 have
been able to estimate the average years of disease for each stage of the Muscular
Impairment Rating Scale (MIRS). Description of muscle strength impairment in DM1
could thus be influenced by phenotypic variability (including CTG repeat length), age of onset, disease duration, and rate of progression.
The second challenge is associated with the different methods used in the international community to assess muscle strength [manual (MMT) or quantitative (QMT) muscle testing]. Several outcome measures are used to describe lower limb function in DM1, but specifically using quantified muscle strength measures allows one to clearly map weakness of all magnitudes in all muscle groups to obtain a better understanding of the related functional deficits and the natural history of the disease over time. On one hand, MMT does not require any equipment and is generally performed according to agreed-upon protocols10. Muscle strength, assessed by MMT, is most often rated on the 5-point scale Medical Research Council (MRC) scale, or its 10-point scale variant, the modified MRC scale11. On the other hand, QMT measures the level of maximum voluntary isometric or isokinetic force of a muscle group in a given position or through a given range of motion, using a force gauge (manual or fixed dynamometers)12. The 2 methods (MMT and QMT) are used in clinical practice and research. However, the methodological properties of QMT and MMT seem to have been explored only modestly in DM1. In addition, although the sensitivity of MMT to changes has been questioned for both clinical outcomes and evaluation of intervention effectiveness13,14, MMT is still used in therapeutic trials.
The workshop on trial readiness in 20095 and the recent report15 on Outcome Measures in Myotonic Dystrophy Type 1 (OMMYD-1) held in 2011 have raised concerns about the availability of methodologically sound outcome measures for muscle strength and the lack of natural history studies. However, no systematic review is available to provide a global picture of evidence-based data on muscle strength impairment in DM1. This is an essential step to clearly identify what is known and what still needs to be done in order to provide proper guidance to clinicians and researchers who are developing programs to document the natural history of the disease and methodological properties of muscle strength assessment procedures.
The objective of this paper is to perform a systematic literature review on lower extremity muscle strength impairment in individuals with the late-onset or adult DM1 phenotype while specifically documenting the variables affecting strength measurement in the DM1 population.
MATERIALS AND METHODS
A systematic literature review was conducted using the PubMed, Medline, and CINAHL databases with the following main keywords (English and French): myotonic dystrophy and strength (see appendices for the full term list). All articles published in French or English between 1980 and 2011 regarding muscular strength impairment in patients with the adult or late-onset DM1 phenotypes were included. The following exclusion criteria were used: 1) absence of muscle strength results, muscle strength results of upper limbs only or respiratory or smooth muscles only; 2) data collected in animals, and; 3) studies with patients with various neuromuscular diseases without specific results for the subsample of participants with DM1.
Three rehabilitation professionals (1 physiotherapist and 2 occupational therapists) performed the first screening of articles, based on title and abstract. Studies that met the inclusion criteria were kept for a further detailed assessment using a standardized extraction grid. The reference lists of retrieved articles were also consulted to cross-reference and find additional papers that also met the inclusion criteria. The extraction grid was developed according to COSMIN guidelines (COnsensus-based Standards for the selection of health Measurements INstruments)16. The COSMIN group developed a critical appraisal tool/checklist containing standards for evaluating the methodological quality of studies on the measurement properties of health measurement instruments (see http://www.cosmin.nl/). The extraction grid focused mainly on the protocol characteristics, methodological properties, and disease characteristics for muscle strength evaluation (phenotypes, disease duration, and selection of muscle groups). Additional information on muscle strength (frequency, severity) and muscle strength impairment rate of progression was also extracted.
RESULTS
The literature review led to a preliminary total of 103 articles. The review of the reference lists led to 6 additional papers (total n = 109). Seventy-seven articles were excluded according to the previously outlined criteria. A total of 32 articles were reviewed and thoroughly analysed by 2 reviewers (EP, CG).
Documentation of disease characteristics for each study is presented in supplementary Tables S1 and S2 (available online)1,13,14,17-46. In terms of muscle weakness distribution, none of the studies described the results according to either adult or late-onset phenotype, and 7 studies also included other additional phenotypes. Overall, muscle strength assessment results are reported by pooled phenotypes. All lower extremity muscle groups (hip flexors, extensors, abductors, knee flexors and extensors, ankle plantarflexors, dorsiflexors, invertors, and evertors) were evaluated in at least 1 study, with the exception of the hip adductors and the internal and external hip rotators (see supplementary Table S3 and S4, available online). The ankle invertors and evertors were also rarely assessed. Muscle strength assessment protocol characteristics are also presented in Tables S1 and S2. A majority of studies used only QMT, or both QMT and MMT, as the main outcome measure for strength. In a little over one-third of the studies, a reproducible protocol was reported (positioning and stabilization of the subject, number of measurements, instructions, encouragement and feedback given, order of tests, instruments used).
