Pépite | Approche intégrée catalyseurs/réacteurs pour une amélioration des performances en oxydation sélective des alcanes
88
0
0
Texte intégral
(2) HDR de Axel Löfberg, Lille 1, 2013. Table des matières Curriculum Vitae .......................................................................................................................5 Activités professionnelles .....................................................................................................5 Etudes .................................................................................................................................5 Enseignement/encadrement ................................................................................................6 Animation et management de la recherche ..........................................................................7 Transfert technologique, relations industrielles et valorisation ..............................................9 I – Introduction générale ...........................................................................................................10 I.1 Intensification des procédés ............................................................................................12 I.2 Application à l’oxydation sélective ...................................................................................15 I.2.1 Introduction au découplage rédox ..............................................................................16 I.2.2 Introduction aux réacteurs structurés .........................................................................18 II – Réacteur Catalytique à Membrane dense ...........................................................................19 II.1 Introduction ....................................................................................................................19 II.2 BIMEVOX ......................................................................................................................22 II.2.1 Perméation et réactivité de membranes polies .........................................................23 II.2.2 Comportement transitoire en RCMD, réactivité de membranes dépolies ..................28 II.2.3 Matériaux composites ...............................................................................................36 II.3 BIMO .............................................................................................................................37 II.3.1 Réactivité des systèmes BiMo en RCMD, oxydation du propène ..............................40 II.4 Conclusions et perspectives...........................................................................................45 III – Réacteurs structurés .........................................................................................................46 III.1 Introduction : microréacteurs et réacteurs structurés .....................................................46 III.1.1 Mise en œuvre de réacteurs à revêtement catalytique .............................................49 III.1.2 Revêtements catalytiques, approche méthodologique .............................................51 III.2 ODH du propane sur VOx/TiO2 ......................................................................................55 III.3 Dépôt contrôlé de VOx / TiO2.........................................................................................57 III.4 Dépôt du support TiO2 ..................................................................................................59 III.4.1 Dépôts de couches par voie sol-gel sur plaques d’aluminium, .................................59 III.4.2 Dépôt sur inox à partir de suspensions ....................................................................61. 3 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(3) HDR de Axel Löfberg, Lille 1, 2013. III.5 Elaboration d’une couche primaire de protection...........................................................63 III.6 Propriétés catalytiques des systèmes structurés ...........................................................66 III.6.1 Plaques VOx/TiO2/Inox .............................................................................................66 III.6.2 Mousses catalytiques...............................................................................................70 III.7 Conclusions et perspectives..........................................................................................73 IV – Reformage sec du méthane ..............................................................................................75 IV.1 Introduction...................................................................................................................75 IV.1.1 Résultats .................................................................................................................76 IV.2 Reformage sec en régime périodique ...........................................................................79 V – Conclusion générale ..........................................................................................................82 Production scientifique .............................................................................................................84. 4 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(4) HDR de Axel Löfberg, Lille 1, 2013. CURRICULUM VITAE LÖFBERG, Axel Kaj Johan Né le 22 juin 1967 à Stockholm (Suède) Double nationalité française et suédoise, marié, sans enfant.. Activités professionnelles ère. 01/2006: Chargé de Recherche 1. Classe au CNRS affecté à l'Unité de Catalyse et de. Chimie du Solide (UMR 8181), issue de la fusion des UMR 8010 et 8012. ère. 10/2001: Chargé de Recherche 1 Classe au CNRS affecté au Laboratoire de Catalyse de Lille (UMR CNRS 8010) de l'Université des Sciences et Technologies de Lille nde. 10/1997: Chargé de Recherche 2 Classe au Centre National de la Recherche Scientifique (CNRS) affecté au Laboratoire de Catalyse de Lille (UPRESA CNRS 8010) de l'Université des Sciences et Technologies de Lille 11/1996-10/97: Post-doctorat au Laboratoire de Catalyse (URA CNRS 402) de l'Université des Sciences et Technologies de Lille dans le cadre d'un projet de collaboration avec FINA Research S.A. (Groupe PetroFina).. Etudes 1990-96 Doctorat en Sciences (chimiques) Université Libre de Bruxelles - 3 octobre 1996 Grade obtenu avec La Plus Grande Distinction Thèse principale: "Le carbure de tungstène en catalyse: Etude de la préparation de catalyseurs massiques et de leurs propriétés pour les réactions alcane-deutérium" Thèse annexe: "Une étude microscopique et spectroscopique en chimie des surfaces permettrait d'élucider l'état chimique de l'oxygène contenu dans le volume du catalyseur lors de la réaction de réduction du NO en régime cinétique oscillatoire." Directeur de thèse: Professeur A. Frennet Lauréat du Prix Stas de l'Académie royale des Sciences, Lettres et Beaux-Arts de Belgique (Décembre 1996) Boursier de la Fondation Universitaire D. et A. Van Buuren (ULB). 1989 Licence en Sciences chimiques Université Libre de Bruxelles Grade obtenu avec Distinction Mémoire: "Analyse du sens thermodynamique des notions d'hydrogène "réversible" et "irréversible", utilisées lors de la caractérisation de catalyseurs métalliques par chimisorption de H2" 1985. Baccalauréat européen Ecole européenne (Bruxelles I), Section francophone options scientifiques. 