SUZIE DUFOUR
MICROSONDES OPTIQUES ET
ÉLECTRIQUES EN NEUROSCIENCES
Thèse présentée
à la Faculté des études supérieures et postdoctorales de l’Université Laval
dans le cadre du programme de doctorat en
neurobiologie
pour l’obtention du grade de
Philosophiae Doctor
(
Ph.D.
)
DÉPARTEMENT D’ANATOMIE ET PHYSIOLOGIE
FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL
QUÉBEC
2012
une science qui utilise les propriétés de la lumière pour sonder le système nerveux. Le développement de plusieurs méthodes de détection optiques, la découverte des protéines fluorescentes, le développement de sondes moléculaires fluorescentes et l’avancement dans le domaine des modifications génétiques fournissent aux neurosciences des outils spécifiques pour l’étude du système nerveux et des différentes populations cellulaires qui le composent. Alors que ces techniques sont pleinement accessibles in vitro, les outils de détection in vivo demeurent limités. En effet, la microscopie est limitée aux couches superficielles du système nerveux et les systèmes endoscopiques sont relativement invasifs.
L’objectif des travaux présentés dans le cadre de cette thèse est de fournir des outils de détection optique multifonctionnels pour la détection de fluorescence ou l’illumination in vivo qui pallient aux limitations des techniques couramment utilisées pour mesurer la fluorescence provenant de cellules du système nerveux ou encor e contrôler (via les outils optogénétiques) l’activité neuronal à l’aide de stimuli optique in vivo.
Cette thèse comprend en premier lieu une revue de littérature et certains aspects théoriques relatifs aux microsondes optiques et électriques. Par la suite, différents types de microsondes sont présentés. Un premier type a été fabriqué à l’aide d’une fibre optique étirée et d’une microélectrode « ion-sensitive » dans le but d’enregistrer les variations intracellulaires de la concentration de l’ion K+, qui jouent
un rôle important pour le maintien du potentiel de membrane cellulaire et pour le niveau d’excitabilité neuronale. Ces sondes ont permis d’enregistrer, pour la première fois, les fluctuations de K+ intracellulaire pendant les crises d’épilepsie chez le chat
anesthésié. Le deuxième type de sonde utilisé fut fabriqué à l’aide d’une fibre à deux cœurs, un cœur optique et un cœur creux servant d’électrode. Ces sondes ont permis d’enregistrer simultanément la concentration calcique intracellulaire et l’activité neuronale extracellulaire. Ces enregistrements ont permis de confirmer le caractère unicellulaire de la résolution de la sonde. Les travaux de cette thèse ont également servi à augmenter les propriétés multifonctionnelles des sondes optiques et éle ctriques
et à élargir la gamme d’expériences auxquelles elles peuvent s’adapter. Nos travaux ont montré que les sondes optiques pouvaient être utilisées avec un système de détection optique multispectrale et combiner plus d’un type d’enregistrement électrophysiologique en utilisant des couches métalliques conductrices. Il a également été montré qu’elles pouvaient être utilisées avec les outils optogénétiques pour contrôler le niveau d’activité neuronale. Les dépolarisations et hyperpolarisations photo-induites avec les sondes ont d’ailleurs permis de développer une nouvelle méthode de mesure permettant de mesurer les propriétés intrinsèques de la membrane neuronale à partir du milieu extracellulaire.
Abstract
Neurophotonic, a science that uses light properties to probe the nervous system, relies on many technological advances. The development of optical detection methods, the discovery of fluorescent proteins, the development of fluorescent molecular probes and the advancement in the field of genetic modification provided neuroscientists specific tools to study the brain and the different cell populations that it includes. While these techniques are fully accessible in vitro, in vivo detection tools remain limited. Indeed, microscopy is limited to superficial layers of the nervous system or is relatively invasive (endoscopic systems).
The objective of this thesis is to provide tools for in vivo fluorescence optical detection or light delivery that overcomes the limitations of commonly used techniques to measure fluorescence arising from neurons or glial cells or control (via optogenetic tools) cellular activity with optical stimuli in vivo.
This thesis first review the optical detection techniques and list theoretical aspects related to optical microprobes. Subsequently, different developed types of microprobes are presented. One type that was developed combines a tapered optical fiber and an ion-sensitive microelectrode to record changes in intracellular K+
concentration, which plays an important role in maintaining cellular membrane potential and the neuronal excitability level. These probes allowed us to record for the first time intracellular K+ fluctuations during epileptic seizures in anesthetised cat.
The second type of probe used in our work was fabricated from a dual core fiber integrating an optical core and a hollow core used as an electrode. Using the latter probe, we measured simultaneously intracellular Ca2+ fluctuations and extracellular
neuronal activity and demonstrated their single-cell resolution capabilities. Our work also involved enhancing the microprobe multifunctional capabilities and the range of experiments that it can be adapted to. We demonstrated that optical microprobes could be used with a multispectral optical detection system and adapted to include thin metallic layers as secondary electrodes to record local field potentials. It was also shown that they could be used with optogenetics tools to control the level of neuronal activity. Finally, photo-induced currents generated with the probes were used to
develop an innovative method to resolve intrinsic membrane properties of neurons from the extracellular medium.
Bien qu’ils ne couvrent pas entièrement l’ensemble des travaux effectués, ces articles résument bien le travail fait au cours des dernières années.
Le premier manuscrit, présenté au chapitre 2, découle des travaux achevés au laboratoire des Profs. Réal Vallée et Florin Amzica et fait suite aux deux comptes rendus de conférences publiés antérieurement. Il décrit en détail la fabrication de micro-optrodes fabriquées à partir de fibres optiques multimodes étirées et d’électrodes ion-sensitives. Ces micro-optrodes ont permis d’enregistrer de manière simultanée et co-localisée la concentration de potassium intracellulaire, extracellulaire et l’activité neuronale dans différentes conditions physiologiques chez le chat in vivo. Sous la supervision de Prof. Amzica et avec les conseils des doctorants Oana Chever et Pascal Dufour, j’ai mis au point le protocole d’expérimentation et la conception de la sonde, j’ai procédé à la totalité des expérimentations et à la rédaction de cet article. Après le départ du Dr. Amzica pour l’Université de Montréal, j’ai joint le groupe du Prof. Yves De Koninck qui s’intéresse également à la conception d’optrode. Une collaboration étroite entre les laboratoires des Profs. Vallée, De Koninck et Deschênes a permis de développer une microsonde optique et électrique pour l’enregistrement neuronale unitaire. Le deuxième article inclus dans cette thèse présente ce nouveau type de sondes optiques fabriquées à l’aide d’une fibre optique à 2 cœurs, un cœur optique et un cœur creux (servant d’électrode). Cette sonde a une résolution spatiale qui lui permet d’identifier des neurones fluorescents in vivo et d’enregistrer leur activité électrique. Yoan LeChasseur, dans le cadre de ses travaux de doctorat, a conçu la sonde et une partie des protocoles d’expérimentation. Ce dernier et Guillaume Lavertu ont fait l’ensemble des enregistrements in vivo dans la moelle de rats. Pour ma part, j’ai fait l’ensemble des enregistrements calciques in vivo et une partie des enregistrements sur les souris transgéniques GAD-65. J’ai fait l’ensemble des mesures sur les souris transgéniques
Thy1::ChR2-YFP.
De plus, j’ai participé aux simulations des propriétés d’excitation et de collection des sondes, aux mesures de résolutions spatiales in vitro et à la rédaction de l’article.Le troisième article, qui est présenté au chapitre 4 et qui sera bientôt soumis pour publication, fait la démonstration du caractère multimodal et flexible de la sonde optique présentée au chapitre 3. On y explique comment les microsondes optiques et le système de détection peuvent être modifiés pour permettre de dupliquer les canaux d’acquisition optique et électrique. Sous la supervision des Profs. Vallée et De Koninck, j’ai mis au point le protocole, la conception des expériences, j’ai effectué les analyses et participer à la rédaction de l’article. J’ai fait la majorité des enregistrements in vivo. Sophie Dufour-Beauséjour, une étudiante au baccalauréat en physique sous ma supervision, a travaillé sur l’enregistrement électrique à l’aide de dépôts métalliques sur fibres optiques et a procédé au double enregistrement électrique in vivo avec l’aide de Guillaume Lavertu. Alexandre Juneau-Fecteau, également étudiant au baccalauréat en physique sous ma supervision, a démontré l’efficacité des sondes pour la détection de cellules de couleurs différentes in vitro.