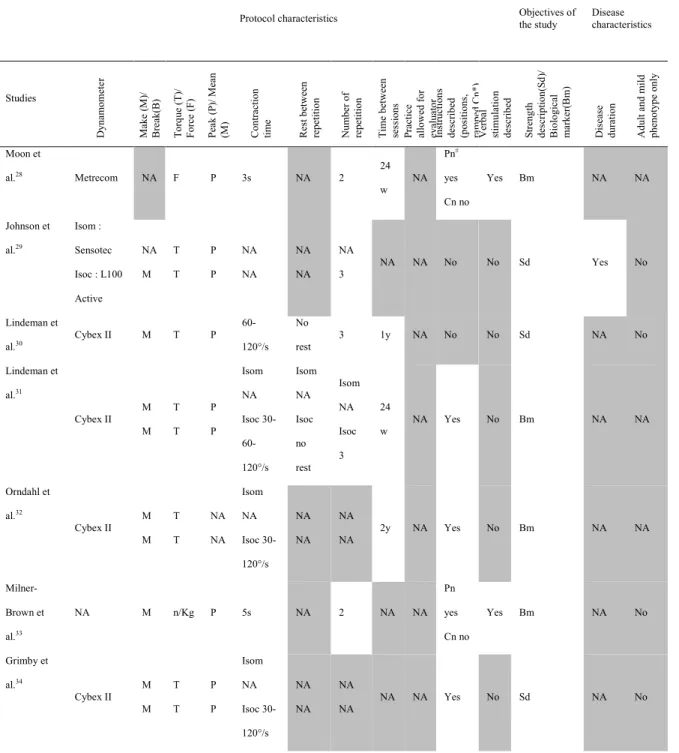
Table S1: Quantitative Muscle Testing Protocol characteristics, Objectives of studies and Disease characteristics summarized
Protocol characteristics Objectives of
the study Disease characteristics Studies D yn amo me te r M ak e (M )/ B re ak (B ) To rq ue (T )/ Fo rc e (F ) Peak (P )/ M ean (M ) C on tra ctio n ti me R es t b et w een re pe titio n N um be r of re pe titio n Ti m e b et w een se ssi on s Pr act ice al lo w ed fo r ev al uat or Ins truc tions des cr ib ed (pos iti ons , ra m pe d C n * ) V er bal stimu la tio n des cr ib ed Str en gth de sc rip tio n( Sd )/ B io lo gic al m ark er (B m ) D is eas e d ur at io n A du lt a nd mild phe not ype onl y Côté et al.17
Chatillon M T M 10s† 60s 2-3 NA Yes Yes Yes Bm Yes NA
Heatwole et
al.18 NA NA NA NA NA NA NA
24w‡
NA No No Bm NA NA
Hébert et al.13 Chatillon M T M 10s 60s 2-3 3 w Yes Yes Yes Sd Yes Yes
Missaoui et
al.19 Cybex M T NA 60°/s
§ No
rest 5 6 w NA Yes No Bm NA NA
Pénisson-Besnier et al.20 Nicholas NA F M NA NA NA 12 w NA No No Bm Yes No
Moxley et al.21 NA NA NA P NA NA 2 NA NA No No Sd NA NA
Wiles et al.22 NA NA F M NA NA 3 - NA No No Bm NA NA
Tarnopolsky et
al.23 NA NA T P 4s 30s 3 16 w NA No No Bm NA NA
Mathieu et
al.24 Nicholas NA F M NA NA 2 NA NA No No Sd Yes No
Lindeman et
al.25 Cybex II M T P 2s NA 1 NA NA Yes No Bm NA No
Tollbäck et
al.26
Kincom
500 H M T M 30°/s NA 3 12 w NA Yes Yes Bm NA Yes
Lindeman et al27 Cybex II M M T T NA NA Isom || NA Isoc ¶ 30-60-20°/s Isom NA Isoc no rest Isom NA Isoc 3 NA NA Yes No Sd NA NA
Table 1: Quantitative Muscle Testing Protocol characteristics, Objectives of studies and Disease characteristics summarized
Protocol characteristics Objectives of the study Disease characteristics
Studies D yn amo me te r M ak e (M )/ B re ak (B ) To rq ue (T )/ Fo rc e (F ) Peak (P )/ M ean (M ) C ont ra ct ion time Res t b et w een re pe titio n N um be r of re pe titio n Ti m e b et w een se ssi on s Pr act ice al low ed f or eva lua tor Ins truc tions des cr ib ed (pos iti ons , ra m pe d C n * ) V er bal stimu la tio n des cr ib ed St re ng th de sc rip tio n( Sd )/ B io lo gic al m ark er (B m ) D is eas e dur at ion A du lt a nd mild phe not ype onl y Moon et al.28 Metrecom NA F P 3s NA 2 24 w NA Pn# yes Cn no Yes Bm NA NA Johnson et al.29 Isom : Sensotec Isoc : L100 Active NA M T T P P NA NA NA NA NA 3 NA NA No No Sd Yes No Lindeman et al.30 Cybex II M T P 60-120°/s No rest 3 1y NA No No Sd NA No Lindeman et al.31 Cybex II M M T T P P Isom NA Isoc 30- 60-120°/s Isom NA Isoc no rest Isom NA Isoc 3 24 w NA Yes No Bm NA NA Orndahl et al.32 Cybex II M M T T NA NA Isom NA Isoc 30-120°/s NA NA NA NA 2y NA Yes No Bm NA NA Milner-Brown et al.33 NA M n/Kg P 5s NA 2 NA NA Pn yes Cn no Yes Bm NA No Grimby et al.34 Cybex II M M T T P P Isom NA Isoc 30-120°/s NA NA NA NA NA NA Yes No Sd NA No