5 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(5) HDR de Axel Löfberg, Lille 1, 2013. Enseignement/encadrement Mes activités d’enseignement et de formation concernent essentiellement l'encadrement d’étudiants en thèse, en stage post-doctoral ou en stage de formation (DEA, Masters, …). Thèses •. Thierry GIORNELLI (Bourse UTC, 2001-2004, soutenue le 29/11/04) : « Optimisation et caractérisation de dépôts de catalyseurs VOx/TiO2 sur parois métalliques pour réacteurs structurés. Application déshydrogénante du propane », Dir. E. BORDES-RICHARD. •. à. l’oxydation. Hervé BODET* (Bourse MERT 2004-2007, soutenue le 13/12/2007) : « Céramiques denses comme réacteur membranaire pour l’oxydation ménagée des hydrocarbures », Dir. E. BORDES-RICHARD. •. Chanapa KONGMARK* (Bourse d'Etat thaïlandais, 2006-2009, soutenue le 7/5/2010) : « Nouvelles céramiques conductrices par ions oxyde pour pile à combustible ou réacteur catalytique à membrane dense », Dir. E . BORDESRICHARD & R.N. VANNIER. •. Adil ESSAKHI (ANR « Millicat », 2007-2010, soutenue le 13/02/2012) : « Réacteurs structurés pour la catalyse. », Dir. E. BORDES-RICHARD et A. LÖFBERG. •. Jesus GUERRERO (Bourse MERT Univ. Lille1, 2012-2015) : « Valorisation du méthane par reformage sec en régime non-stationnaire, application du réacteur à alimentation périodique » Dir. L. JALOWIECKI-DUHAMEL et A. LÖFBERG. Post-doctorants •. Philippe MALFOY (1997-1998) post-doctorat, contrat industriel PétroFina. •. Hui LU (2005-2006), “Bismuth-based ionic conductors and their applications as membrane reactors for propane conversion”. •. Svetlana HEYTE (2009-2010) post-doctorat, contrat industriel Adisseo.. •. Stéphane HONNART (2011-2012), contrat industriel Dexera.. •. Alexey TYUNYAEV (2012-2013) post-doctorat, contrat industriel Adisseo.. Stages de formation •. Anne-Sophie MAMEDE (DEA, USTL 2000). •. Fabien NEGRIER (Thèse, Univ. Paris VI, 6 mois, 2001). •. Samir BENAISSA (DEA, UTC, 2002). •. Mohamed TAOUTI (Thèse, Univ. de Laghout - Algérie, 3 mois, 2002). 6 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(6) HDR de Axel Löfberg, Lille 1, 2013. •. Salaheddine BOUJMIAI (DEA, USTL, 2003). •. Siham BARAMA (Thèse, USTBH – Algérie, 1 an, 2006/7). •. Alin-Alexandru PARASCHIVU (Erasmus-ENSCL, 6 mois, 2010). •. Rafik BENRABAA (Thèse, USTBH – Algérie, 6 mois, 2011). •. Liu CHANG (Master 2, Univ. Lille1, 2011). •. Gaëtan PERRUSSEL (Master 2, Ecole Centrale Lille, 2012). Animation et management de la recherche Activités internes à l’UCCS Depuis 1998 : gestion informatisée des gaz dans le Laboratoire (équipe Catalyse Hétérogène) 1999-2008 : gestion du site web du Laboratoire 1999-2008 : gestion des ressources bibliographiques du laboratoire, correspondant PubliCNRS puis HAL. 2001-2005 : membre du Conseil de Laboratoire 2001-2008 : correspondant formation du Laboratoire Activités externes à l’UCCS Membre extérieur de la commission de spécialistes de la section 31, Université des Sciences et Technologies, de 2001 à 2004 puis de 2007 à 2009. Membre extérieur de la commission unique de spécialistes de l'Ecole Centrale de Lille de 2007 à 2009. Membre élu (SNCS-FSU) de la section 14 « Chimie de coordination, catalyse, interfaces et procédés » du Comité national (CoNRS) depuis septembre 2012, mandat 2012-2016. Membre élu (SNCS-FSU) de la CID 50 « Gestion de la Recherche » du Comité national (CoNRS) depuis février 2013, mandat 2012-2016. Depuis juin 2012, correspondant scientifique pour le compte de l’Institut de Chimie du CNRS auprès de l’ERA-Net « CAPITA - Catalytic Processes for Innovative Technology Applications » [1]. Membre du Conseil de Développement de Lille Métropole au titre du SNCS-FSU (collège économique) depuis 2010 (mandat 2010-2014); co-pilote du groupe de travail. 1 - http://www.era-capita.eu/. 7 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(7) HDR de Axel Löfberg, Lille 1, 2013. « Intersyndicats » du Conseil de Développement, membre du Conseil de Gouvernance du Plan Métropolitain de Développement Economique (2010-2012). Organisation de manifestations scientifiques J’ai assuré le Secrétariat général du Comité d’organisation et du Comité scientifique du th. 6 World Congress on Oxidation Catalysis. Ce congrès s’est tenu à Lille en juillet 2009 [2] (500 participants). Il a été organisé conjointement par l’UCCS (E. Bordes-Richard, cochairperson) et le Laboratoire de Catalyse de l’Université catholique de Louvain (P. Ruiz, cochairperson). Après la tenue du congrès une centaine de communications ont été sélectionnées et invitées à soumettre un manuscrit pour publication dans un volume spécial de la revue Catalysis Today dont je suis co-éditeur (avec E. Bordes-Richard, E. Gaigneaux, E. Payen, P. Ruiz). Au terme de la procédure habituelle d’examen, 78 articles ont été retenus dans ce volume spécial [3]. J’ai également participé au Comité d’organisation de la seconde édition de la Conférence Internationale « A Greener Chemistry for Industry » (GCI 2011) qui s’est tenue à Villeneuve d’Ascq du 10 au 14 Décembre 2012 [4]. L’UCCS a organisé à Munich (Allemagne) en juillet 2012 un symposium international intitulé Synfuel2012 [5] comme colloque « satellite » à l’International Congress on Catalysis (ICC) qui se tenait dans cette même ville les jours suivants. J’ai activement contribué à l’organisation de cette manifestation qui a obtenu un grand succès (210 participants, 53 communications orales donc 5 conférences invitées et 10 keynotes, 100 posters). J’ai également participé au comité d’organisation du XIème Symposium International sur « Environnement, Catalyse et Génie des Procédés » (ECGP’11) qui a réuni à Villeneuve d’Ascq près de 120 participants les 26, 27 et 28 Juin 2013 [6]. Enfin, j’ai coordonné la préparation de la candidature de la Section régionale de la Société Chimique de France pour l’organisation du congrès SCF’2015, candidature qui a été retenue par le bureau du Conseil d’Administration de la SCF. Je suis donc membre du Comité d’organisation de SCF’15.. 2 - http://www.6wcoc.org 3 - “Towards an integrated approach in innovation and development” , Proceedings of the 6th World Congress on Oxidation Catalysis Lille, France, 5-10 July 2009, Catal Today, Volume 157, Vol. 1-4, pages 1-466 (2010) Edited by Elisabeth Bordes-Richard, Eric M. Gaigneaux, Axel Löfberg, Edmond Payen and Patricio Ruiz 4 - http://www.ensc-lille.fr/actu/GCI2011/index_fr.html 5 - http://www.synfuel2012.org/ 6 - http://www.amcemorocco.ma/ECGP11/FR/. 8 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(8) HDR de Axel Löfberg, Lille 1, 2013. Transfert. technologique,. relations. industrielles. et. valorisation - Projet de recherche industriel tripartite (Adisseo, UCCS, LGPC-Villeurbanne) En 2009, nous avions effectué une étude exploratoire de faisabilité de dépôts de matériaux catalytique sur des parois métalliques. Ces travaux ont permis de définir les conditions de prétraitement des matériaux substrats (plaques et mousses) afin de stabiliser la phase active. Des essais comparatifs ont été réalisés afin de déterminer le gain potentiel offert par la structuration du réacteur catalytique. Interrompu en 2011 en attente de évaluation économique effectuée par le partenaire industriel, ce projet a été repris en 2012 afin d’apporter des données complémentaires, avant d’engager une étude plus approfondie du système (Adisseo, UCCS). - Projet de développement d’un catalyseur (DEXERA) Depuis 2011, je suis responsable d’un projet de développement d’un catalyseur d’oxydation sélective d’un composé aromatique substitué, intermédiaire de fabrication d’un polymère. Cette collaboration comporte la mise à disposition d’un test catalytique et l’assistance dans la mise au point du catalyseur industriel ainsi que celle des conditions opératoires. Cette collaboration a été renouvelée jusqu’en mai 2013 (Convention DEXERA - Univ. Lille1), et fait actuellement l’objet d’une évaluation pour un projet de plus grande envergure. - Demande de brevet Un brevet est en cours d’évaluation auprès de la SATT Nord pour la valorisation des travaux réalisés par M. J. Guerrero (Thèse en cours) sur le reformage sec du méthane.. 