Finalement, le quatrième article présenté dans cette thèse démontre comment la combinaison des outils optogénétiques et des microsondes optiques peut être utilisée
pour sonder de manière non-invasive (sans perforer la membrane cellulaire et affecter la composition du milieu intracellulaire) les propriétés de la membrane cellulaire. Cette technique a été utilisée pour évaluer des changements de résistance membranaire dus à l’application de drogues, pour mettre en évidence des courants ioniques et des événements post-synaptiques, ainsi que pour estimer leur potentiel d’inversion. Encore une fois, en étroite collaboration avec les Profs. De Koninck, Vallée et Deschênes, j’ai mis au point le protocole, la conception des expériences, j’ai effectué les expériences, les analyses et rédigé l’article sous leur supervision.
Remerciements
J’ai eu le privilège d’avoir, pas un, ni deux, mais trois superviseurs pour mes travaux! Alors en 3 courts paragraphes…
Merci à Florin Amzica, qui a toujours été patient avec la physicienne de son laboratoire. Florin est un passionné qui m’a transmis une passion pour les neurosciences. Je voudrais le remercier pour un million de chose s, mais pour faire court, je le remercie du temps qu’il m’a consacré pour m’apprendre à présenter mes résultats. C’est essentiel en sciences!
Merci à Réal Vallée, avec qui c’est toujours un plaisir de parler et de faire des mises au point. Réal est un excellent physicien et ses conseils m’ont beaucoup aidé à faire avancer mes projets et à garder le cap.
Merci à Yves De Koninck, qui m’a accueilli pour les dernières années de ma thèse. Cela a été un plaisir de connaître Yves, il a une manière particulière d’aborder les sciences, sans œillère, avec l’enthousiasme d’un enfant, mais avec un impressionnant bagage de connaissances et une capacité incroyable de tout mettre en concept.
D’autres chercheurs m’ont beaucoup aidé dans mes travaux de recherches et je tiens à les remercier chaleureusement. Le Prof. Igor Timofeev, qui m’a permis d’utiliser ses installations après le déménagement du Prof. Amzica. Le Prof. Martin Deschênes, qui a mis à ma disposition beaucoup de ressources matérielles et humaines. Le Prof. Deschênes est une source impérissable de bonnes idées. Je remercie également les Drs. Paul De Koninck et Daniel Côté à qui j’ai souvent demandé des conseils ou de l’aide. Je tiens également à remercier mes collègues étudiants et stagiaires post-doctoraux : Oana Chever, Daniel Kröger, Daniel Gingras, Alex Fraser, Yoan LeChasseur, Guillaume Lavertu, Cyril Bories, Sophie Laffray, Annie Castonguay, Simon Labrecque, Christian Tardif, Stéphane Pagès, Sophie Dufour-Beauséjour et Alexandre Juneau-Fecteau. Je suis également reconnaissante envers le personnel de laboratoire qui fait un travail extraordinaire : Maxime Demers, qui m’a beaucoup aidé, Marc D’auteuil, Hugues Dufour, Pierre Giguère, Sergiu Ftomov, Sylvain Côté, Karine Bachand, Karen Vandal, Jacqueline Turmel, Francine Nault et Marie-Ève Paquet.
Les chances qu’ils lisent cette thèse sont infinitésimales, mais un gros merci au pianiste Yiruma et à Jason Mraz, c’est tellement agréable de préparer des sondes optiques en leur compagnie! Maxime, je suis désolée que tu aies dû m’entendre chanter si souvent!
Je remercie finalement, ma famille, mes parents, Marie-France Cauchon et Marcel Dufour pour leur soutien, mon conjoint et physicien Pascal Dufour, qui doit « obligatoirement » écouter tout ce que j’ai à raconter sur mes expériences. Il le fait toujours avec intérêt et je l’apprécie beaucoup. Et pour finir, mes enfants, Anabelle, Félix-Olivier et Marianne qui ensoleillent mes journées.
Je dédis cette thèse à ma famille :
Pascal, Anabelle, Félix-Olivier et
Marianne mes rayons de soleil.
Abstract ... iii
Avant-Propos... v
Remerciements ... vii
Table des matières... ix
Liste des tableaux...xiii
Liste des figures... xiv
Chapitre 1 : Introduction ... 1
1.1 La lumière comme outil de détection in vivo ... 3
1.1.1 Interaction lumière-matière ... 3
1.1.2 Les différents outils fluorescents ... 6
1.1.3 Utilisation de la fluorescence in vivo ... 11
1.1.3 Spécificité ... 15
1.2 La lumière comme outil de contrôle... 16
1.2.1 Les outils optogénétiques pour contrôler l’activité neuronale... 16
1.2.2 Drogues encapsulées ... 19
1.3 Techniques de détections optiques en neurosciences ... 19
... 23
1.4 Problématique : limitations des outils de détection ... 28
1.5 Microsondes optiques et électriques ... 29
1.6 Concepts théoriques en optique ... 30
1.6.1 La fibre optique ... 30
1.6.2 Quelques concepts d’optique des tissus ... 36
1.7 Concepts théoriques : électrophysiologie in vivo ... 39
1.7.1 Enregistrement extracellulaire unitaire ... 42
1.7.2 Enregistrement intracellulaire ... 44
1.7.3 Flux ioniques ... 46
1.8 Différentes microsondes utilisées ... 52
1.8.1 Sonde avec fibre multimode étirée et électrode K+-sensitive... 54
1.8.2 Microsonde avec électrode intégrée (fibre à deux cœurs) ... 56
1.8.3 Fibre à 2 cœurs version électrode double ... 58
1.9 Division de la thèse ... 59
Chapitre 2 : In vivo simultaneous intra- and extracellular potassium recordings using a micro-optrode ... 61
Résumé : ... 61
Abstract : ... 62
2.1 Introduction ... 63
2.2. Materials and methods ... 64
2.2.1. Micro-optrode preparation... 64
2.2.2. Experimental optical setup (Fig. 2.2) ... 67
2.2.4 Animal preparation ... 68
2.2.5 Cell loading ... 68
2.2.6 Seizures and coma induction... 69
2.2.7 Histological analysis ... 69
2.2.8 Data recording and analysis... 70
2.3.1. Database and cellular identification... 71
2.3.2. Simultaneous extra- and intracellular potassium concentration recordings during normal, sleep-like patterns ... 73
2.3.3. Simultaneous extra- and intracellular K+ recordings during paroxysmal seizures ... 76
2.3.4. Simultaneous extra- and intracellular K+ recordings during coma ... 80
2.3.5. Recording stability over time ... 82
2.4. Discussion ... 82
2.4.1. The micro-optrode ... 82
2.4.2. Cell loading and correlation between intra- and extracellular K+ recordings ... 83
2.4.3. Technical improving possibilities... 85
2.4.4 Biological applications of the technique ... 85
2.5. Acknowledgements... 86
Chapitre 3 : A microprobe for parallel optical and electrical recordings from single neurons in vivo... 87 Résumé : ... 87 Abstract: ... 88 3.1 Introduction ... 89 3.2 Results ... 90 3.2.1 The microprobe ... 90
3.2.2 Optical resolution and single cell detection ... 92
3.2.3 Recording from single retrograde-labeled neurons in vivo ... 94
3.2.5 Recording from single eGFP-expressing GABAergic neurons... 96
3.2.7 Optogenetic activation of single cells... 100
3.3 Discussion ... 101
3.4 Methods ... 102
3.4.1Microprobe fabrication ... 102
3.4.2 Optical setup ... 103
3.4.3 Brain slice preparations ... 104
3.4.4 In vivo extracellular single unit recording ... 105
3.4.5 Single cell Ca2+ measurements... 106
3.4.6 Photostimulation in Thy1::ChR2-YFP transgenic mice ... 106
3.4.7 Histology ... 107
3.4.8 Simulations and data presentation ... 107
3.4.9 Satistical analysis ... 107
Acknowledgements ... 107
Author Contributions ... 108
Supplementary data ... 108
Chapitre 4 : A monostructure micro-optrode combining field and single unit recording, multispectral detection and photolabeling capabilities ... 118
Résumé : ... 118
Abstract : ... 119
4.1 Introduction ... 120
4.2 Methods: ... 121
4.2.1 Microprobe and optical setup ... 121
4.2.2 Quantification of light collection through the microprobe wall... 122
4.2.3 Animals ... 123
4.2.5 Cell cultures ... 124
4.2.6 Multicolor transgenic mice ... 124
4.2.7 Photoconversion... 125
4.2.8 Data analysis ... 125
4.3 Results ... 126
4.3.1 Light collection through the microprobe wall ... 126
4.3.2 Side collection ... 127
4.3.3 In vivo simultaneous field potential and single unit recordings ... 129
4.3.4 Multispectral detection ... 130
4.3.5 Photoconversion... 132
4.4 Discussion ... 133
Author contributions: ... 134
Acknowledgment: ... 134
Chapitre 5 : In vivo monitoring of membrane electrical properties using extracellular optical microprobes and optogenetic ... 135