Table 2: Manual Muscle Testing Protocol characteristics, Objectives of studies and Disease characteristics summarized
Protocol characteristics Objectives of
the study Disease characteristics
Studies Practice allowed
for evaluator Instructions described (positions, uni or bilateral evaluation) Verbal stimulation described Time between S1-S2* Strength description (Sd) / biological marker (Bm) Disease duration (dd) or phenotype (p) Adult and mild phenotype only Côté et al.17 NA No No - Bm Yes dd NA Heatwole et al.18 NA No No - Bm NA NA
Hermans et al.35 NA experienced
examiner No No - Bm Yes dd No
Hamano et al.36
NA No No - Sd NA NA
Hébert et al.13 NA experienced
pt† Yes No 3 w ‡ Sd Yes dd Yes Pruna et al.37 NA No No - Sd NA NA Pénisson-Besnier et al.20 NA No No - Bm Yes dd No Moxley et al.19 NA No No - Sd NA NA Sansone et al.38 NA No No 1 to 10 y§ Sd Yes dd Yes Whittaker et al.14 NA No No 1 to 8 y Sd NA NA Tarnopolsky et al.23 NA No No - Bm NA NA Mathieu et al.24
NA Yes (Brooke 1983) No - Sd Yes dd NA
Mathieu et al.39
NA No No - Bm Yes p No
Hedberg et al.40
Table 2: Manual Muscle Testing Protocol characteristics, Objectives of studies and Disease characteristics summarized
Protocol characteristics Objectives of
the study Disease characteristics
Studies Practice allowed for evaluator Instructions described (positions, uni or bilateral evaluation) Verbal stimulation described Time between S1-S2* Strength description (Sd) / biological marker (Bm) Disease duration (dd) or phenotype (p) Adult and mild phenotype only Mathieu et al.41 NA No No - Bm Yes p No Nitz et al.42
experienced pt Yes (Daniels and
Worthingham1986) No ≈ 1 y Sd NA NA
Sugino et al.43
NA No No - Bm NA NA
Nitz et al.44
experienced pt Yes (Daniels and
Worthingham 1986) No - Bm NA NA
Johnson et al.29 experienced pt Yes (Brooke 1981) No - Sd Yes p No
Vlachopapadopoulou et al.45 NA No No - Bm NA NA Mathieu et al.1 NA No No - Sd Yes dd NA Lord et al.46 registered pt No No - Sd Yes dd NA
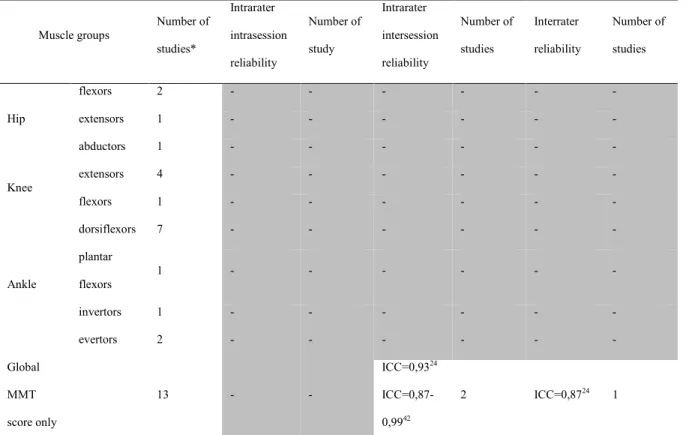
Table 3: Ranges of Reliability types per muscle group outlined against the number of studies - QMT
Muscle groups Number of studies* Intrarater intrasession reliability Number of study Intrarater intersession reliability Number of studies Interrater reliability Number of studies Hip flexors 2 - - - - extensors 2 - - - - abductors 2 - - - - Knee extensors 12 - - - - flexors 8 - - - - Ankle dorsiflexors 6 - - R 2 :0,90-0,9613 1 rp: 0,70-0,9313 1 plantar flexors 3 - - - - invertors 1 - - - - evertors 2 - - R 2: 0,89-0,9413 1 rp: 0,72-0,9413 1 Global QMT score only 6 - - - -