9 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(9) HDR de Axel Löfberg, Lille 1, 2013. I – INTRODUCTION GENERALE Depuis quelques décennies, on assiste à une progressive prise de conscience des limites d’un développement économique basé exclusivement sur l’utilisation de ressources non renouvelables, que ce soit en matières premières qu’en énergie, et sans considération sur l’impact environnemental et sociétal d’un procédé de transformation. Le plus souvent, les solutions restent toutefois basées sur des solutions « end of pipe » visant à corriger l’impact du procédé de manière ponctuelle. Au mieux, les procédés sont adaptés pour réduire certains impacts à la source. Une approche holistique de cette question a été proposée à la fin des années 80, en particulier par Frosch et Gallopoulos [7], développée par la suite notamment par Allenby [8], et reprise dans l’ouvrage de Erckman [9]. Cette approche consiste à ne pas considérer un processus industriel comme cloisonné mais comme un véritable écosystème dont les flux de matière et d’énergies sont comparables à ceux d’un écosystème biologique, d’où la terminologie d’écologie industrielle. Ainsi, les systèmes industriels actuels peuvent être décrits par un écosystème de type I (Figure 1) dans lequel ressources et déchets sont illimités. En revanche, un système biologique se caractérise par un écosystème de type III, dans lequel seule une source d’énergie extérieure (typiquement l’énergie solaire) est nécessaire pour alimenter un écosystème dans lequel les déchets des uns sont les matières premières des autres. Si un processus industriel peut difficilement aboutir à un écosystème parfait de type III, il peut néanmoins s’approcher de celui de type II moyennant une interaction forte entre les différents intervenants de ce processus, depuis le producteur de ressources premières aux consommateurs en passant par les processeurs de déchets et des matériaux.. Figure 1 - Différents types d’écosystèmes d’après Erckman et Allenby [9]. 7 - R.A. Frosch, N. E.Gallopoulos, «Strategies for Manufacturing», Scientific American, vol. 261, n°3, (1989) 94 8 - B. Allenby, Progress in Industrial Ecology - An International Journal, Vol. 3, Nos 1/2 (2006) 28 9 - S. Erckman, Vers une écologie industrielle, Editions-Diffusion Charles Léopold Mayer, Paris (2004). 10 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(10) HDR de Axel Löfberg, Lille 1, 2013. Figure 2 – Ecosystème industriel idéal d’après Erckman et Allenby [9]. Cette vision basée sur l’interaction des différents intervenants, définie ensuite comme « symbiose industrielle », a été mise en pratique bien avant qu’elle ne soit conceptualisée. La ville de Kalundborg au Danemark constitue l’exemple type de cette démarche d’écologie industrielle. Au-delà de cet exemple pratique, la notion d’écologie industrielle peut paraître très conceptuelle. On constate néanmoins qu’une telle approche holistique est de plus en plus en vogue, notamment dans des études émanant des milieux industriels. Ainsi, le CEFIC (Conseil Européen des Industries Chimiques) a produit en 2011 un rapport intitulé « The chemical industry in Europe: Towards Sustainability » [10] qui base le futur développement de l’industrie chimique sur trois piliers, celui de la « planète », donc sur des considérations environnementales, celui des « gens » avec des considérations sociétales et enfin celui du « profit » avec des enjeux purement économiques. L’association SPIRE (Sustainable Process Industry through Resource and energy Efficiency) qui regroupe les plus grands industriels européens, notamment de la chimie, ainsi que des centres de recherche a également produit très récemment [11] une « feuille de route » particulièrement intéressante en identifiant les grands défis que l’industrie de transformation en général devra relever dans un avenir très proche. Sans jamais citer l’écologie industrielle proprement dite − dont la terminologie a sans doute une connotation devenue entretemps trop « politique » − ce rapport inclut nommément la symbiose industrielle comme un des éléments clés du futur développement de cette industrie. De fait, cette feuille de route revendique une véritable vision holistique et intersectorielle sur la manière de surmonter ces défis sociétaux en considérant notamment les producteurs de ressources, les aspects logistiques, le recyclage des déchets ainsi que le. 10 - http://www.cefic.org/Documents/Learn%20and%20Share/Cefic_Sutainability_Report20112012.pdf 11 - http://www.spire2030.eu/uploads/Modules/Documents/spireroadmap_september_2013_pbp_03.pdf. 11 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(11) HDR de Axel Löfberg, Lille 1, 2013. comportement des consommateurs en plus des aspects plus traditionnels du développement industriel.. Figure 3 – Vision de l’évolution de l’industrie de transformation (SPIRE Roadmap [11]). De manière plus concrète, elle décrit une évolution de l’industrie de transformation (Figure 3) impliquant une réduction progressive des ressources employées (REDUCE), le recyclage croissant (RE-USE), le remplacement progressif des ressources (REPLACE), notamment pétrolières, vers des ressources renouvelables. Mais plus important, elle postule la nécessité aussi de « réinventer » les procédés existant par le développement de nouveaux matériaux et de nouvelles technologies de transformation de ceux-ci (REINVENT). Evidemment, l’industrie chimique n’aura pas attendu ce rapport pour réinventer des procédés afin de les rendre, notamment, plus efficaces, plus sûrs et plus économes en ressources. Si l’approche est souvent plus pragmatique et moins globale elle se base entre autres sur un concept qui s’en approche sur de nombreux aspects : l’intensification des procédés.. I.1 Intensification des procédés Le développement d’un procédé chimique industriel débute en général par la mise au point d’une nouvelle réaction qui, par la matière première mise en œuvre, ou le produit formé, présente un avantage par rapport à un procédé existant ou répond à un nouveau besoin/marché. Souvent cette réaction est mise au point à l’échelle du laboratoire dans un premier temps. Il s’en suit une étape d’up-scaling vers une production à l’échelle industrielle faisant intervenir tout le savoir-faire en génie chimique et catalytique pour faire face aux. 12 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(12) HDR de Axel Löfberg, Lille 1, 2013. conditions de transferts de matière et de chaleur souvent drastiquement différents en comparant les réacteurs de laboratoire et ceux de production. Lorsqu’un procédé aboutit à la phase d’exploitation industrielle et commerciale, le développement peut se poursuivre en vue d’une amélioration de la productivité. Elle peut être obtenue en intervenant sur la chimie de la réaction, et notamment par le développement de nouveaux catalyseurs, ou sur le procédé industriel, ces deux aspects étant évidemment très étroitement liés. Toutefois, une fois le procédé opérationnel, le type de réacteur employé est rarement remis en cause car cela reviendrait à développer un tout nouveau procédé. Les marges de manœuvre sont donc limitées. Il est toutefois possible d’envisager cette démarche autrement, en considérant la façon de mettre en œuvre des réactions à l’échelle industrielle dès l’étape des recherches en laboratoire. Cette approche permet d’intégrer le réacteur chimique dans un ensemble plus vaste de contraintes concernant l’ensemble du procédé. Celui-ci n’est plus vu comme une somme d’opérations unitaires ayant chacune son efficacité ou sa productivité mais comme un tout. C’est le sens général du concept d’intensification des procédés. Bien que n’étant pas nouveau en soi – les bases de cette démarche ont été posées à partir des années 80 – il prend tout son sens si l’on considère les préoccupations actuelles pour une utilisation rationnelle de l’espace, pour un environnement plus sain et pour une croissance économique forte et durable. On peut résumer le concept en se référant à la définition donnée par Stankiewicz et al. [12] : « L’intensification d’un procédé consiste à développer des appareils et des techniques innovantes qui, comparés à ceux utilisés de manière courante de nos jours, sont susceptibles d’apporter des améliorations substantielles dans la fabrication et le fonctionnement des installations. Il s’agit notamment de diminuer fortement le rapport entre la taille des équipements et les capacités de production, et de diminuer également la consommation d’énergie ainsi que la quantité de déchets produits afin, à terme, de résulter en une technologie plus économique et plus durable ». L'intensification des procédés vise donc à assurer le développement de procédés plus efficaces, plus sûrs et moins coûteux en énergie comme en atomes. Il s’agit d’une démarche complexe qui doit prendre en considération l’ensemble du procédé lui-même mais aussi tout son environnement économique, écologique et social et s’inscrit donc parfaitement dans la notion de symbiose industrielle décrite plus haut à une échelle plus globale. Elle nécessite donc une forte multidisciplinarité et donne une vision plus générale du concept que celle, initiale, qui visait plus spécifiquement la miniaturisation des procédés et des réacteurs [13, 14].. 