Résumé : ... 135
Abstract : ... 136
5.1 Introduction ... 137
5.2 Results ... 138
5.2.1 Extracellular manipulation and quantification of membrane potential ... 138
5.2.2 Concordance between intracellular and extracellular recordings ... 140
5.2.3 Measuring conductance change induced by neurotransmitters... 145
5.2.4 Revealing differences in reversal potentials of synaptic events... 147
5.3 Discussion ... 148 5.4 Methods ... 149 5.4.1 Microprobe ... 149 5.4.2 Optical setup ... 150 5.4.3 Animal preparation ... 151 5.4.4 Extracellular recordings ... 151
5.4.5 Intra and extracellular recordings... 151
5.4.6 Iontophoresis ... 152
5.4.7 Whisker stimulation ... 152
5.4.8 Simulation... 152
Acknowledgment: ... 154
Chapitre 6: Conclusion ... 155
6.1 Résumé des résultats obtenus avec les microsondes... 155
6.2 Importance de ces résultats pour le domaine des neurosciences ... 160
6.2.1 Combinaison des enregistrements électriques et optiques ... 161
6.2.2 Enregistrement de neurones ciblés... 161
6.2.3 Étude des fluctuations ioniques intracellulaires ... 162
6.2.4 Un outil de contrôle spécifique ... 162
6.3 Comparaison entre les différentes méthodes utilisées... 163
6.4 Optimisation du rapport S/N... 164
6.5 Un outil multifonctionnel ... 165
6.6 L’avenir des microsondes ... 165
6.6.1 De nouvelles configurations pour les prochaines générations de microsondes ... 166
6.6.2 Développement de nouvelle sonde moléculaire et de nouvelles protéines fonctionnelles ... 170
6.7 Épilogue... 170 Bibliographie... 171
Liste des tableaux
Tableau 1.1: Principaux marqueurs calciques disponibles chez Invitrogen. Source [8]..9 Tableau 1.2 : Principaux outils optogénétiques. ...17 Tableau 1.3 : Coefficients d’absorption, de diffusion et facteur d’anisotropie pour le
cerveau pour des valeurs de longueurs d’onde autour de 500 nm [93]. ...38 Tableau 1.4 : Indices de réfraction de différents tissus...38 Tableau 1.5 : Concentration intra et extracellulaires des différentes espèces ioniques
pour des neurones de mammifères [94]. ...40 Tableau 1.6 : Comparaison entre les paramètres physiques des différents types
d’enregistrement in vivo et les bandes de fréquences généralement utilisées pour chacune d’entre elles [95]...41 Tableau 1.7 : Principaux canaux potassiques [103], [106], [107]. ...48 Tableau 1.8 : Principaux canaux calciques [106]. ...50 Tableau 1.9: Types de sondes qui ont été fabriquées dans le cadre des présents travaux de doctorat...54 Tableau 1.10 : Caractéristiques de la fibre à électrode intégrée fournie par l’Institut
national d’optique. ...57 Table 5.1: Optical fiber properties ...149
Liste des figures
Figure 1.1 : Diagramme de Jablonski ...5
Figure 1.2: Agents de contraste fluorescents. ...7
Figure 1.3: Fluorescence de solutions contenant différentes protéines fluorescentes [24]. ...10
Figure 1.4: Souris transgéniques. ...13
Figure 1.5: Schématisation de la structure générale d'un virus. ...14
Figure 1.6: Schématisation de l’introduction d’un gène à l’aide de la recombinaison CRE-LOX...15
Figure 1.7: Mécanismes d’action des canaux ioniques channelrhodopsin et des pompes ioniques halorhodopsin et archaerhodopsin...18
Figure 1.8: Type d’imagerie par fluorescence. ...20
Figure 1.9 : Microendoscopie. ...23
Figure 1.10 : Sondes spectroscopiques...24
Figure 1.11 : Optrodes pour optogénétique. ...27
Figure 1.12: Sondes spectroscopiques...28
Figure 1.13 : La fibre optique. ...30
Figure 1.14 : Modes LPlm. ...32
Figure 1.15 : Sortie d’une fibre multimode...33
Figure 1.16 : Forme du profil de sortie d’une fibre multimode...34
Figure 1.17 : Effet de l’étranglement. ...35
Figure 1.18 : Coefficients d’absorption de différents absorbants présents dans le tissu [92]. ...37
Figure 1.19 : Champ extracellulaire. ...40
Figure 1.20 : Différentes méthodes d’enregistrement extracellulaire et intracellulaire [95]. ...41
Figure 1.21 : Le potentiel d’action...43
Figure 1.22 : Différents courants observés dans un neurone d’hippocampe ainsi que leurs comportements [103]. ...47
Figure 1.23 : Différents composantes d’une électrode ion-sensitive. ...51
Figure 1.24 : Caractéristiques de l’indicateur potassique PBFI. ...55
Figure 1.25 : Schéma de la liaison entre l’électrode et la fibre optique multimodale étirée. ...56
Figure 1.26 : Fibre et Microsonde. ...57
Figure 2.1 : ...65
Figure 2.2 : Experimental setup...67
Figure 2.3: PBFI immunohistological analysis.. ...72
Figure 2.4 : Cortical intracellular and extracellular K+ variations during spontaneous sleep-like patterns. ...75
Figure 2.5 : Cortical intracellular and extracellular K+ variations during epileptic seizures induced by topic application of penicillin on the cortex.. ...77
Figure 2.6 : SW seizures induced in some cells decreases of the intracellular K+. ...79
Figure 2.7: Cortical intracellular and extracellular K+ variations during induction of deep coma with isoflurane. ...81
Figure 3.1 : The microprobe...91
Figure 3.3 Single-unit recordings from neurons in vivo.. ...95
Figure 3.4: Optical and electrical signal profiles of single cells in vivo. ...97
Figure 3.5 : Monitoring Ca2+ fluctuations confirm optical and electrical recording from the same neuron in vivo.. ...99
Figure 3.6 : Detection and activation of single ChR2-expressing neurons in vivo. ...101
Figure 3.7 : Photomicrographs of microprobe core and tips. ...108
Figure 3.8 : Parameters affecting microprobe optical resolution. ...109
Figure 3.9 : Electrical properties of the Microprobes. ...110
Figure 3.10 : Size distribution and detection of GFP-labeled reticular thalamic (RT) neurons in mice...110
Figure 3.11 : Schematic representation of a probe tip.. ...112
Figure 3.12 : Illumination model.. ...116
Figure 4.1: Optical and electrical microprobes. ...122
Figure 4.2 : Side acceptance of tapered waveguides...127
Figure 4.3 : Impact of fluorescence collection through the microprobe walls. ...128
Figure 4.4 : Coated glass microprobes enable dual electrical recordings. ...130
Figure 4.5 : Multispectral detection allows identifying dopaminergic cells in vitro and in vivo. ...131
Figure 4.6: Photoconversion of mEOS. ...132
Figure 5.1: Expression characterization...138
Figure 5.2: Cell control and recording with a microprobe.. ...139
Figure 5.3: Concordance between intra and extra-cellular in vivo recorded field.. ...142
Figure 5.4: Calcium spikes. ...144
Figure 5.5: Revealing membrane changes and intrinsic properties. ...146
Figure 5.6: Subliminal pulses during AHP. ...148
Figure 6.1 : Tétrodes. ...167
Figure 6.2 : Fibre optique à cœur métallique [205]. ...168
Figure 6.3: Micrographie électronique d’une coupe transversale d’une fibre optoélectronique composée d’un cœur creux entourée d’un miroir composé de couches successives de As2Se3 et de polyetherimide, d’un microfilment de Sn (sections pâles) et d’une gaine de polymère [208]. ...169
Figure 6.4 : a) Micrographie d’une coupe d’une sonde optique. Plusieurs segments ont été coupés. b) Le degré de liberté de l’appareil permet de façonner la pointe de la sonde. ...169
Le domaine des neurosciences a connu au cours des dernières décennies un essor remarquable. Cet essor est directement relié à l’avancement de différentes disciplines scientifiques et parmi celles-ci on compte la photonique. Le microscope optique avait déjà permis de faire plusieurs observations au niveau cellulaire et même sous-cellulaire de l’organisation du système nerveux, mais avec l’arrivée des sources lasers à impulsions ultra-brèves (permettant, entre autre, le développement de la microscopie confocale [1] et multi-photonique [2]), les techniques d’imagerie et de détection se sont raffinées et succédées à une vitesse incroyable. Il est maintenant possible grâce à la résolution microscopique de ces techniques d’observer en temps réel le fonctionnement d’une cellule nerveuse du point de vue moléculaire. Un des avantages des techniques optiques, comparativement à d’autres techniques de microscopie comme la microscopie électronique, est qu’elle ne requiert pas la déshydratation des tissus qui peuvent ainsi être maintenus vivants.