Table 4: Ranges of Reliability types per muscle group outlined against the number of studies - MMT
Muscle groups Number of studies* Intrarater intrasession reliability Number of study Intrarater intersession reliability Number of studies Interrater reliability Number of studies Hip flexors 2 - - - - extensors 1 - - - - abductors 1 - - - - Knee extensors 4 - - - - flexors 1 - - - - Ankle dorsiflexors 7 - - - - plantar flexors 1 - - - - invertors 1 - - - - evertors 2 - - - - Global MMT score only 13 - - ICC=0,9324 ICC=0,87-0,9942 2 ICC=0,8724 1
* Number of studies where results were provided for each specific muscle group and not only a global MMT score.
Concerning severity of muscle strength impairment, all studies that reported an overall assessment of muscle strength by MMT showed an average score of 4 or less on the MRC scale for all tested muscular groups1,21,22,24,29,38,39,41. According to these studies, the weakness profile in DM1 progresses from distal to proximaland does so in a symmetrical manner. One study suggested that significant proximal weakness was common in DM142. For QMT, there is great variability among studies of muscle strength parameters (variations within the same muscle group, different units of measurement, and number of trials). In addition, no comparative values such as normative data or control groups were used except for 1 study29.
Rate of progression was documented in most studies using a cross-sectional design with the exception of 6 longitudinal studies, including 2 over a one-year period30,42, 1 over
a two-year32, 1 over a ten-year29, and 2 over a period varying between 1 to 10 years14,38. From cross-sectional studies, Mathieu et al.24 found a 0.95% decrease of MMT per year of disease duration (1.2% distal muscles, 0.7% proximal muscles / men = women), and a QMT decrease of 1.2 - 1.6% per year of disease duration for proximal muscles and of 2.0 - 3.0% per year for distal muscles (women < men). Hébert et al.13 similarly reported an initial decline in the first 2 decades of the disease of 2.5% and 3.2% in QMT per year for the ankle evertors and dorsiflexors, respectively, followed by a rate of progression of about 1.5% (dorsiflexors) to 2.2% (evertors) for the subsequent years. A similar loss was reported by Whittaker et al.14 (1.0 to 1.2%, and 0.2 to 0.4% in MMT per year for distal and proximal muscles, respectively). Using a longitudinal design, Sansone et al.38 reported a 1.2% MMT decrease per year using a global score. The loss of muscle strength is described as linear, slow, and faster for distal muscles compared to proximal muscles24. Most people with DM1 progress to mild (44.7%) or moderate (50.9%) myopathy, and a lower proportion of people affected progress to a severe level of impairment (4.4%)1.
