Silica reinforced triblock copolymer gels
Texte intégral
Figure
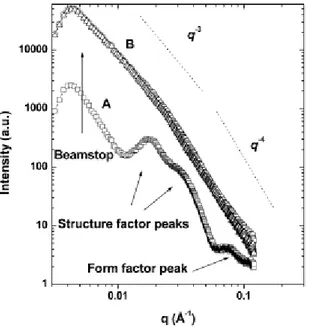
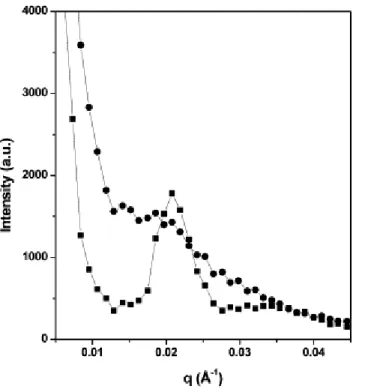
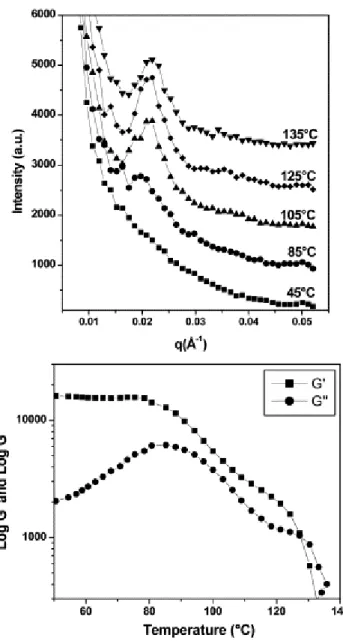
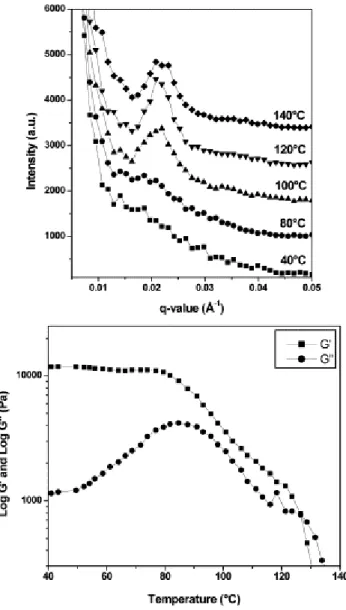
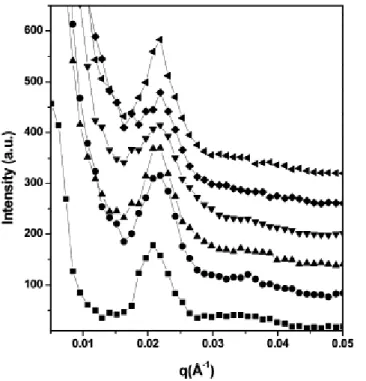
Documents relatifs
As explained in Section 2.3, one first determines the number n(, P ) of Ne or He atoms per unit volume of solid penetrating a realistic model of silica as a function of the
In addition, Fanger played a decisive role in founding the International Society of Indoor Air Quality and Climate (ISIAQ), the International Academy of Indoor Air Sciences (IAIAS),
To confirm the strengthening of the filler-matrix interaction, non-irradiated and irradiated E- SiU samples have been subjected to cycle tensile tests at 80°C (cf. Insert in figure
slip is measured at the rotor that can be linked to an in- flexion point in the flow curve, and (ii) at T = 39.4 ◦ C the stress plateau in the flow curve is associated to station-
Effective heat of combustion and combustion ef ficiency Elemental analysis of C11SiP residue obtained from the cone calorimeter test is shown in Table 4.. As the results show,
The comparison of these values with those obtained by small angle neutron scattering (SANS) on the same series of samples after hypercritical drying, i.e.. aerogels,
Abstract. 2014 We present an analysis of the critical behaviour of viscosity and elastic modulus during the gelation of silica particles. Two systems are studied :
Schematic representation of two extreme models for the triblock copolymer I : looping occurs only in the surface layer, bridging everywhere else ; II : looping occurs in all