De concert avec l’avancement de la photonique, ou plus précisément de la biophotonique, l’avancement en génétique et en chimie a permis de développer plusieurs outils qui aident à augmenter, par différentes techniques, le contraste optique des cellules ou d’une sous-section de celles-ci, et cette augmentation peut même être spécifique et ne cibler qu’un seul sous type, ou sous-section, cellulaire. Mais comme nous le verrons, les techniques de détection optique développées jusqu’ici comportent certaines limitations en raison des propriétés de la lumière et des tissus vivants et elles sont limitées à la surface tissulaire. Ces outils sont tout-à-fait indiqués pour l’expérimentation in vitro, mais lorsqu’il est nécessaire d’étudier le système nerveux dans son ensemble chez l’animal vivant leurs limitations peuvent parfois être gênantes.
Les expérimentateurs peuvent se tourner vers les guides d’ondes optiques, incluant les fibres optiques, pour l’expérimentation in vivo en profondeur. Les guides d’ondes peuvent être utilisés pour l’imagerie [3] ou encore comme simples sondes [4–6]. Il s’agit d’un excellent moyen de guider la lumière à travers le tissu jusque dans une
région d’intérêt et d’en retirer l’information optique voulue. Dans cette thèse, nous présentons une nouvelle manière, encore peu populaire mais prometteuse, d’utiliser les fibres optiques en neurosciences.
Les objectifs poursuivis par ces travaux de doctorat sont de fournir des outils optiques pour la détection de fluorescence et l’illumination in vivo qui pallient aux limitations des techniques couramment utilisées et de démontrer l’utilité et la multifonctionnalité de ceux-ci. Ces outils ont été développés pour répondre aux objectifs suivants:
i) être très fins pour permettre une résolution qui atteint le niveau cellulaire en limitant les dommages aux tissus et d’accéder à des régions profondes.
ii) permettre l’excitation lumineuse et la collection de fluorescence via le même dispositif afin d’être utilisables en profondeur dans les tissus in vivo.
iii) combiner l’enregistrement optique aux méthodes d’enregistrements électrophysiologiques conventionnelles pour permettre de mesurer les fluctuations de fluorescence ou d’identifier les cellules par fluorescence tout en mesurant leur activité. Ceci permet de bénéficier à la fois de la spécificité des outils optiques et de la stabilité et la résolution temporelle des mesures électriques extracellulaires.
En gardant ces objectifs en tête, nous avons développé différents types de microsondes optiques. Elles sont fabriquées à partir de fibres optiques étirées. La même sonde est utilisée dans le but de guider la lumière d’excitation jusqu’aux tissus fluorescents et de collecter la fluorescence qui en émane. Plusieurs méthodes ont été utilisées pour coupler ces sondes optiques aux enregistrements électrophysiologiques conventionnels et rendre ces sondes multifonctionnelles.
Le présent chapitre couvre quelques éléments théoriques et travaux préliminaires et a pour but d’introduire les publications incluses dans les chapitres successifs et de faciliter leur compréhension. La première partie décrit les principaux agents de contraste qui utilisent le phénomène de la fluorescence ainsi que les méthodes utilisées pour les introduire dans les organismes vivants. La seconde partie de ce chapitre fait la description des techniques de détection de fluorescence in vivo les plus
populaires. Les systèmes de microscopie, de microendoscopie et les sondes optiques y seront abordés. Cette section mettra en évidence les limitations de ces techniques, notamment en ce qui à trait à l’expérimentation in vivo. Ceci permettra de bien mettre en évidence l’importance des microsondes optiques dans le domaine des neurosciences, le sujet principal de cette thèse. Finalement, nous y présentons quelques aspects de théorie relatifs à la fibre optique, l’optique des tissus et les mesures électrophysiologiques.
1.1 La lumière comme outil de détection in vivo
Lorsqu’on veut examiner un objet, on se doit de le faire avec les bons outils et choisir notre sonde en fonction de la résolution que l’on veut obtenir. On ne pourrait dire si un grain de sable est rugueux simplement en le touchant du doigt, mais ce constat est possible pour une pierre. Similairement, si on veut pouvoir tirer des conclusions sur la morphologie ou l’état d’un objet microscopique on doit choisir une sonde de taille microscopique ou sous-microscopique (quelques centaines ou dizaines de nanomètres). C’est un problème auquel les biologistes sont confrontés quotidiennement et les photons du spectre visible sont tout indiqués pour sonder des objets microscopiques puisque la résolution théorique qu’ils offrent est de l’ordre de quelques centaines de nanomètres (nm). De plus, puisque les biologistes s’intéressent souvent à des systèmes dynamiques et non statiques, ils doivent disposer d’une sonde avec une excellente résolution temporelle qui leur permet de suivre l’évolution de ces systèmes. Encore une fois la lumière est tout indiquée pour ce type d’applications puisqu’elle voyage rapidement (près de 3 ∙ 108 m/s dans l’air et 2 ∙ 108 m/s dans le tissu vivant).
Cependant, il faut noter que la faible efficacité de recollection de certaines configurations de détection optique fait en sorte que les signaux optiques doivent être intégrés sur de plus longues périodes de temps causant un compromis au niveau de la résolution temporelle. La section qui suit fait un bref rappel de quelques notions concernant l’interaction de la lumière avec la matière.
1.1.1 Interaction lumière-matière
Dans une section subséquente de ce même chapitre, l’interaction de la lumière avec les tissus vivants fera l’objet d’une brève revue, mais auparavant, l’interaction de la lumière avec la matière au sens large sera brièvement décrite. Lorsqu’un photon entre
en collision avec la matière, il sera soit diffusé (de manière élastique on non) soit absorbé par l’atome où la molécule avec lequel il entre en collision.
1.1.1.1 Diffusion
Lorsqu’un photon est dévié suite à l’interaction de celui-ci avec une particule se trouvant sur son chemin, on parle de diffusion. Dans le cas où la diffusion est élastique, c’est-à-dire, dans le cas ou l’énergie du photon est conservée, on parle surtout de diffusion de Rayleigh ou de Mie. Lorsque la diffusion est inélastique on parle alors de diffusion Raman ou encore de Brillouin. Dans la diffusion Raman la différence d'énergie entre le photon absorbé et le photon réémis est égale à la différence d'énergie entre deux états de rotation ou de vibration de l'objet diffusant (phonon optique). Ce type de diffusion estlargement utilisé en spectroscopie pour identifier des molécules qui ont un spectre Raman connu. La diffusion Brillouin est le résultat de l’interaction d’un photon avec les ondes acoustiques (phonons acoustiques) qui se propagent dans un milieu. La perte d’énergie du photon dans ce cas est minime comparativement à ce qu’on observe dans la diffusion Raman. Pour les liquides, le décalage correspondant en longueur d’onde se traduit par quelques picomètres dans la région visible du spectre (400-800 nm).
La diffusion donne naissance à trois phénomènes :
- La réflexion : une diffusion dans la direction opposée à celle d’incidence.
- La réfraction : en partie due à la diffusion dans le sens de propagation et qui a pour effet de réduire la vitesse de l’onde-électromagnétique dans un milieu par rapport à la vitesse qu’elle aurait dans le vide.
- La diffraction : une superposition d’onde diffusée qui, lorsque le milieu est bien organisé, génère des patrons de diffraction dont on peut prédire la périodicité.
1.1.1.2 Absorption
Dans le cas ou le photon est absorbé, l’énergie de ce dernier sera transférée à un électron qui changera d’état. Celui-ci passera à un état d’énergie supérieur (excité). Éventuellement, l’électron excité retournera à son état initial (dit fondamental). Se faisant, un photon dont l’énergie correspond à la transition entre les deux états
d’énergie (excité et fondamental) sera émis. Le retour à l'état fondamental peut alors se faire de différentes manières : par relaxation non-radiative (sans émission de photon), par fluorescence ou par phosphorescence. Pour la phosphorescence, l’électron met beaucoup de temps (jusqu’à des heures) à revenir à l’état fondamental car il doit passer par un état interdit illustré à la figure 1.1.
Figure 1.1 : Diagramme de Jablonski illustrant les transitions électroniques donnant lieux aux phénomènes de fluorescence et de phosphorescence (version adaptée de [7]).