12 - A.I. Stankiewicz, J.A. Moulijn, Chem. Eng. Prog. Vol. 96/1 (2000) 22 13 - J.R. Burns, C. Ramshaw, Trans. IchemE, 77 (1999) 206 14 - C. Ramshaw, Green Chem. 1 (1999) 15. 13 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(13) HDR de Axel Löfberg, Lille 1, 2013. D’autres définitions de l’intensification des procédés existent [15,16] mais toutes ont en commun les mots-clés : innovation et substantiel. D’une part il doit se distinguer d’une approche conventionnelle de génie chimique pour l’optimisation et la conception du procédé (innovation) et, d’autre part, l’objectif n’est pas d’obtenir une amélioration de quelques pourcents de rendement mais un véritable saut qualitatif et quantitatif dans la gestion et l’optimisation d’une unité industrielle (substantiel). Si on décline ces principes pour un procédé chimique, on peut développer ces différents aspects de la manière suivante : Efficacité : Elle est directement liée à un meilleur contrôle de l’environnement de la réaction, en particulier des transferts de chaleur et de matière et donc de la température, permettant de meilleurs rendements, conversions et sélectivités. Les pertes de matières premières et la consommation d’énergie doivent être limitées ainsi que les besoins de purification et de gestion des déchets. Sécurité : La sécurité des procédés chimiques peut être considérablement améliorée dans le cas d’utilisation de réacteurs de plus petite taille. Le meilleur contrôle de la température doit permettre de limiter les risques d’emballement de la réaction et donc d’explosion dans le cas de réactions fortement exothermiques. En cas d’incident, les conséquences peuvent également être réduites par la mise en œuvre de quantités de matières inférieures. Ainsi, il a été calculé [17] que l’application des principes de miniaturisation et d’intensification des procédés auraient permis de faire fonctionner l’usine de fabrication de pesticides de Bhopal en Inde avec la même productivité et, à tout instant, un stock de seulement 10 kg d’isocyanate de méthyle, à comparer aux 40 tonnes libérées lors de la catastrophe de 1984 ! Coûts : L’objectif ici est d’assurer une optimisation des coûts sur l’ensemble du procédé depuis celui des terrains nécessaires (surfaces au sol), des investissements en matériel et de la maintenance du système, sans oublier le coût de la matière première, de l’énergie consommée et de la gestion des déchets nécessaires à son fonctionnement. En pratique, l’intensification des procédés peut se concrétiser essentiellement [18] de deux manières. D’une part par l’intégration fonctionnelle, à savoir la combinaison de plusieurs opérations (mélange, séparation, échange de chaleur, …) au sein d’une seule unité avec, par exemple, des réacteurs-échangeurs [19] ou des réacteurs permettant l’apport contrôlé des réactifs ou la séparation d’un produit (réacteurs à membrane catalytique [20],. 15 - A.I. Stankiewicz, J.A. Moulijn, Process intensification, Ind. Eng. Chem. Res. 41 (2002) 1920-1924 16 - T. Van Gerven, A. Stankiewicz, Ind. Eng. Chem. Res. 48 (2009) 2465 17 - D.C. Hendershot, Chem. Eng. Prog. Vol. 96/1 (2000) 35 18 - P. Lutze, R. Gani, J.M. Woodley, Chem. Eng. Process. 49 (2010) 547 19 - Z. Anxionnaz, M. Cabassud, C. Gourdon, P. Tochon, Heat exchanger/reactors (HEX reactors): Concepts, technologies: State-of-the-art, Chemical Engineering and Processing: Process Intensification, 47 (2008), 2029. 20 - Exemples: “Catalysis in membrane reactors. Proceedings of the 9th International Conference on Catalysis in Membrane Reactors.” (France), 2009, A. Giroir-Fendler, N. Guilhaume, J.-A. Dalmon, D. Farrusseng and M. Pera-Titus Eds, Catalysis Today 156 3-4 (2010). 14 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(14) HDR de Axel Löfberg, Lille 1, 2013. ...). L’autre approche est la réduction drastique de la taille des réacteurs (microréacteurs). Souvent ces deux approches sont effectuées simultanément (microéchangeurs [21], micromélangeurs, …).. I.2 Application à l’oxydation sélective Depuis 50 ans la catalyse d'oxydation sélective des hydrocarbures (C1-C8) représente environ 25 % des procédés catalytiques en pétrochimie et chimie de base. Elle intervient dans notamment dans la synthèse de monomères et d'intermédiaires chimiques destinés à l’industrie des polymères. A noter que l’on utilise la terminologie d’oxydation « sélective » par opposition à l’oxydation totale en COx (CO2 et/ou CO). Toutefois plusieurs autres terminologies sont utilisées dans ce domaine en particulier celles d’oxydation partielle et d’oxydation ménagée. D’autres réactions entrant dans cette catégorie des oxydations sélectives ont une appellation plus spécifique en fonction du type de produit obtenu : époxydation, oxydation déshydrogénante, ammoxydation (insertion de N), oxychlorination (insertion de Cl). Enfin, le couplage oxydant se caractérise par une augmentation du nombre de carbone dans le produit hydrocarbure par formation de liaisons C-C. Plus récemment, les alcanes (C1-C4), obtenus lors du raffinage du pétrole, ont fait l'objet d'études poussées dans le but de les substituer aux alcènes correspondant. Mais cela demeure encore aujourd’hui un défi. A ce jour, seul quelques exemples de développement industriels ont vu le jour. L’oxydation du n-butane en anhydride maléique constitue sans aucun doute le plus bel exemple. L’oxydation du propane en acide acrylique [22] connait de ce point de vue un intérêt croissant de même que celle de l’éthane en acide acétique puisqu’elles ont atteint le stade pilote [23]. Le couplage oxydant du méthane en éthane et éthylène, comme l'oxydation déshydrogénante (ODH) des C2-C4 ne sont pas industrialisés car les catalyseurs ne sont pas assez performants. Pourtant les réactions d'ODH présentent l'avantage d'être exothermiques et directes comparées aux déshydrogénations simples (endothermiques et équilibrées). La principale difficulté des réactions d’oxydation sélective est la réactivité importante des produits de réaction, souvent supérieure à celle des réactifs de départ. L’oxydation totale en COx, notamment lorsque O2 est utilisé comme oxydant, est souvent favorisée et il en découle que, typiquement, la sélectivité diminue avec l’augmentation du degré de conversion du réactif. Les meilleurs rendements en produits sont donc rarement obtenus à fort taux de conversion.. 21 - J.J. Brandner, L. Bohn, T. Henning, U. Schygulla, K. Schubert, Heat Transfer Engineering, 28:8-9 (2007), 761 22 - A. Godefroy, G.S. Patience, T. Tzakova, D. Garrait, J.-L. Dubois, Chem. Eng. Technol., 32 No. 3 (2009) 373–379 23 - E. Bordes-Richard, J. Védrine, Techniques de l’Ingénieur, Catalyse et procédés catalytiques, J1215 (2013). 