With regards to methodological properties, only 2 muscle groups have documented intrarater and interrater reliability (good to excellent13) for QMT and only a total score for MMT (good to excellent24,42) (see supplementary Table S3 and S4, available online). The challenge associated with responsiveness to change of both methods has been discussed in 2 studies13,14 but no data are available. The smallest mean difference using QMT was calculated in 1 study and reported to be roughly twice the standard error of measurement13. DISCUSSION
This systematic literature review of lower extremity muscle strength impairment in individuals with late-onset and adult DM1 phenotypes has allowed us to develop a global picture of the evidence-based knowledge of muscle strength. However, these findings raise a few relevant observations that question our current understanding of muscle strength impairment in DM1. We will first discuss observations related to muscle strength assessment more specifically, and then we will discuss potential implications of our findings with regard to characterization of muscle strength impairment.
Muscle strength assessment
Regarding the selection of muscle strength assessment methods, there is great variability in the choice of methods used. As seen in Tables S1 and S2, the QMT and MMT protocols vary considerably between studies, and they are often not sufficiently detailed to them to be reproduced. The protocols used also are fundamentally dissimilar (make/break test, peak/mean, contraction time, rest between repetitions, number of repetitions, positioning, and type of verbal stimulation), and that could influence the measurements obtained. Also, MMT protocols, when described, vary on many important key points such as positioning, uni- or bilateral assessment, and verbal stimulation that could significantly influence the result. In addition, the lack of agreement between studies with regard to the choice of the assessment protocols limits the comparisons considerably and prevents pooling of results to increase sample size.
Another issue that should be addressed is standardization of the strength assessment protocols. Complete information was only given in 1 paper (standardization of the protocol and training process)13. Developing and standardizing the administration protocol and the rater training process for each selected outcome measure are also major issues. Standardization of outcome measures and structured/systematic training for testing have previously been emphasized for clinical trials and natural history studies in neuromuscular disorders12,13,47. Considering the relatively low worldwide prevalence of DM1, a multicenter approach will be necessary to develop therapeutic trials. This may introduce additional challenges related to maintaining a good to excellent interrater reliability; however, with appropriate and standardized, structured training, the sample size of clinical trials can be decreased significantly in some cases without any lessening of statistical power48.
Although MMT and QMT have been described as acceptable methods for measuring muscle strength in individuals diagnosed with DM15, our results clearly show the lack of documented methodological properties.
Muscle strength impairment in relation to population characteristics and methodological properties
All studies have pooled the findings for both late-onset and adult phenotypes, and sometimes for the congenital and infantile phenotypes also. This pooling of results could lead to an over- or under-estimation of muscle strength impairments among the different phenotypes. Also, for natural history studies, it is essential to describe the phenotypes separately, as we do not know whether the rate of progression is the same. In this data analysis from a pool of 198 patients with DM1, we have found that the mean strength of ankle dorsiflexors, as assessed by QMT, was quite different between the adult (n = 158; 94.6 N) and the late-onset (n = 40; 167.5 N) phenotypes. Therefore, if we had pooled the data (109.4 N), muscle strength in the late-onset phenotype would have been clearly underestimated.
Although several studies describe muscle strength in DM1, the objective of only a few studies was to characterize the profile of muscle strength1,13,14,21,22,24,27,29,30,34,39,41,42. Other studies published results using muscle strength as biological markers for treatment efficacy (medication or exercise) or as part of a validation process of new techniques to assess muscle damage (e.g., magnetic resonance imaging) or functional status (e.g., Motor Function Measure). This may partly explain why some mildly affected muscles (e.g., knee flexors) have been assessed frequently while other key muscles have not been as frequently evaluated despite their known key functional role in other populations (e.g. hip stabilizers). In addition, only 1 study was designed clearly to document muscle strength as an outcome measure for clinical trials13.
The progression pattern of muscle strength impairment from distal to proximal in late-onset and adult phenotypes is essentially supported by the results of studies that have used MMT24,39,40. In contradistinction, Nitz et al.42 reported significant and common proximal weakness among DM1 patients. However, they did not report disease duration or age of onset. Reasons why proximal weakness has not been observed early on in the disease may be explained by the use of protocols that did not allow a valid measurement of proximal muscle strength and from the inability of MMT to detect mild to moderate weakness in proximal muscle groups that are among the strongest muscles, such as the hip.