Par contre, pour la fluorescence l’état excité a un temps de vie très court; souvent de l’ordre de quelques nanosecondes. Avant de retomber à l’état fondamental l’électron subit des pertes d’énergie non-radiatives, par conséquent, le photon émis a une énergie légèrement inférieure à celle du photon incident. La différence d’énergie entre le photon absorbé et celui émis est appelé le décalage de Stokes. La figure 1.1 montre un diagramme de Jablonski illustrant les transitions électroniques donnant lieu aux phénomènes de fluorescence et de phosphorescence.
0
1
2
5
0
1
2
5
0
1
2
5
S
1S
2S
0 États d’énergie vibrationnelsT
1État Triple excité
0 1 2 5 Absorption (10 -15 s) Fluorescence (10-9 – 10-7 s) Conversion interne et relaxation vibrationnelle (10-14 – 10-11 s) Relaxation non-radiative Fluorescence tardive Échange de système État fondamental États excités
Les molécules fluorescentes sont des agents de contraste couramment utilisés en imagerie microscopique in vitro et in vivo, et ils sont utilisés dans le cadre de cette thèse. L’absorption d’un agent fluorescent va varier selon la longueur d’onde. Il est possible de mesurer le spectre d’absorption d’un tel agent et ce spectre nous renseigne sur les niveaux d’énergie de la molécule. De même, il est possible d e mesurer le spectre d’émission d’un agent fluorescent avec un spectromètre suite à l’excitation de celui-ci. Connaissant ces spectres, on peut établir le décalage de Stokes. En microscopie, il est souvent recommandé d’optimiser le décalage de Stokes afin que la fluorescence soit facilement séparée de la lumière d’excitation, ce décalage est donc une propriété importante lorsqu’on choisit un fluorophore. La section efficace d’un fluorophore est régie par la probabilité que ce dernier absorbe un photon incident (d’énergie et de polarisation donnée) et l’efficacité quantique, ɸ, quantifie l’efficacité du processus de fluorescence (équation 1.1).
ɸ
(1.1)
Lorsque l’intensité du signal d’excitation est très grande, il peut y avoir des phénomènes d’absorption à 2 photons ou plus. Dans le cas de l’absorption à 2-photons, deux photons (ayant chacun une énergie équivalente à la moitié de la transition d’énergie vers le niveau excité) seront simultanément absorbés. Il s’agit d’un phénomène non linéaire et la probabilité d’émission de fluorescence est alors proportionnelle à l’intensité du signal d’excitation élevé e à la puissance 2. L’absorption multiphotonique dépasse le cadre de cette thèse et n’est donc pas abordée davantage.
1.1.2 Les différents outils fluorescents
Bien que différentes techniques optiques soient fréquemment utilisées en biophotonique nous nous contenterons ici de présenter celles ayant rapport à la fluorescence puisque ce sont celles-ci qui seront utilisées avec les sondes optiques présentées dans cette thèse. Le terme agent de contraste est surtout utilisé en imagerie par résonnance magnétique, mais il s’applique tout aussi bien à l’imagerie par fluorescence. L’agent de contraste est inséré dans les cellules, ou encore, dans des sections de celles-ci pour augmenter le contraste entre ces cellules (ou
sous-sections) et leur environnement. La figure 1.2 montre un exemple d’une cellule ou trois agents avec des spectres d’émission et d’excitation différents ont été utilisés pour marquer trois sous-sections cellulaires différentes (mitochondries, tubules et noyau). Elle témoigne bien de la spécificité et de la résolution des techniques de détection optiques utilisant la fluorescence.
Figure 1.2: Agents de contraste fluorescents. Cellule endotheliale bovine dont les tubules ont été marqués en vert (Alexa Fluor 488), les mitochondries ont été marquées en rouge (Alexa Fluor 555) et le noyau a été marqué en bleu (DAPI) [8].
Pour qu’un agent puisse être utilisé in vivo, il doit pouvoir être introduit dans les cellules ou encore produit par celles-ci, il doit y rester et demeurer fonctionnel pour la durée de l’expérience qui peut durer de quelques dizaines de minutes à des semaines. Celui-ci se doit également d’être le moins toxique possible. Certains agents fluorescents, ou encore, les produits nécessaires à leur dissolution ou leur insertion dans le tissu peuvent avoir des effets toxiques sur la cellule. Par exemple, la solution utilisée pour marquer les cellules in vivo avec les marqueurs de type AM (abordés à la section 1.1.3.2) contient des agents connus pour avoir des effets sur la membrane cellulaire, soient le dimethylsulfoxide et l’acide pluronique [9], [10]. De plus, l’illumination des fluorophores peut entraîner la production de radicaux libres, des substances qui sont à la base d’une chaîne de réaction chimique menant à la photo-toxicité [11], [12]. Le spectre d’excitation d’un agent fluorescent est aussi un enjeu majeur dont il faut tenir compte pour l’expérimentation in vivo, par exemple, la lumière UV est connue pour provoquer davantage de dommages cellulaires comme la production de radicaux libres et la dégradation de l’ADN [13–15]. Plusieurs types d’agents fluorescents peuvent être utilisés in vivo et les sections qui suivent en
présentent quelques-uns dont les molécules organiques fluorescentes incluant les protéines fluorescentes et les molécules non-organiques.
1.1.2.1 Molécules organiques fluorescentes
Il existe plusieurs familles de molécules organiques fluorescentes, les rhodamines en étant un exemple. Cette famille compte entre autres la rhodamine 6G, la rhodamine B ou encore la rhodamine 123. Plusieurs dérivés de ces composés chimiques sont disponibles, comme la sulforhodamine 101 souvent utilisée pour marquer les astrocytes in vivo, le Texas red, souvent utilisé pour cibler les protéines, les Alexa Fluor ou les DyLight Fluor. Les dérivés de cette famille sont en général non-toxiques et l’ensemble de leurs spectres d’émission et d’absorption couvrent la totalité du spectre visible [16]. Les chaînes carbocyanines (DiI, DiO, DiD et DiR) représentent un autre exemple de famille de molécules organiques fluorescentes. Ce sont des marqueurs lipophiles utilisés pour marquer les membranes ou autres structures hydrophobes). Les molécules fluorescentes présentées ci-dessus peuvent être utilisées seules ou encore être encapsulées dans des microsphères dont le diamètre peut varier de plusieurs micromètres à 0,04 µm.
i) Caractères fonctionnels
Dans plusieurs cas, les molécules fluorescentes sont souvent des marqueurs morphologiques (ou anatomiques) et leur fluorescence et location dans la cellule demeurent stables au cours de la période d’enregistrement. Ce sont des colorants qui servent à caractériser de manière anatomique le tissu et ses composantes. Cependant, plusieurs molécules fluorescentes ont des modalités fonctionnelles, c’est-à-dire que leur émission va dépendre de la présence ou de la concentration d’un autre agent (qui peut être une molécule, une composante cellulaire, ou bien un ion en solution) ou encore d’une caractéristique physique susceptible de changer telle la polarité ou bien la température. Ainsi, en étudiant l’évolution temporelle de la fluorescence, on peut en retirer l’information sur la présence de l’agent ciblé et son évolution temporelle. Les marqueurs fonctionnels les plus communs sont les marqueurs calciques. Il existe plusieurs familles de marqueurs calciques avec des spectres d’émission qui peuvent être aussi bien dans la région bleue du spectre optique que dans la région rouge. Le calcium est un important second messager qui prend part à la signalisation
intracellulaire, il n’est donc pas étonnant qu’il y ait autant de marqueurs calciques fluorescents. Le tableau qui suit fait la liste des marqueurs calciques disponibles chez Invitrogen, l’un des principaux fournisseurs.
Marqueur calcique Mode § Kd (nM) **
Bis-fura Excitation (Ex) 340/380 370
BTC Ex 400/480 7000
Calcium Green-1 Émission (Em) 530 190
Calcium Green-2 Em 535 550 Calcium Green-5N Em 530 14,000 Calcium Orange Em 575 185 Calcium Crimson Em 615 185 Fluo-3 Em 525 390 Fluo-4 Em 520 345 Fluo-5F Em 520 2300 Fluo-4FF Em 520 9700 Fluo-5N Em 520 90,000 Fura-2 Ex 340/380 145 Fura-4F Ex 340/380 770 Fura-6F Ex 340/380 5300 Fura-FF Ex 340/380 5500 Fura Red Ex 420/480 140 Indo-1 Em 405/485 230 Mag-fluo-4 Em 520 22,000 Mag-fura-2 Ex 340/380 25,000 Mag-indo-1 Em 405/485 35,000 Magnesium Green Em 530 6000
Oregon Green 488 APTA-1 Em 520 170
Oregon Green 488 BAPTA-2 Em 520 580
Oregon Green 488 BAPTA-6F Em 520 3000
Oregon Green 488 BAPTA-5N Em 520 20,000
Quin-2 Em 495 60 Rhod-2 Em 580 570 Rhod-3 Em 580 570 Rhod-FF Em 580 19,000 Rhod-5N Em 580 320,000 X-rhod-1 Em 600 700 X-rhod-5F Em 600 1600 § Mesures en nm.