15 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(15) HDR de Axel Löfberg, Lille 1, 2013. C’est donc dans le domaine des réactions d’oxydation sélective que des avancées majeures peuvent être attendues en modifiant le procédé, tant en terme d’augmentation de productivité pour des réactions existantes qu’en terme d’évolution des sources de carbone pour, entre autres, la pétrochimie. Plusieurs pistes peuvent être explorées. D’une part, la voie « traditionnelle » consiste à améliorer les formulations des catalyseurs utilisés dans des réacteurs catalytiques habituels (lit fixes ou fluidisés). L’autre voie, répondant au concept d’intensification des procédés, consiste à explorer d’autres façon de mettre en œuvre la réaction catalytique afin d’optimiser, voire de supprimer, certaines contraintes néfastes à la productivité du système. Cette approche nécessite d’intégrer le mode de mise en contact du catalyseur dans le réacteur dès la conception, que ce soit à l’échelle du laboratoire ou à celle du développement industriel. C’est cette approche intégrée catalyseur-réacteur qui est au cœur de ma démarche scientifique. Elle sera illustrée dans la suite de ce manuscrit de deux façon : par le découplage rédox et par l’utilisation de réacteurs structurés. I.2.1 Introduction au découplage rédox La plupart des réactions d’oxydation sélective obéissent au mécanisme redox de Mars et Van Krevelen [24]: HC + KO → PO + K (+ H2O). k1, Vred-K. (1). K + O (air) → KO. k2, Vox-K. (2). où KO et K représentent les formes respectivement oxydée et réduite du catalyseur, HC l’hydrocarbure et PO le produit d'oxydation ménagée. Dans un réacteur traditionnel co-alimenté en oxygène et en hydrocarbure, ces deux étapes ont lieu simultanément, et à l’état stationnaire le catalyseur (volume et surface) se trouve dans un état plus ou moins réduit en fonction des conditions opératoires. Compte tenu des limites d’explosibilité des mélanges, les marges de travail sont étroites et conditionnent de manière importante l’activité et, surtout, la sélectivité et la productivité en PO des catalyseurs. 2-. L’espèce oxygène réputée sélective est l’oxygène nucléophile (O ) provenant du -. 2-. -. réseau du catalyseur. Par opposition, les espèces électrophiles (O2 , O2 , O ) adsorbées à la surface du catalyseur conduisent en règle générale à la formation de CO et CO2 [25].. 24 - P. Mars, D.W. van Krevelen, Chem. Eng. Sci. 3 (1954) 41–59 25 - J. Haber, in Perspectives in Catalysis, J.M. Thomas, K.I. Zamaraev (Eds.), Blacwell Science Publishers, Oxford (1992). 16 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(16) HDR de Axel Löfberg, Lille 1, 2013. Le découplage rédox consiste à dissocier les deux étapes du mécanisme, et présente des avantages importants compte tenu de l'absence d'oxygène gazeux lors de la mise en contact du catalyseur avec l'hydrocarbure : •. la teneur en hydrocarbure n'est plus limitée par l'explosibilité du mélange,. •. l'hydrocarbure non converti est plus facilement recyclé,. •. la sélectivité peut être améliorée en contrôlant la quantité et la qualité (plus ou moins nucléophile) de l’oxygène à la surface du solide.. Ce découplage peut être réalisé de plusieurs manières : •. Découplage spatial : les deux réactions sont mises en œuvre dans des réacteurs différents et le catalyseur est transporté d’un réacteur à l’autre (réacteur à lit circulant, RLC). Il s’agit du seul cas de découplage rédox ayant à ce jour connu un développement industriel, qui a malheureusement été interrompu faute de rentabilité économique (oxydation du n-butane en anhydride maléique par DuPont de Nemours) [26, 27].. •. Découplage temporel : Plus abordable à l’échelle du laboratoire, cette méthode consiste à alimenter un réacteur à lit fixe (ou fluidisé) de manière périodique en alternant les flux réducteurs et oxydants. Difficilement réalisable à l’échelle industrielle, ce type de découplage permet néanmoins d’étudier les catalyseurs utilisés en lit circulant.. •. Découplage en continu : Le catalyseur, sous forme de membrane dense, fait office de séparateur entre deux compartiments du réacteur, l’un alimenté avec l’hydrocarbure, l’autre avec l’oxygène. Ce dernier est dissocié par le catalyseur, 2-. diffuse sous forme ionique (O ) au travers de la membrane et réagit sur la face opposée. Dans les deux premiers cas le catalyseur fonctionne en régime transitoire ou transitoire forcé. Une bonne compréhension des mécanismes d’oxydo-réduction des matériaux catalytiques est donc primordiale afin d’éviter une réduction ou une oxydation trop importante pouvant irréversiblement altérer le solide et les performances du système. Par ailleurs, le solide étant continuellement recyclé et régénéré, de nouveaux matériaux peuvent être considérés puisqu'un des critères majeurs de sélection en régime stationnaire, la stabilité dans le temps, n'est pas primordial dans un système découplé. Ainsi, des solides qui présentent des propriétés catalytiques intéressantes mais qui seraient écartés parce que trop "instables" peuvent donc être considérés. Par contre, de nouvelles contraintes apparaissent au niveau du choix du catalyseur. Il doit être capable de céder une partie de l'oxygène du réseau superficiel de manière sélective et réversible. Dans le cas du RLC il doit de plus présenter une bonne résistance à l'attrition permettant sa circulation entre les réacteurs. La reproductibilité de son comportement est essentielle. La notion de reproductibilité, voire de répétabilité, se substitue à celle de stabilité.. 26 - R.M. Contractor, A.W Sleight, Catal. Today 3 (1988) 175 27 - E. Bordes, R.M. Contractor, Topics in Catalysis 3 (1996) 365. 17 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(17) HDR de Axel Löfberg, Lille 1, 2013. D’autres contraintes existent pour l’utilisation d’un réacteur à membrane dense mais seront développés dans le Chapitre II de ce manuscrit consacré à ce sujet. En effet, le découplage au moyen d’une membrane dense a été l’objet d’un long travail mené en collaboration avec l’équipe animée par Rose-Noëlle VANNIER au sein de l’UCCS. L’utilisation d’un réacteur à alimentation périodique sera évoquée dans le cadre du reformage sec du méthane dans le Chapitre IV. I.2.2 Introduction aux réacteurs structurés Une des caractéristiques communes à toutes les réactions d’oxydation sélective est leur exothermicité. Qui plus est, ces réactions sont toujours accompagnées d’oxydation totale, encore plus exothermique. Il en résulte que la maîtrise du comportement thermique du réacteur est cruciale dans l’étude et surtout le développement de ce type de réaction. Les réacteurs traditionnels présentent en général des profils/gradients de température plus ou moins importants le long du lit catalytique ou de manière radiale entre le cœur du réacteur et le bord en contact avec les parois. Typiquement, les solutions pour éviter ces gradients consistent à effectuer des remplissages sélectifs du réacteur avec, par exemple des concentrations de catalyseurs différentes le long du lit catalytique, ou à optimiser les échanges thermiques en réduisant le diamètre des tubes. Une mauvaise maîtrise de cet aspect peut conduire à la formation de points chauds au sein du lit catalytique avec des conséquences néfastes. Du fait de la température élevée, le catalyseur à cet endroit est très actif pouvant conduire localement à de très fortes conversions du réactif et donc à de plus faibles sélectivités en produit d’oxydation sélective. Cette suractivité peut même conduire à un emballement proprement dit de la réaction avec des conséquences en termes de sécurité du procédé. D’autre part les conditions particulières de fonctionnement dans ces points chauds peuvent entraîner la désactivation du catalyseur. Les catalyseurs d’oxydation sélective sont pour la plupart des oxydes, donc typiquement des matériaux réfractaires, ce qui ne contribue pas à améliorer les propriétés de transfert thermique du lit catalytique. Une solution, qui consiste à diluer le catalyseur avec un autre matériau inerte mais bon conducteur de chaleur, est régulièrement utilisée pour tenter de pallier ces difficultés. Toutefois, dans ce genre de système le matériau catalytique n’est pas en contact « intime » avec le diluant, ce dernier n’étant pas lui-même en contact « intime » avec les surfaces du réacteur par lequel les échanges de chaleur avec l’extérieur peuvent être optimisés. De fait, les transferts de chaleurs doivent systématiquement passer par la phase gaz. L’utilisation de réacteurs structurés dans lesquels le catalyseur est déposé sous forme de revêtement sur des substrats conducteurs de chaleur constitue une option possible pour améliorer les performances catalytiques en oxydation sélective. Mes travaux dans ce domaine sont décrits dans le Chapitre III de ce manuscrit.. 