Assuming that the progression of disease is from distal to proximal, muscle strength impairment will inevitably reach the proximal muscles as disease duration increases. Thus, if the sample in the Nitz, et al. study consisted mostly of patients in an advanced stage of the disease, the authors could have consequently observed proximal weaknesses43. But leaving aside the duration of disease issue, the responsiveness and discriminative properties of MMT seem insufficient to identify proximal muscle strength impairment unless it is of a significant magnitude13. Therefore, this leaves the perception that the pattern of progression is systematically and always from distal to proximal for all DM1 patients, which may not be the case; this still needs to be validated with protocols and instruments that have proper methodological properties. Concerning QMT, studies do not allow any conclusion regarding the relative severity of impairment for each muscle group over time on account of the great variability of parameters and the lack of comparison to normative data or a control group in most studies.
The protocols used to measure muscle strength in the vast majority of these studies are either insufficiently described or have significant methodological flaws, which in both cases does not allow one to draw firm conclusions on the profile of muscle strength impairment. The lack of documented methodological properties could influence results in many ways. For example, MMT has been reported to be less sensitive, especially in weak patients13, and would not properly convey the slow progression pattern of the muscle strength impairment14. The results of Hebert et al.13, Whittaker et al.14, and Johnson29 raise questions about the use of MMT in monitoring the clinical course of patients and in assessing the effectiveness of interventions because of its low sensitivity to detect changes.
Finally, few studies have described the progression of muscle strength impairment over time14,29,30,32,38,42,24,13. The small number of longitudinal studies further limits the knowledge regarding the rate of progression of muscle strength impairment. Furthermore, none have categorized the myopathy according to each phenotype. The ability to generalize results to all DM1 patients is often also limited because of small sample sizes. A better characterization over time of lower extremity muscle strength impairment in DM1 patients according to phenotype is essential in order to facilitate clinical decision making with regards to the monitoring of these patients. Additionally, further studies are needed to
identify which specific lower extremity muscle strength impairments most contribute to the decline of functional autonomy in these patients in order to justify their use in clinical trials. Therefore, although several studies report data on muscle strength impairment in DM1, most could be partly misleading in their message, as several key variables where not taken into consideration in the design of the study.
The results show that, although previous studies have contributed significantly to our knowledge of muscle strength impairment in DM1, their findings must be interpreted with caution and within the limitations of the protocols used, including unknown or questionable methodological properties and strength protocols that have not considered all muscle groups. In DM1, the choice of the measurement method to assess muscle strength impairment should be based on specific criteria, including the one developed by the COSMIN initiative16: 1) known and acceptable psychometric qualities, including validity, reliability, and responsiveness; 2) feasible for a broad choice of muscles (proximal versus distal, upper versus lower limb versus spine); 3) clinical or research goals (assessing, treating, exercise program, gene therapy, etc.); and 4) type of study (cross sectional, longitudinal, randomized clinical trial, etc.). Other considerations related to the transfer of knowledge (feasibility of using the measures in a clinical setting; who will be the primary evaluator, a physician, physiotherapist, occupational therapist, or others) and the availability of equipment may also be taken into account.
Study limitations
The risk for selection bias was minimized in this review by using 3 independent reviewers to screen articles. However, articles published in English and French only were reviewed. In addition, only published papers were reviewed and not thesis or conference proceedings. Also, as we have chosen to focus on muscle strength impairments, other relevant studies using other outcome measures of lower limb function such as timed function tests or performance tests were not considered. Therefore, these findings are specific to muscle strength and are not inclusive of all the research that has been conducted on lower limb functional deficits in DM1.
CONCLUSION
This literature review illustrates the wide variability in the methods used to assess muscle strength. In addition, key variables that need to be taken into account while designing a study to document muscle strength impairment in DM1 have been outlined. The major issue to be addressed by future studies is documentation of methodological properties for muscle strength assessment, which is lacking at the moment. To overcome this situation, there is an urgent need to adopt an international consensus on the use of a standardized muscle strength assessment protocol with documented methodological properties to permit effective and efficient knowledge sharing among clinicians and researchers.
REFERENCES
1. Mathieu J, De Braekeleer M, Prévost C, Boily C. Myotonic dystrophy: clinical assessment of muscular disability in an isolated population with presumed homogeneous mutation. Neurology 1992;42(1):203-208.