** Constante de dissociation du Ca2+ mesurée in vitro à 22°C dans100 mM KCl, 10 mM
MOPS et pH 7.2.
Tableau 1.1: Principaux marqueurs calciques disponibles chez Invitrogen. Source [8].
D’autres indicateurs sont également disponibles comme les indicateurs chloriques, sodiques, potassiques et de pH [17–20].
ii) Protéines fluorescentes
Parmi les molécules organiques fluorescentes, on peut inclure les protéines fluorescentes. Elles sont empruntées de la nature et souvent améliorées pour mieux répondre aux besoins des chercheurs. La plus populaire des ces protéines est la GFP (Green Fluorescent Protein) qui provient de la méduse Aequorea victoria. La découverte de la GFP a eu un impact si important pour les sciences biomédicales qu’elle a valu aux chercheurs Osamu Shimomura, Martin Chalfie et Roger Y. Tsien le prix Nobel de chimie en 2008. Maintenant, plusieurs dérivés de la GFP et d’autres protéines fluorescentes sont couramment utilisés et ils couvrent tout le spectre visible[21], [22]. La figure 1.3 en témoigne. Le groupe de Roger Y. Tsien a publié en 2005 un guide pour faciliter la sélection des protéines fluorescentes [23].
Figure 1.3: Fluorescence de solutions contenant différentes protéines fluorescentes [24].
Les protéines fluorescentes ont l’avantage de pouvoir être fabriquées par la cellule elle-même, du moment que celles-ci en possède la recette (séquence d’ADN, appelée gène, qui code pour une protéine). Un organisme peut être génétiquement modifié pour inclure dans son ADN un gène fluorescent. Notez que les protéines fluorescentes peuvent également avoir une modalité fonctionnelle. Elles peuvent, par exemple, être utilisées pour étudier l’interaction entre 2 protéines grâce à la méthode du FRET (Förster Resonance Energy Transfer) [25], ou encore pour mesurer une concentration ionique. La construction GCaMP, par exemple, change de conformation en présence de calcium et est donc un indicateur calcique qui peut être introduit génétiquement [26]. Ces protéines, bien qu’elles soient plus grosses que les molécules organiques fluorescentes vues précédemment, ont beaucoup d’avantages sur ces dernières et le plus important est que l’animal peut être programmé génétiquement pour les produire. Cela évite l’introduction dans le tissu d’un agent exogène qui doit parfois
être administré plusieurs jours, voir semaines, avant l’expérimentation. L’animal doit donc subir une chirurgie préparatoire qui peut parfois être très lourde (ex : laminectomie). Un autre avantage certain est le gain en spécificité obtenu en associant la séquence du gène de la protéine fluorescente à un gène spécifique qui ne s’exprime que dans la cellule d’intérêt.
1.1.2.3 Particules non-organiques
Il existe également des particules fluorescentes non-organiques. Les points quantiques (quantum dots) en sont un exemple. Ils présentent certains avantages par rapport aux molécules organiques. Ceux-ci sont plus stables (pas de photoblanchiment), ont un spectre d’absorption plus large et un spectre d’émission facilement adaptable (ce dernier varie avec les tailles des points quantiques). Le principal inconvénient de ce type de particules est leur toxicité et leur faible solubilité dans l’eau. Ces problèmes ne sont cependant pas sans solutions, par exemple les points quantiques peuvent être entourés de micelles, une couche mono-lipidique qui les rend solubles, non-toxiques et adéquats pour la fonctionnalisation à l’aide d’anticorps [27]. D’autres nanoparticules telles que les «nanorods » ont un avenir prometteur en biologie, bien que leur biocompatibilité soit toujours un enjeu, leur taille nanométrique et leur stabilité en font de bons candidats [28].
1.1.3 Utilisation de la fluorescence in vivo
Avoir une panoplie de particules fluorescentes est peut-être commode pour l’imagerie et la physiologie in vivo, mais encore faut-il les introduire dans les cellules. Il est important de garder la cellule dans un état sain pour que les résultats obtenus à partir de celle-ci soient physiologiquement pertinents. La section qui suit trace un court résumé des méthodes utilisées pour introduire les molécules fluorescentes dans les cellules vivantes. Elles peuvent être introduites par des méthodes mécaniques, chimiques, électriques ou encore génétiques.
1.1.3.1 Méthodes mécaniques
Parfois une simple injection mécanique à l’aide d’une micro-seringue ou un système de micro-injection automatisé est nécessaire pour marquer les cellules. C’est le cas lorsqu’on utilise des marqueurs membranaires, qui une fois dans le tissu, vont s’introduire et diffuser à l’intérieur de la membrane cellulaire [29], [30].
1.1.3.2 Méthodes chimiques
Pour le marquage in vivo, il est souvent très utile de rendre les molécules non-polaires de sorte qu’elles puissent passer librement à travers les membranes cellulaires. C’est dans ce but que des groupements carboxyl, comme les esters acetoxymethyl (AM) sont souvent ajoutés aux molécules fluorescentes. Une fois dans le milieu intracellulaire les enzymes estérases clivent les groupements AM et les molécules redeviennent polaires et imperméables pour les membranes. On peut colorer un tissu en injectant de petites quantités de solution contenant le fluorophore AM à l’aide d’un système de micro-injection [31], [32] ou encore, pour les tissus superficiels on peut faire une application topique de la solution [33]. Cette méthode permet de cibler un grand nombre de cellules, et peut parfois être spécifique à certains types cellulaires. Par exemple, in vivo, le SR101 et le fluo4, semblent ne marquer que les astrocytes [33]. Pour obtenir de meilleurs résultats avec cette méthode il est important de laisser le temps aux molécules de diffuser à travers la membrane et d’avoir subi la dé-estérification. Le temps d’attente (généralement fourni par le fournisseur) peut varier entre quelques dizaines de minutes et plusieurs heures selon les molécules utilisées.
1.1.3.3 Méthodes électriques
On peut également perméabiliser la membrane cellulaire en appliquant transitoirement un champ électrique à l’aide d’électrodes. Cette technique, appelée électroporation, est utilisée pour introduire dans le milieu intracellulaire des drogues, des bouts d’ADN et aussi des marqueurs fluorescents. Les protocoles d’électroporation varient et peuvent être utilisés pour marquer une seule cellule [34], ou encore, plusieurs [35].
Lorsqu’on utilise une électrode d’enregistrement intracellulaire, on peut ajouter à la solution d’enregistrement un fluorophore qui peut diffuser dans la cellule et ainsi la marquer. Cette méthode est souvent couplée à une méthode électrique que l’on appelle iontophorèse. Elle consiste à injecter, via de courtes impulsions de courant, le fluorophore [36]. Cette dernière méthode est très spécifique puisque seulement la cellule enregistrée est marquée, mais elle est peu efficace puisqu’elle ne permet pas de cibler un grand nombre de cellules [37]. Notez qu’un procédé similaire est aussi possible avec un enregistrement du type juxta-cellulaire [38].
1.1.3.4 Méthodes génétiques
Comme mentionné précédemment, chaque gène est un morceau d’ADN qui est une recette pour synthétiser une protéine donnée. La cellule est équipée pour décoder cette recette et assembler les acides aminés qui composeront la protéine. Plusieurs séquences d’ADN codant pour des protéines fluorescentes ont été isolées dans les dernières années, mais pour qu’il y ait traduction de ces protéines dans une cellule, elle doit en posséder la recette (sous forme de molécule d’ADN ou encore d’ARN). La section suivante résume les principales méthodes pour introduire un gène dans une ou plusieurs cellules d’un organisme.
i) Animaux transgéniques
Plusieurs espèces d’animaux transgéniques peuvent maintenant être produites, mais les animaux transgéniques utilisés dans le cadre de cette thèse sont exclusivement des souris. Sans entrer dans les détails, il est possible d’introduire un gène exogène dans l’ADN d’un organisme de sorte que celui-ci exprime une protéine particulière. Le nouveau bagage génétique sera transmis aux descendants de l’organisme. Il existe plusieurs protocoles pour produire une lignée de souris transgéniques [39]. La figure suivante montre un exemple d’une souris transgénique exprimant la protéine fluorescente DsRed.T3.