18 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(18) HDR de Axel Löfberg, Lille 1, 2013. II – REACTEUR CATALYTIQUE A MEMBRANE DENSE II.1 Introduction Ainsi qu’il a été dit dans l’introduction, le découplage rédox peut être réalisé à l’aide d’un réacteur catalytique à membrane dans lequel le catalyseur sous forme de membrane dense fait office de séparateur entre deux compartiments du réacteur, l’un alimenté avec l’hydrocarbure, l’autre avec l’oxygène (Figure 4). Ce dernier est dissocié par le catalyseur, 2-. diffuse sous forme ionique (O ) au travers de la membrane et réagit sur la face opposée. La force motrice de cette diffusion est la différence de pression partielle en oxygène entre les deux compartiments. Dans une configuration idéale, la membrane est à la fois le séparateur des compartiments mais aussi le catalyseur. Il est donc constitué d’un matériau unique qui doit présenter simultanément un ensemble de caractéristiques fondamentales : •. être un bon catalyseur d’oxydation sélective (tant du point de vue de l’activité que de la sélectivité),. •. être un bon conducteur ionique par O afin d’assurer le transfert de l’oxygène d’un compartiment à l’autre. •. être suffisamment conducteur électronique afin d’assurer la migration en sens inverse des électrons en l’absence d’un circuit électrique extérieur. •. enfin, être mécaniquement et chimiquement stable malgré l’exposition des deux faces à des atmosphères très différentes.. 2-. Figure 4 - Schéma de principe d’un RCMD. 19 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(19) HDR de Axel Löfberg, Lille 1, 2013. Il faut noter que dans un RCMD, l’objectif est la réaction entre l’oxygène de réseau superficiel avec l’hydrocarbure et non la recombinaison et la formation de O2 dans le compartiment basse pression oxygène. Si cela était le cas, la membrane se comporterait comme un diffuseur d’oxygène moléculaire à l’instar des membranes poreuses, par exemple. Si ce mode d’alimentation contrôlée, ou étagée, en oxygène peut conduire à une amélioration de la sélectivité pour certaines réactions d’oxydation sélective, il n’est aucunement basé sur le principe de découplage rédox proprement dit. Les premiers travaux présentant un réacteur à membrane dense avec diffusion ionique de l’oxygène remontent au début des années 80 par Stoukides et al [28, 29] à la nuance près que ces travaux faisaient usage de matériaux conducteurs purement ioniques, d’où la nécessité d’ajouter un circuit électrique extérieur pour le transfert des électrons d’une face à l’autre de la membrane. D’une manière générale, l’utilisation d’un circuit extérieur permet soit 2-. d’aider le transfert d’O en imposant un courant électronique de manière similaire à une pompe électrochimique, ou de récolter le courant issu de cette migration d’ions, à l’instar d’une pile à combustible. Les systèmes sont dans ce cas plus complexes car ils nécessitent d’intégrer des électrodes afin de collecter les électrons aux deux faces de la membrane. Ces électrodes doivent donc être constituées de matériaux conducteurs mixtes ionique et électronique. La première publication posant le principe du RCMD au sens strict est celle de Di Cosimo et al [30] en 1986 qui proposaient de réaliser la déshydromérisation oxidante du propène en hexadiène et benzène avec une membrane de Bi2O2-La2O3 faisant office de séparateur des compartiments (conducteur mixte) et de catalyseur pour la réaction. Depuis, de nombreux travaux ont été réalisés dans ce domaine et d’excellentes revues ont été publiées. On notera, entre-autres, celles de Bouwmeester et al, qui abordent la question de manière plus théorique notamment sur la chimie du solide des catalyseurs d’oxydation et plus particulièrement dans les réacteurs à membrane dense [31, 32]. Parmi les autres revues intéressantes on notera celles de Sundmacher [33] et de Julbe et al [34]. Très récemment, Wei et al ont également proposé une revue utile dans le domaine [35]. Même s’ils ne sont pas exclusivement consacrés au RCMD, les recueils des publications aux différents congrès internationaux de catalyse par les membranes (ICCMR) constituent une source documentaire intéressante [36].. 28 - M. Stoukides, C.G. Vayenas J. Catal. 82 (1983) 45 29 - M. Stoukides, C.G. Vayenas J. Catal. 70 (1981) 137 30 - R. Di Cosimo, J.D. Burrington, R.K. Grasselli, J. Cat. 102 (1986) 234 31 - H.J.M. Bouwmeester, Catal. Today 82 (2003) 141 32 - P.J. Gellings, H.J.M. Bouwmeester Catal. Today 58 (2000) 1 33 - K. Sundmacher, L.K. Rihko-Struckmann, V. Galvita, Catal. Today 104 (2005) 185 34 - A. Julbe, D. Farrusseng, C. Guizard, Catal. Today 104 (2005) 102 35 - Y Wei, W. Yang, J. Caro, H. Wang, Chem. Eng. J. 220 (2013) 185 36 - International Conferences on Catalysis in Membrane Reactors, Cat Today, volumes 104 (2005), 118 (2006), 156 (2010), 193 (2012). 20 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(20) HDR de Axel Löfberg, Lille 1, 2013. Sans refaire une bibliographie exhaustive du sujet, il convient de souligner que les travaux sur l’utilisation de réacteurs à membrane dense en oxydation sélective peuvent être classés en deux groupes, ceux visant à la valorisation du méthane et ceux concernant des hydrocarbures plus complexes. En effet, en raison des températures de fonctionnement plutôt élevées nécessaires pour obtenir une conductivité ionique suffisante, beaucoup de travaux se sont focalisés sur la réactivité du méthane. Ainsi, le reformage oxydant du méthane en gaz de synthèse CO + H2 a été étudié notamment par Balachandran et al [37] depuis le milieu des années 90. Il a depuis été l’objet de nombreux travaux en particulier en vue de l’intégration de cette réaction comme source d’hydrogène pour des piles à combustible (internal reforming). L’utilisation du CO2 comme oxydant (reformage sec) a également été envisagé plus récemment, par exemple par Slade et al [38]. Il convient toutefois d’être vigilant car compte tenu des températures en jeu (>800~1000°C) et de la thermodynamique des réactions de reformage, il n’est pas évident que l’on soit réellement en présence de découplage rédox au sens strict. En effet, l’oxydation totale du méthane dans le compartiment à basse pression partielle d’oxygène produit CO2 et H2O qui peuvent être responsables du reformage sec et/ou du vaporeformage du méthane non réagi. D’ailleurs, Cheng et al [39] proposent un procédé dans lequel ces deux étapes sont explicitement exploités. Le couplage oxydant constitue la seconde voie de valorisation du méthane envisageable dans un RCMD qui a été explorée notamment par Van Veen et al [40, 41] ainsi que Zeng et al [42], et Bhatia et al [43], par exemple. La principale difficulté de cette réaction réside dans le délicat compromis à trouver entre les vitesses de transfert d’oxygène au travers de la membrane, les réactions de surface et les réactions radicalaires afin d’obtenir une réactivité suffisante sans désactivation notamment par cokage. A noter également que faisant suite aux développements des piles à combustible, des procédés originaux combinant une valorisation du méthane (par exemple par reformage) et la production d’énergie par courant électrique ont également été proposés, par exemple par Semin et al [44]. C’est dans la transformation d’hydrocarbures plus longs que l’application du principe de découplage rédox du mécanisme de Van Krevelen a son plus fort potentiel, suite aux premiers travaux de Grasseli et al [30]. Wang et al ont comparé les performances d’un réacteur à membrane dense à base de Ba0.5Sr0.5Co0.8Fe0.2O3-δ (BSCF) et celles de réacteurs. 37 - U. Balachandran, J.T. Dusek, R.L. Mieville, R.B. Poeppel, M.S. Kleefisch, S. Pei, T.P. Kobylinski, C.A. Udovich, A.C. Bose, Appl. Catal. A 133 (1995) 19 38 - D.A. Slade, Q. Jiang, K.J. Nordheden, S.M. Stagg-Williams, Catal. Today 148 (2009) 290 39 - C.-S. Chen, S.-J. Feng, S. Ran, D.-C. Zhu, W. Liu, H.J.M. Bouwmeester, Angew. Chem. Int. Ed. 2003, 42, 5196 40 - S. Haag, A.C. van Veen, C. Mirodatos, Catal. Today 127 (2007) 157 41 - L. Olivier, S. Haag, C. Mirodatos, A.C. van Veen, Catal. Today 142 (2009) 34 42 - Y. Zeng, Y. S. Lin, J. Catal. 193 (2000) 58 43 - S. Bhatia, C. Yen Thien, A.R. Mohamed, Chem. Eng. J. 148 (2009) 525 44 - G.L. Semin, V.D. Belyaev, A.K. Demin, V.A. Sobyanin, Appl. Catal. A: Gen. 181 (1999) 131. 21 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