2. Fu YH, Friedman DL, Richards S, Pearlman JA, Gibbs RA, Pizzuti A , et al. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science (New York, NY 1993;260(5105):235-238.
3. Harley HG, Rundle SA, MacMillan JC, Myring J, Brook JD, Crow S , et al. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet 1993;52(6):1164-1174.
4. Harper P. Myotonic dystrophy. London: WB Saunders; 2001. 436 p. p.
5. Thompson R, Schoser B, Monckton DG, Blonsky K, Lochmüller H. Patient Registries and Trial Readiness in Myotonic Dystrophy--TREAT-NMD/Marigold International Workshop Report. Neuromuscular Disorders 2009;19(12):860-866.
6. Gagnon C, Mathieu J, Jean S, Laberge L, Perron M, Veillette S , et al. Predictors of disrupted social participation in myotonic dystrophy type 1. Arch Phys Med Rehabil 2008;89(7):1246-1255.
7. Kierkegaard M, Harms-Ringdahl K, Widen Holmqvist L, Tollback A. Functioning and disability in adults with myotonic dystrophy type 1. Disabil Rehabil 2011:1-11.
8. Kierkegaard M, Harms-Ringdahl K, Widén Holmqvist L, Tollbäck A. Perceived functioning and disability in adults with myotonic dystrophy type 1: a survey according to the International Classification Of Functioning, Disability and Health. Journal Of Rehabilitation Medicine 2009;41(7):512-520.
9. Arsenault ME, Prévost C, Lescault A, Laberge C, Puymirat J, Mathieu J. Clinical characteristics of myotonic dystrophy type 1 patients with small CTG expansions. Neurology 2006;66(8):1248-1250.
10. Daniels L, Worthingham C. Muscle testing : Technique of manual examination. Philadelphia Pa: WB Saunders Co; 1986.
11. Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, Miller JP , et al. Clinical investigation in duchenne dystrophy: 2. Determination of the "power" of therapeutic trials based on the natural history. Muscle & Nerve 1983;6(2):91-103.
12. Hogrel JY, Ollivier G, Desnuelle C. [Manual and quantitative muscle testing in neuromuscular disorders. How to assess the consistency of strength measurements in clinical trials?]. Rev Neurol (Paris) 2006;162(4):427-436.
13. Hebert LJ, Remec JF, Saulnier J, Vial C, Puymirat J. The use of muscle strength assessed with handheld dynamometers as a non-invasive biological marker in myotonic dystrophy type 1 patients: a multicenter study. BMC Musculoskelet Disord 2010;11:72. 14. Whittaker RG, Ferenczi E, Hilton-Jones D. Myotonic dystrophy: practical issues relating to assessment of strength. J Neurol Neurosurg Psychiatry 2006;77(11):1282-1283. 15. Gagnon C, Meola G, Hebert LJ, Puymirat J, Laberge L, Leone M. Report of the first Outcome Measures in Myotonic Dystrophy type 1 (OMMYD-1) international workshop: Clearwater, Florida, November 30, 2011. Neuromuscul Disord 2013.
16. Terwee CB, Mokkink LB, Knol DL, Ostelo RW, Bouter LM, de Vet HC. Rating the methodological quality in systematic reviews of studies on measurement properties: a scoring system for the COSMIN checklist. Qual Life Res 2012;21(4):651-657.
17. Cote C, Hiba B, Hebert LJ, Vial C, Remec JF, Janier M , et al. MRI of tibialis anterior skeletal muscle in myotonic dystrophy type 1. Can J Neurol Sci 2011;38(1):112-118.
18. Heatwole CR, Eichinger KJ, Friedman DI, Hilbert JE, Jackson CE, Logigian EL , et al. Open-label trial of recombinant human insulin-like growth factor 1/recombinant human
insulin-like growth factor binding protein 3 in myotonic dystrophy type 1. Arch Neurol 2011;68(1):37-44.
19. Missaoui B, Rakotovao E, Bendaya S, Mane M, Pichon B, Faucher M , et al. Posture and gait abilities in patients with myotonic dystrophy (Steinert disease). Evaluation on the short-term of a rehabilitation program. Ann Phys Rehabil Med 2010;53(6-7):387-398.
20. Penisson-Besnier I, Devillers M, Porcher R, Orlikowski D, Doppler V, Desnuelle C , et al. Dehydroepiandrosterone for myotonic dystrophy type 1. Neurology 2008;71(6):407-412.