Figure 1.4: Souris transgéniques. a) Image en fluorescence d’une souris exprimant la protéine fluorescente DsRed.T3 sous le promoteur « CMV enhancer/chicken actin promoter » (gauche) et d’une souris contrôle (droite). b) Photographie standard des mêmes souris [40].
ii) Vecteurs viraux
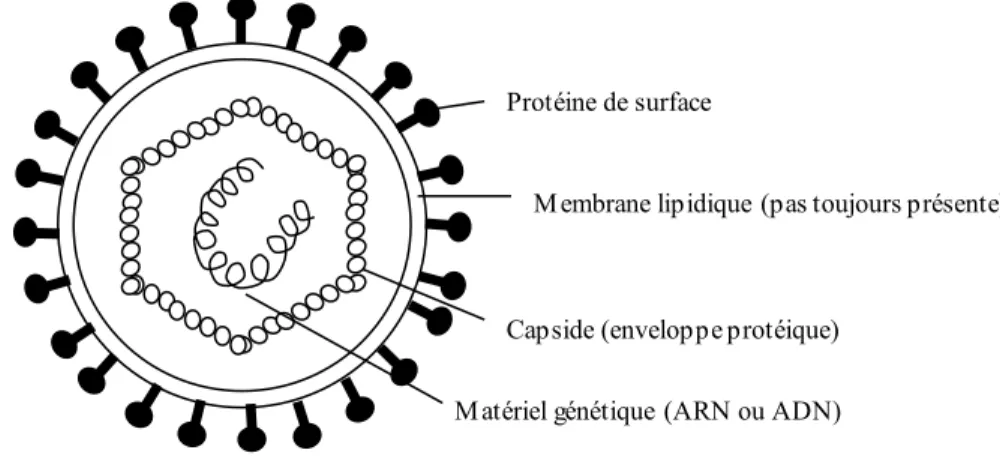
La figure 1.5 montre les constituants de base des virus. Ceux-ci transportent à l’intérieur de leur capside une molécule d’ADN (ou ARN). Normalement, cette
molécule d’ADN contient les gènes qui produisent les protéines nécessaires à la reproduction du virus.
Figure 1.5: Schématisation de la structure générale d'un virus.
Dans la plupart des cas, lorsqu’ils infectent une cellule, les virus se répliquent et peuvent ainsi infecter d’autres cellules. Notez qu’en neurosciences, on a généralement recours à des virus non-réplicatifs. Les virus peuvent être utilisés comme vecteurs, une sorte de cheval de Troie, qui peuvent pénétrer une cellule et y livrer le matériel génétique de notre choix. En choisissant bien le type de virus qu’on utilise et le matériel génétique qu’on y introduit, on peut obtenir un marquage fluorescent très spécifique, ciblant seulement un type de cellule et/ou une composante de celle-ci. Notez que les virus ne peuvent infecter toutes les cellules, seulement celles pouvant être reconnues par leurs protéines de surface et que cette propriété peut parfois servir de technique de ciblage.
Il existe d’autres stratégies très efficaces pour introduire un gène dans une population ciblée de manière très spécifique, notamment celles utilisant le système de recombinaison CRE-LOX. CRE est une enzyme qui catalyse la recombinaison entre deux sites de type LOXP. Ce système est largement utilisé pour produire des lignées transgéniques avec déplétion ou introduction d’un certain gène à un endroit précis [41]. Il existe maintenant plusieurs lignées de souris CRE disponibles commercialement. En utilisant un vecteur conçu pour insérer un gène CRE-dépendant (incluant une cassette de type LOXP-flanked-STOP ou LOXP-flanked-INVERSE),
Matériel génétique (ARN ou ADN) Protéine de surface
Membrane lipidique (pas toujours présente)
seulement les cellules CRE-positives vont activer l’expression du gène [42]. La figure suivante montre un exemple d’un tel système.
Figure 1.6: Schématisation de l’introduction d’un gène à l’aide de la recombinaison CRE-LOX.
iii) Électroporation
Tout comme pour les molécules fluorescentes, le bagage génétique peut être électroporé à l’intérieur de neurones in vivo [34], [43].
1.1.3 Spécificité
L’avantage des marqueurs fluorescents pour l’expérimentation in vivo est la spécificité qu’ils procurent. Lorsqu’il veut connaître le rôle précis d’une population cellulaire donnée dans une pathologie ou encore dans un état physiologique quelconque, l’électrophysiologiste dispose de peu de moyens pour identifier précisément le type de cellule qu’il enregistre. Il peut se référer à son patron de décharge et aux propriétés électrophysiologiques de la cellule, mais ces propriétés ne permettent que de différencier quelques grands sous-types cellulaires. Il peut marquer une cellule in vivo, préserver le tissu et confirmer le type de cellule enregistrée par sa morphologie, ses sites de connections ou son phénotype (e.g. les récepteurs et type de neurotransmetteur qu’elle exprime) sous microscope après le sacrifice de l’animal. Mais cela demande beaucoup de temps et empêche l’expérimentation sur un très grand nombre de cellules. En effet, pour arriver à identifier une cellule enregistrée in vivo, l’expérimentateur doit utiliser un marqueur intracellulaire lors de ses enregistrements et pour éviter la confusion, il doit souvent limiter ses enregistrements à une ou très peu de cellule(s) chez le sujet. Il doit ensuite récupérer le tissu cérébral,
le fixer, le sectionner et effectuer des analyses immunohistologiques. De plus, lorsque la population ciblée ne représente qu’un faible pourcentage de la population totale le taux de récolte est extrêmement faible. Les outils optiques procurent la spécificité dont l’électrophysiologiste a besoin. On peut, par exemple, trouver une protéine spécifique aux cellules qui nous intéressent et y associer une protéine fluorescente. Ainsi, seules les cellules ciblées exprimeront la protéine fluorescente et on peut restreindre les enregistrements à ces dernières, voir leur fonction dans certains mécanismes pathologiques, ou bien, vérifier leur réponse face à une drogue ou un traitement. Il est aussi possible d’injecter un marqueur rétrograde (qui remonte le long de l’axone pour colorer le corps cellulaire) et ainsi pouvoir différencier les cellules qui projettent à un site précis de celles qui n’y projettent pas.
De plus, les marqueurs fluorescents peuvent être utilisés comme sondes et fournir une spécificité fonctionnelle, comme dans le cas des marqueurs calciques. En utilisant les techniques optiques, l’électrophysiologiste peut joindre à ses enregistrements électriques conventionnels l’enregistrement des flux ioniques.
1.2 La lumière comme outil de contrôle
1.2.1 Les outils optogénétiques pour contrôler l’activité
neuronale
Jusqu’ici nous avons vu que la lumière peut être utilisée pour sonder le vivant, mais elle peut aussi être utilisée pour le contrôler. Les outils optogénétiques peuvent être des canaux membranaires photoactivables similaires à ceux que l’on retrouve dans les cellules de la rétine (cônes et bâtonnets) tout au fond de la cavité oculaire ou bien des pompes ioniques modifiées de sorte que leur activité puisse être induite ou inhibée par un stimulus lumineux. Contrôler l’activité d’une population bien définie de neurones, sans activer les autres cellules nerveuses adjacentes dans le tissu était auparavant difficile, voire impossible. Une électrode de stimulation électrique produit un champ qui a un effet sur toutes les cellules indépendamment de leur type. De plus, elles produisent des artéfacts de stimulations qui peuvent souvent gêner l’analyse des signaux enregistrés. Plusieurs stratégies ont été mises de l’avant pour contrôler l’activité électrique des neurones à l’aide de stimuli lumineux. En 2004, le groupe de
Kramer a mis au point un canal potassique pouvant être ouvert avec un stimulus lumineux à 380 nm et pouvant être fermé avec un deuxième signal lumineux à 500 nm [44]. Peu de temps après, en 2005, le groupe de Trauner a développé un récepteur de glutamate (un important neurotransmetteur excitateur) sensible à une stimulation lumineuse [45]. Finalement, plusieurs chercheurs dont Karl Deisseroth, Gero Meisenboch et Georg Nagel ont proposé la modification et l’introduction de la protéine Channelrhodopsin-2 (CHR2) dans la membrane neuronale, un canal perméable aux cations provenant d’une algue, afin de dépolariser les neurones et les activer [46]. Tout l’appareillage nécessaire à l’activation de la cellule est contenu dans une seule protéine (nécessitant un seul gène) rendant cette approche simple et universelle. De plus, la précision temporelle de CHR2 rapportée par le groupe de Deisseroth, offre la possibilité de contrôler la génération de potentiel d’action de manière fiable même à haute fréquence (100 Hz). Cette découverte a été le catalyseur d’une série de publications qui a donné un essor au domaine de l’optogénétique. Notez que le terme optogénétique fait souvent référence aux outils génétiques utilisant la lumière pour contrôler l’activité neuronale mais, selon certaines définitions [47], ce domaine inclut également les outils génétiques utilisant la lumière pour sonder cette activité.