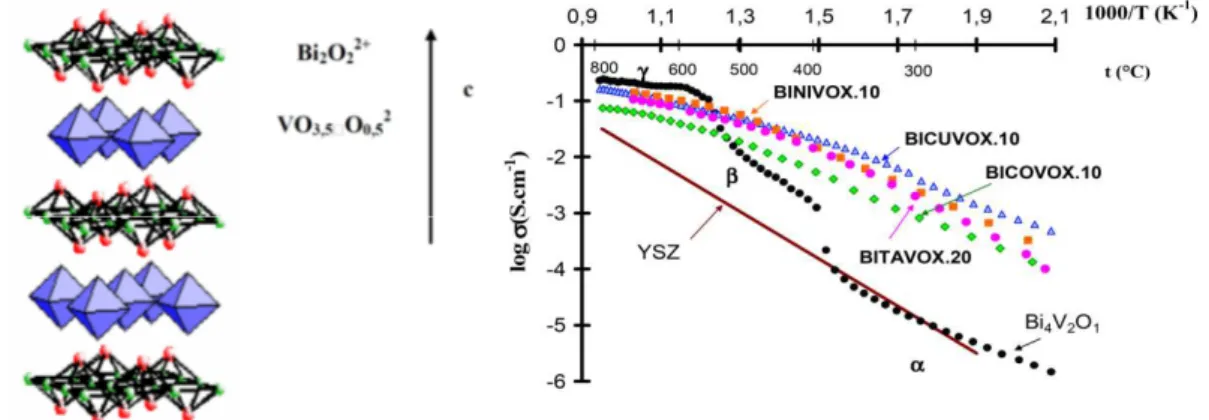
(21) HDR de Axel Löfberg, Lille 1, 2013. co-alimenté et pulsés pour la réaction de déshydrogénation oxydante (ODH) de l’éthane [45, 46] et du propane [47] et ont ainsi démontré l’intérêt du RCMD pour cette réaction. Rebeillau et al ont également étudié la réaction d’ODH de l’éthane sur des matériaux de membrane similaire, mais leur approche diffère dans la mesure où la face en contact avec l’hydrocarbure est modifiée par l’ajour d’un catalyseur (cluster de Pd ou V/MgO) [48, 49]. Si la présence d’un catalyseur améliore effectivement les performances d’un tel système, la question demeure de savoir si les espèces oxygènes responsables de l’ODH proviennent 2-. effectivement du transfert des espèces O depuis la membrane vers la phase active ou si une étape de recombinaison en O2 suivie de la réadsorption sur la surface du catalyseur a lieu. Dans le second cas, on assisterait plutôt à une distribution contrôlée de O2 moléculaire qu’à un véritable découplage rédox. Une autre possibilité à envisager serait que la réaction procède par une déshydrogénation simple dont l’équilibre serait favorisé par l’oxydation de H2 en H2O successivement comme l’ont proposé Czuprat et al [50]. Notre approche RCMD a consisté avant tout à privilégier dans un premier temps des systèmes utilisant le matériau de membrane directement comme catalyseur. Ensuite nous avons développé des systèmes composites comportant des surfaces poreuses mais composées du même matériau que la membrane dense afin de favoriser la migration de l’oxygène ionique vers la surface du catalyseur. Ces travaux ont été réalisés sur deux familles de matériaux, les BIMEVOX et les BIMO, et sont décrits dans la suite de ce chapitre.. II.2 BIMEVOX De nombreux conducteurs par ions oxyde ont été découverts et étudiés au sein de l’équipe « Chimie du Solide » de l’UCCS (anciennement Laboratoire de Cristallochimie et Physicochimie du Solide, LCPS). A la fin des années 80, une nouvelle famille présentant d’excellentes propriétés de conduction ionique par oxygène à basse température, les BIMEVOX [51, 52], a été mise en évidence. Ces matériaux dérivent du composé parent Bi4V2O11 (Figure 5a). Ils sont obtenus par substitution partielle du vanadium par un métal. On obtient donc des composés de formule Bi2V1-xMexOy. BICUVOX.10 par exemple est l’acronyme correspondant au remplacement de 10% de vanadium par du cuivre (+II) dans la structure, soit Bi2V0,9Cu0,1O5,35. Cette phase présente à 300°C une conductivité comparable à celle de la zircone à 700°C. Ces matériaux 45 - H.H. Wang, Y. Cong, W.S. Yang, Chem. Comm. 14 (2002) 1468 46 - H.H. Wang, Y. Cong, W.S. Yang, Catal. Lett. 84 (2002) 101 47 - H.H. Wang, Y. Cong, X.F. Zhu, W.S. Yang, React. Kinet. Catal. Lett. 79 (2003) 351 48 - M. Rebeilleau, A.C. van Veen, D. Farrusseng, J. Rousset, C. Mirodatos, Z.P. Shao, G.X. Xiong, Stud. Surf. Sci. Catal. 147 (2004) 655 49 - M. Rebeilleau, S. Rosini, A.C. van Veen, D. Farrusseng, C. Mirodatos, Catal. Today 104 (2005) 131 50 - O. Czuprat, S. Werth, T. Schiestel, J. Caro, AIChE J. 56 (2010) 2390 51 - F. Abraham, J.C. Boivin, G. Mairesse, G. Nowogrocki, Solid State Ionics 40/41(1991) 934 52 - J.C Boivin, G. Mairesse, Chem. Mater. 10 (1998) 2870. 22 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(22) HDR de Axel Löfberg, Lille 1, 2013. sont encore reconnus comme les meilleurs conducteurs par ions oxyde à température modérée 300-600°C, et leur découverte a contribué à la reconnaissance internationale du LCPS. La structure cristalline des BIMEVOX est isotype de celle de γ-Bi4V2O11. Elle repose sur 2+. 2-. des feuillets bismuthyles Bi2O2 , qui alternent avec des couches d’octaèdre VO3,5. lacunaires en oxygène. La présence des lacunes d’oxygène dans les feuillets permet la diffusion des ions oxyde et la conductivité qui en résulte est bidimensionnelle. Les BIMEVOX sont des conducteurs essentiellement ioniques mais certains présentent également une faible conductivité électronique de type p (par exemple BICOVOX) ou n (BICUVOX).. Figure 5 – Conductivités des BIMEVOX comparées à celles de Bi4V2O11 et de YSZ (zircone dopée à l’yttrium). Leurs conductivités sont comparées à celle du composé parent, Bi4V2O11 et à celle de la zircone stabilisée à l’yttrium (Figure 5b). Les propriétés de BICOVOX et BITAVOX sont comparables et ils sont cent fois plus conducteurs que la zircone yttriée à 600°C (matériaux de référence dans les piles à combustibles). BICUVOX présente des propriétés de conduction encore meilleures que les autres. Leurs propriétés catalytiques en tant que matériaux pulvérulents pour l’oxydation du propène en réacteur traditionnel ont fait l'objet d'une thèse de doctorat à l'Université des Technologies de Compiègne (A. Chetouani – 2003 – UTC). II.2.1 Perméation et réactivité de membranes polies Les premiers travaux au Laboratoire sur l’utilisation de ces matériaux remontent à 2002, époque où un nouveau réacteur catalytique a été conçu et élaboré dans le cadre du DEA de Samir Benaissa [53]. Celui-ci permettait de mesurer les caractéristiques de semiperméabilité à l'oxygène sur des membranes denses au moyen d'une jauge électrochimique à oxygène ainsi que l'activité catalytique pour la conversion du propène par analyse en ligne par spectrométrie de masse quadripolaire. Outre la mise au point de. 53 - S. Benaissa – DEA UTC Septembre 2002. 23 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
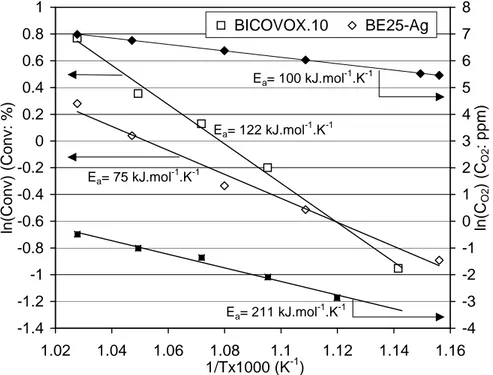
(23) HDR de Axel Löfberg, Lille 1, 2013. l’appareillage, M. Benaissa a comparé le comportement de membranes de type BiCoVOx, avec celles composéés d’oxydes mixtes de bismuth et d’erbium (Bi2O3-Er2O3) et de cermet (Bi2O3-Er2O3 avec ajout de 40% d’Ag). Des améliorations de l’installation ont été ensuite réalisées lors du stage de DEA de M. Salaheddine Boujmiai [54] en 2003 avec, notamment, l’ajout de l ‘équipement nécessaire au suivi de la polarisation de la surface sous l’effet des différentes atmosphères en contact avec les deux faces de la membrane (Figure 6). Ar or He + C3H6. TC. Gold tip (WE). Pyrex seal Membrane. Mullite tubes Alumina tube. N2. O 2 + N2 TC. Gold grid (CE). Figure 6 - Schéma de principe du réacteur catalytique à membrane dense.. 2-. Malgré leurs propriétés conductrices par ion O exceptionnelles, les flux de perméation de O2 sont très faibles sur les matériaux BiMeVOx (Me = Cu ou Co). Des travaux effectués au LCPS par échange isotopique. 18. 16. O/ O couplé à la spectroscopie SIMS ont montré [55] 2-. que l’étape de recombinaison des ions O en O2 (2O. 2-. -. → O2 + 2e ) du côté basse pression. d’oxygène (BPO) de la membrane est l’étape limitante du processus de perméation.. 54 - S. Boujmiai – DEA USTL Juillet 2003 55 - R.N. Vannier, S.J. Skinner, R.J. Chater, J.A. Kilner, G. Mairesse, Solid State Ion. 160 (2003) 85. 24 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(24) HDR de Axel Löfberg, Lille 1, 2013. 1. 8. BICOVOX.10. 0.8. BE25-Ag. 7. 0.6. 6 Ea= 100 kJ.mol-1.K-1. 0.2. 4. Ea= 122 kJ.mol-1.K-1. 0 -0.2. 5. 3 2. Ea= 75 kJ.mol-1.K-1. ln(CO2) (CO2: ppm). ln(Conv) (Conv: %). 0.4. -0.4. 1. -0.6. 0. -0.8. -1. -1. -2. -1.2 -1.4 1.02. -3. Ea= 211 kJ.mol-1.K-1. 1.04. 1.06. 1.08 1.1 1/Tx1000 (K-1). 1.12. 