21. Moxley RT, 3rd, Logigian EL, Martens WB, Annis CL, Pandya S, Moxley RTt , et al. Computerized hand grip myometry reliably measures myotonia and muscle strength in myotonic dystrophy (DM1). Muscle Nerve 2007;36(3):320-328.
22. Wiles CM, Busse ME, Sampson CM, Rogers MT, Fenton-May J, van Deursen R. Falls and stumbles in myotonic dystrophy. J Neurol Neurosurg Psychiatry 2006;77(3):393-396.
23. Tarnopolsky MA, Mahoney DJ, Vajsar J, Rodriguez C, Doherty TJ, Roy BD , et al. Creatine monohydrate enhances strength and body composition in Duchenne muscular dystrophy. Neurology 2004;62(10):1771-1777.
24. Mathieu J, Boivin H, Richards CL. Quantitative motor assessment in myotonic dystrophy. Can J Neurol Sci 2003;30(2):129-136.
25. Lindeman E, Spaans F, Reulen J, Leffers P, Drukker J. Progressive resistance training in neuromuscular patients. Effects on force and surface EMG. J Electromyogr Kinesiol 1999;9(6):379-384.
26. Tollback A, Eriksson S, Wredenberg A, Jenner G, Vargas R, Borg K , et al. Effects of high resistance training in patients with myotonic dystrophy. Scand J Rehabil Med 1999;31(1):9-16.
27. Lindeman E, Leffers P, Reulen J, Spaans F, Drukker J. Quadriceps strength and timed motor performances in myotonic dystrophy, Charcot-Marie-Tooth disease, and healthy subjects. Clin Rehabil 1998;12(2):127-135.
28. Moon JH, Na YM, Kang SW, Lee HS. The changes in muscle strength and relaxation time after a comprehensive rehabilitation program for patients with myotonic dystrophy. Yonsei Med J 1996;37(4):237-242.
29. Johnson ER, Abresch RT, Carter GT, Kilmer DD, Fowler WM, Jr., Sigford BJ , et al. Profiles of neuromuscular diseases. Myotonic dystrophy. Am J Phys Med Rehabil 1995;74(5 Suppl):S104-116.
30. Lindeman E, Leffers P, Spaans F, Drukker J, Reulen J. Deterioration of motor function in myotonic dystrophy and hereditary motor and sensory neuropathy. Scand J Rehabil Med 1995;27(1):59-64.
31. Lindeman E, Leffers P, Spaans F, Drukker J, Reulen J, Kerckhoffs M , et al. Strength training in patients with myotonic dystrophy and hereditary motor and sensory neuropathy: a randomized clinical trial. Arch Phys Med Rehabil 1995;76(7):612-620.
32. Orndahl G, Grimby G, Grimby A, Johansson G, Wilhelmsen L. Functional deterioration and selenium-vitamin E treatment in myotonic dystrophy. A placebo-controlled study. J Intern Med 1994;235(3):205-210.
33. Milner-Brown HS, Miller RG. Myotonic dystrophy: quantification of muscle weakness and myotonia and the effect of amitriptyline and exercise. Arch Phys Med Rehabil 1990;71(12):983-987.
34. Grimby G, Hedberg M, Henriksson KG, Johansson G, Wigerstad-Lossing I, Sellden U , et al. Muscle function and morphology in myotonic dystrophy. Acta Med Scand 1988;224(4):349-356.
35. Hermans MC, Faber CG, Vanhoutte EK, Bakkers M, De Baets MH, de Die-Smulders CE , et al. Peripheral neuropathy in myotonic dystrophy type 1. Journal of the Peripheral Nervous System 2011;16(1):24-29.
36. Hamano T, Kawamura Y, Mutoh T, Hirayama M, Kuriyama M. Muscle MRI in myotonic dystrophy type 1 with foot drop. Eur Neurol 2010;63(3):144-148.
37. Pruna L, Machado F, Louis L, Vasse G, Kaminsky P. Fonction musculaire et atteinte d'organes dans la dystrophie myotonique de type 1. Rev Neurol (Paris) 2010.
38. Sansone V, Gandossini S, Cotelli M, Calabria M, Zanetti O, Meola G. Cognitive impairment in adult myotonic dystrophies: a longitudinal study. Neurol Sci 2007;28(1):9-15.