Aujourd’hui, plusieurs versions de canaux ou pompes photosensibles permettant l’activation ou l’inhibition d’une cellule sont disponibles ou en voie de l’être. Le tableau 1.2 fait la liste des principaux outils de contrôle optique développés dans les dernières années.
Protéine Effet Références
Channelrhodopsin (CHR2) activation [46]
Red-shifted channelrhodopsin (VChR1) activation [48]
Halorhodopsin (NpHR) inhibition [49]
Enhanced halorhodopsin (eNpHR3.0) inhibition [50]
ArCH inhibition [51], [52]
ChETA activation [53]
OptoXRs Contrôle de la signalisation
intracellulaire (Protéine G)
[54]
Step Function Opsins (SFOs) activation [55]
ChEF activation [56]
ChiEF activation [56]
CatCh activation [57]
Chacun de ces outils étant des protéines connues, il est possible de les introduire par des méthodes génétiques tout comme les protéines fluorescentes. L’action de tous les outils présentés ci-haut nécessite un stimulus lumineux. Le mécanisme d’action de quelques-uns de ces outils est représenté à la figure suivante.
Figure 1.7: Mécanismes d’action des canaux ioniques channelrhodopsin et des pompes ioniques halorhodopsin et archaerhodopsin.
Les dépolarisations ou hyperpolarisations cellulaires peuvent être entrainées par différents mécanismes et il est important de tenir en compte du potentiel d’inversion, les ions en jeux et les propriétés d’inactivation des canaux (ou pompes) pour
interpréter les résultats obtenus. L’enjeu de ces mécanismes est extrêmement important pour la technique de mesure des propriétés membranaires présentée au chapitre 5. Par exemple, les mécanismes d’action de l’halorhodopsin et de l’archaerhodopin sont très différents, le premier utilise le Cl- alors que le deuxième
utilise l’ion H+. Ceci a une implication importante pour les phénomènes impliquant le
neurotransmetteur GABA puisque ce dernier active des canaux ioniques perméables aux ions Cl-. En utilisant la halorhodopsin, par exemple, on ne peut mesurer le
potentiel d‘inversion du récepteur GABA que par extrapolation puisqu’il est impossible d’hyperpolariser la cellule au-delà du potentiel d’inversion du Cl- (environ -65 mV).
Les microsondes optiques dans certains protocoles d’expérimentation s’avèrent être un choix judicieux pour amener la lumière dans le tissu et activer localement ces outils. Les microsondes optiques présentées dans cette thèse combine nt l’enregistrement optique à l’enregistrement électrique et peuvent donc être utilisées à la fois pour détecter les cellules, délivrer le stimulus et en enregistrer l’effet sur la cellule.
1.2.2 Drogues encapsulées
Dans le même ordre d’idées, ces mêmes sondes peuvent être utilisées pour libérer localement des drogues encapsulées. Il est, en effet, possible d’encapsuler ou d’inactiver des drogues et de les injecter dans l’organisme. Les drogues restent inactives tant qu’un stimulus lumineux ne leur est pas fourni [58]. Les sondes optiques présentées dans cette thèse, de par leur taille microscopique, sont peu invasives (elles ont une pointe de quelques micromètres et une forme effilée qui s’apparente à celle des microélectrodes de verre) et seraient un outil de choix pour libérer ou activer les drogues de manière locale en profondeur dans le tissu.
1.3 Techniques de détections optiques en
neurosciences
Comme les sections précédentes le démontrent, plusieurs types d’agents de contraste fluorescents sont disponibles et il existe plusieurs manières d’introduire ceux-ci dans des organismes vivants. Mais comment accéder à cette information optique et à cette spécificité in vivo? Évidemment, les microsondes optiques sont une manière d’y parvenir, mais avant de les présenter, nous verrons une brève description des
principaux outils de détections optiques utilisés in vivo pour détecter la fluorescence. La figure 1.8 montre les principaux types d’imagerie par fluorescence.
Figure 1.8: Type d’imagerie par fluorescence. a) Microscopie à fluorescence conventionnelle. b) Microcopie confocale. c) Schéma optique de la microscopie photons. d) Comparaison entre les volumes d’excitation 1-photon (droite) et 2-photons (gauche) [59].
1.3.1 Microscopie conventionnelle
Un microscope à fluorescence conventionnelle utilise une lampe blanche comme source d’excitation (figure 1.8a). Généralement, il s’agit d’une lampe au mercure ou au xénon. Un filtre d’excitation transmet seulement la plage de longueurs d’onde qui correspond au spectre d’absorption de l’échantillon fluorescent. La lumière transmise est ensuite réfléchie sur un miroir dichroïque et collimée à l’aide d’un objectif. Sous excitation,
l’échantillon émet de la fluorescence qui est collectée par l’objectif, transmise par le miroir dichroïque et filtrée par le filtre d’émission. Ce type de microscope, quoique parfois utilisé in vivo [60], présente certains désavantages. Lorsqu’utilisé avec des échantillons épais, la fluorescence provenant des couches situées au-dessus et en dessous du plan focal de l’objectif peut être collectée, ce qui a pour effet de réduire la qualité des images (elles paraissent floues).
1.3.2 Microscopie confocale
La microscopie confocale (figure 1.8b) utilise une source monochromatique, généralement un laser. Le faisceau laser est réfléchi par le miroir dichroïque et excite l’échantillon. Tout comme pour la microscopie conventionnelle, la fluorescence est collectée par l’objectif et traverse le miroir dichroïque. La lumière collectée est incidente sur une lentille, et un sténopé est placé au point focal de cette lentille. Ainsi, la fluorescence émise à une profondeur différente du point focal de l’objectif est bloquée par le sténopé et n’atteint pas le détecteur. En balayant le faisceau laser sur l’objectif, on peut reconstruire l’image d’un plan mince, dont l’épaisseur varie avec la taille du sténopé. Il s’agit donc d’une méthode plus adéquate que la microscopie conventionnelle pour les échantillons épais ou l’expérimentation in vivo [1].
1.3.3 Microscopie à 2-photons
La microscopie à 2-photons est basée sur le principe de l’absorption à 2-photons brièvement abordée à la fin de la section 1.1. Le principe est similaire à la microscopie laser, mais dans ce cas, puisque l’absorption 2-photons est proportionnelle à l’intensité de la lumière élevée à la puissance deux (I2), le volume d’excitation est très restreint et
l’utilisation du sténopé devient inutile. La figure 1.8c montre le schéma de la microscopie à 2-photons et la figure 1.8d compare le volume d’excitation 1-photon (microscopie conventionnelle et confocale) et 2-photons. Comme l’absorption 2-photons est observable à forte intensité, ce type de microscopie utilise des lasers impulsionnels générant de très courtes et intenses impulsions. Les longueurs d’onde utilisées en microscopie 2-photons (proche infrarouge) sont moins diffusées dans le tissu et permettent d’imager à des profondeurs atteignant 1 mm [61]. Cela en fait une technique très populaire pour l’imagerie in vivo.
![Figure 1.1 : Diagramme de Jablonski illustrant les transitions électroniques donnant lieux aux phénomènes de fluorescence et de phosphorescence (version adaptée de [7])](https://thumb-eu.123doks.com/thumbv2/123doknet/3280922.94141/22.918.156.808.281.742/diagramme-jablonski-illustrant-transitions-électroniques-phénomènes-fluorescence-phosphorescence.webp)
![Figure 1.2: Agents de contraste fluorescents. Cellule endotheliale bovine dont les tubules ont été marqués en vert (Alexa Fluor 488), les mitochondries ont été marquées en rouge (Alexa Fluor 555) et le noyau a été marqué en bleu (DAPI) [8]](https://thumb-eu.123doks.com/thumbv2/123doknet/3280922.94141/24.918.364.596.274.444/figure-contraste-fluorescents-cellule-endotheliale-marqués-mitochondries-marquées.webp)




![Figure 1.14 : Modes LP lm . Profils transverses de différents modes de type LP (« linearly polarised ») observables dans une fibre optique multimodale[82] (l = nombre azimutal et m = nombre radial)](https://thumb-eu.123doks.com/thumbv2/123doknet/3280922.94141/49.918.263.695.578.901/profils-transverses-linearly-polarised-observables-optique-multimodale-azimutal.webp)