1.14. -4 1.16. Figure 7 - Conversion du propène et flux de perméation de O2 sur BiCoVOx et Bi2O3-Er2O3 40% Ag (BE25-Ag). « HPO » : air ; « BPO » : 1% C3H6 dans He (100cc/min) [56]. Avec BiCoVOx et BiCuVOx, les conversions sont de l’ordre de 1 à 2 % dans le domaine de température exploré (600-700°C). Ces valeurs sont évidemment très faibles mais résultent du polissage « miroir » de la membrane qui limite le nombre de site actifs. Ce polissage (effectué au papier SiC "4000") était, à ce stade, nécessaire à une bonne caractérisation de la perméabilité et de la polarisation des membranes. Néanmoins, dans les conditions opératoires utilisées, une conversion de 1 % du propène (soit 100 ppm) nécessite une quantité d’oxygène équivalant à 300 ppm de O2 (pour une transformation en 3CO + 3H2O). Or, dans les mêmes conditions mais en absence de propène, la concentration de O2 dans le compartiment BPO résultant de la perméation est inférieure à 10 ppm. D’ailleurs, les conversions de propène mesurées pour des membranes présentant des flux de perméation élevés (Cermet Bi2O3-Er2O3 / Ag, noté BE25-Ag) sont du même ordre de grandeur que pour BiCuVOx ou BiCoVOx (Figure 7). Il apparait donc clairement que la réaction avec l’hydrocarbure fournit une voie 2-. 2-. alternative à l’extraction d’O de la surface du catalyseur, et que le flux global de O diffusant au travers de la membrane est accéléré en présence de propène. De bonnes performances en termes de perméation de O2 ne sont donc pas requises pour une utilisation du matériau pour la catalyse pourvu que cette perméation soit limitée par des phénomènes de surface et non par la diffusion ionique dans le solide.. 56 - A. Löfberg, S. Boujmiai, E. Capoen, M.C. Steil, C. Pirovano, R.N. Vannier, G. Mairesse, E. Bordes-Richard, Catal. Today 91-92 (2004) 79. 25 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
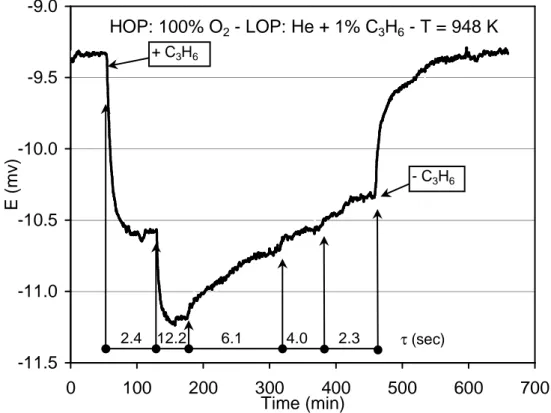
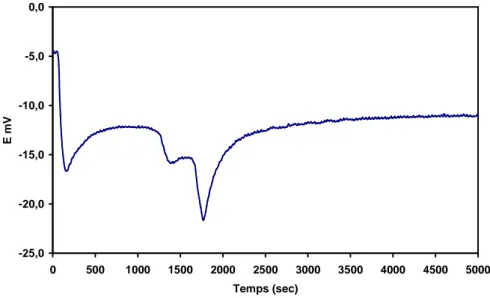
(25) HDR de Axel Löfberg, Lille 1, 2013. D’ailleurs, lors du suivi de la polarisation de la membrane au cours du test catalytique (Figure 8) on constate que la différence de potentiel (∆E) mesurée entre les deux faces de la membrane est faible en absence de propène. La relation de Nernst permet de calculer le ∆E théorique que l’on devrait mesurer compte tenu des pressions partielles d’oxygène auxquelles sont exposées les deux faces de la membrane : BPO R.T pO2 ln ∆E = 4.F pOHPO 2 . Avec le compartiment haute pression d’oxygène (HPO) alimenté avec de l’oxygène pur et le compartiment BPO contenant environ 3ppm de O2, le ∆E théorique est de -295.5 mV. Ceci indique donc que, dans les conditions utilisées, la surface de la membrane du côté BPO est loin d’être en équilibre avec la phase gazeuse environnante. Inversement, avec cette même équation de Nernst, nous pouvons calculer l’activité thermodynamique de l’oxygène du côté BPO correspondante aux polarisations mesurées. Exprimée en terme de pression de O2, une différence de potentiel de –9.5 mV correspondrait à une pression d’équilibre de O2 de 0.6 atm. Cette valeur est proche de la pression appliquée du côté HPO (1 atm) et reflète l’important déséquilibre de la face BPO avec l’atmosphère environnante due au blocage de l’oxygène à la surface (surtension).. -9.0. HOP: 100% O2 - LOP: He + 1% C3H6 - T = 948 K + C3H6. -9.5. E (mv). -10.0 - C3H6. -10.5. -11.0 2.4 12.2. 6.1. 4.0. 2.3. τ (sec). -11.5 0. 100. 200. 300 400 Time (min). 500. 600. 700. Figure 8 – Evolution de la différence de potentiel mesurée entre les deux faces de la membrane BiCoVOx en absence, puis en présence de propène, ainsi qu’au cours de la variation du temps de contact [56].. 26 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
(26) HDR de Axel Löfberg, Lille 1, 2013. La différence de potentiel diminue (ainsi que la polarisation) lorsque le propène est introduit. Ce phénomène est accentué si l’on diminue le débit de gaz dans le compartiment BPO, et donc en augmentant le temps de séjour et la conversion du propène. Inversement, la diminution du temps de séjour conduit à une augmentation de ∆E. Enfin, le phénomène est parfaitement réversible puisque les valeurs initiales de polarisation sont retrouvées lorsque l’on arrête le flux de réactif. Les mesures de polarisation montrent que l’activité thermodynamique de l’oxygène à la surface BPO est proche de celle de la face HPO à cause de la limitation cinétique de la 2-. recombinaison de O en O2. Il en découle qu’en faisant varier la pression partielle de O2 dans le compartiment HPO on peut modifier l’activité de l’oxygène du côté de la réaction catalytique, et donc espérer modifier les propriétés catalytiques du matériau notamment du point de vue de la sélectivité. De fait, la Figure 9 montre qu’en baissant la pression de O2 dans le comportement HPO, la sélectivité en produits d’oxydation partielle est effectivement améliorée au détriment de la production de CO. Les conversions obtenues sont faibles (de l’ordre de 1-3%) à cause du polissage de la membrane, et les sélectivités restent largement en faveur de l’oxydation totale. Néanmoins, l’utilisation de ces matériaux catalytiques dans un réacteur à membrane est envisageable puisque, contrairement à ce qui pouvait être craint, leur forte polarisation empêche leur réduction au contact de l’hydrocarbure. La perspective de pouvoir modifier l’activité thermodynamique d’un des réactifs à la surface du catalyseur, pratiquement en absence de ce réactif dans la phase gazeuse, est également particulièrement intéressante.. Hexadiene. Benzene. Acrolein. CO. 16. 100. 14. 90 80. Selectivity (%). 12. 70. 10. 60. 8. 50. 6. 40 30. 4. 20. 2. 10. 0 0.001. Selectivity in CO (%). CO2. 0 0.01 0.1 O2 partial pressure (atm). 1. Figure 9 - Evolution de la sélectivité pour l’oxydation du propène en fonction de la pression d’O2 du côté HPO, Membrane BiCuVOx, T=675°C, pC3H6=1%patm, Conversion = 0.6-0.7%. 27 © 2014 Tous droits réservés.. doc.univ-lille1.fr.
Figure
![Figure 3 – Vision de l’évolution de l’industrie de transformation (SPIRE Roadmap [11])](https://thumb-eu.123doks.com/thumbv2/123doknet/3524381.103129/11.892.198.730.162.550/figure-vision-l-évolution-industrie-transformation-spire-roadmap.webp)
+7

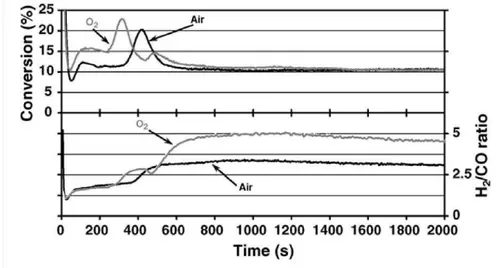
![Figure 10 - Conversion et produits de réaction de l’oxydation du propène sur membrane BiCuVOx dépolie (700°C, pC 3 H 6 =1%p atm introduit à t = 480s, 50cc/min) [57]](https://thumb-eu.123doks.com/thumbv2/123doknet/3524381.103129/27.892.159.747.690.1007/conversion-produits-réaction-oxydation-propène-bicuvox-dépolie-introduit.webp)
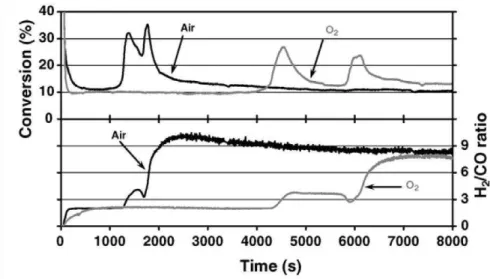
![Figure 19 - Pressions de CO lors de l'oxydation de C 2 H 6 et de C 3 H 8 sur membrane BiTaVOx dépolies (T = 700°C, pC 3 H 8 = 1%p atm dans He, D = 50cc/min, HPO = air) [59]](https://thumb-eu.123doks.com/thumbv2/123doknet/3524381.103129/34.892.215.709.492.763/figure-pressions-co-oxydation-membrane-bitavox-dépolies-hpo.webp)
Documents relatifs
L’objet de ce chapitre entre dans le cadre global de la lutte contre la pollution causée par les effluents spécifiques chargés en phosphates présents dans les eaux
Dans le contrôle DTC classique, il ya deux vecteurs par secteur qui présente une ambiguïté dans le contrôle de couple, donc (V i et V i+3 ) ne sont pas utilisés
Par contre il n’existe que des modestes indications sur les courbes de remous et de dépression, et le ressaut hydraulique dans cette section en forme ‘‘U’’ n’a pas fait
Le volume de contrôle de type rectangulaire nous permet de modéliser les phénomènes électromagnétiques que pour des géométrique simples, elle consiste à
Cette partie de l’étude vise à établir une relation approchée explicite au calcul du taux de remplissage de la conduite, impliquant ainsi celui de la
ℎ , = ℎ = 5.67 + 3.86 ⋅ (II-36) II.4.3.2- Echanges convectifs entre l’absorbeur et la vitre : Le transfert thermique dans l’espace compris entre la vitre et
Les produits de départ sont pesés dans les proportions désirées, broyés intimement pour permettre une homogénéisation et placés dans le creuset, ce dernier est chauffé à
Par ailleurs, il est intéressant de noter que les résultats que nous avons obtenus au cours de la chloration du phénol et du résorcinol dans les différents milieux de dilutions
