NIPBL et le complexe cohésine lient
l’organisation 3D des gènes à la régulation
transcriptionnelle
Thèse
Imène Boudaoud
Doctorat en biologie cellulaire et moléculaire
Philosophiae Doctor (Ph.D.)
Québec, Canada
NIPBL et le complexe cohésine lient
l’organisation 3D des gènes à la régulation
transcriptionnelle
Thèse
Imène Boudaoud
Sous la direction de :
Steve Bilodeau, directeur de recherche
Résumé
En réponse à des signaux environnementaux, la cellule module son programme transcriptionnel afin de mener à une expression spatio-temporelle adéquate des gènes. L’orchestration d’une telle adaptation repose entre autres sur la séquence primaire du génome, son organisation au sein de la chromatine, ainsi que sa structure tridimensionnelle au sein du noyau. De plus, de nombreux régulateurs permettent d’intégrer ces différents niveaux de régulation afin de contrôler l’activité de l’ARN polymérase II. Dans ce contexte, le complexe cohésine et son facteur de charge sur l’ADN, NIPBL, jouent un rôle clé dans l’interconnexion fonctionnelle entre l’organisation 3D du génome et la transcription. En effet, ces facteurs modulent l’activation de la transcription en rapprochant des régions enhancers de promoteurs et participent à la formation de domaines d’interactions chromosomiques. Par ailleurs, des mutations de NIPBL et du complexe cohésine sont associées au Syndrome de Cornelia de Lange (CdLS), une pathologie caractérisée par une altération de l’expression des gènes. Toutefois, les mécanismes moléculaires impliqués dans la régulation de la transcription par NIPBL et cohésine sont encore méconnus. L’objectif général de mon projet de doctorat est de définir le rôle de NIPBL et du complexe cohésine dans la régulation du lien entre la topologie du génome et le contrôle de l’expression des gènes. Dans un premier temps, nous montrons que les gènes dérégulés dans le CdLS sont préférentiellement organisés au sein de communautés de gènes, des structures formées par des interactions d’éléments régulateurs non codants ainsi que de gènes dans l’espace chromosomique tridimensionnel. Au sein de cette organisation, les gènes affectés par des mutations de NIPBL ou de la sous-unité SMC1A du complexe cohésine sont retrouvés positionnés à portée de régions occupées par cohésine et NIPBL et interagissent par l’intermédiaire de contacts promoteur-promoteur. Dans un second temps, nous présentons des données suggérant un rôle de cohésine dans la régulation de l’initiation de la transcription et un rôle de NIPBL dans le contrôle de la relâche de la pause. Enfin, nous apportons des évidences d’une fonction de NIPBL et cohésine dans la régulation du niveau basal et de l’activation des gènes dont l’expression est stimulée par des hormones. Dans leur ensemble, ces travaux contribuent à l’amélioration des connaissances sur la contribution de l’architecture des chromosomes aux mécanismes généraux de la régulation de la transcription.
Abstract
In response to environmental signals, the cell modulates its transcriptional program in order to carry out appropriate spatiotemporal gene expression. The orchestration of this adaptation relies on the primary sequence of the genome, its organization into chromatin, and its tridimensional structure inside the nucleus. Moreover, multiple regulators integrate these different regulation levels in order to control the activity of RNA polymerase II. In this context, the cohesin complex and its DNA loader, NIPBL, play a pivotal role in the functional interconnection between the 3D organization of the genome and transcription. Indeed, these factors modulate the activation of transcription by bringing enhancers and promoters into close proximity and participate in the formation of chromosome interaction domains. Moreover, mutations in NIPBL and the cohesin complex are associated with the Cornelia de Lange Syndrome (CdLS), a pathology characterized by gene expression changes. However, the exact molecular mechanisms involved in the regulation of transcription by NIPBL and cohesin are still not understood. The general aim of my doctoral research is to define the role of the cohesin complex and NIPBL in the regulation of the connection between genome topology and gene expression control. First, we show that genes deregulated in CdLS are preferentially organized into connected gene communities, structures emerging from the interactions of noncoding regulatory elements and genes in the three-dimensional chromosomal space. Within this organization, genes affected by mutations in NIPBL and the SMC1A subunit of the cohesin complex are positioned within reach of NIPBL- and cohesin-occupied regions through promoter- promoter interactions. In addition, we present data suggesting a role of the cohesin complex in the initiation of transcription and a role of NIPBL in the control of pause release. Finally, we show evidence of a function of NIPBL and cohesin in the regulation of the basal level and the activation of genes stimulated by hormones. Ultimately, this work aims to gain insight into the contribution of the architecture of chromosomes to the general mechanisms of transcriptional regulation.
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des tableaux ... ix
Liste des figures ... x
Abréviations ... xii
Épigraphe ... xvi
Remerciements ... xvii
Avant-‐propos ... xix
CHAPITRE 1 : Introduction ... 1
1.1
Régulation de la transcription par l’ARN polymérase II ... 1
1.1.1 Les éléments régulateurs de la transcription ... 2
1.1.1.1
Le promoteur ... 2
1.1.1.2
Les séquences amplificatrices distales ou enhancers ... 4
1.1.1.3
Les autres éléments régulateurs distaux ... 6
1.1.2 La machinerie transcriptionnelle eucaryote ... 8
1.1.2.1
Les facteurs généraux de la transcription ... 9
1.1.2.2
Les activateurs ... 10
1.1.2.3
Les coactivateurs ... 11
1.1.3 Les étapes de la transcription par l’ARN polymérase II ... 14
1.1.3.1
Le CTD de l’ARN polymérase II ... 15
1.1.3.2
L’initiation ... 16
1.1.3.3
L’élongation ... 18
1.1.3.4
La terminaison ... 23
1.1.4 La répression de la transcription ... 26
1.2
La chromatine ... 27
1.2.1 Organisation de la chromatine ... 27
1.2.1.1
Différents niveaux de compaction de la chromatine ... 27
1.2.1.2
L’unité structurale de la chromatine : le nucléosome ... 28
1.2.1.3
Organisation fonctionnelle de la chromatine ... 30
1.2.2 Hétérogénéité structurale de la chromatine ... 31
1.2.2.1
Les modifications post-‐traductionnelles des histones ... 31
1.2.2.2
Les modifications de l’ADN ... 34
1.2.2.3
Les variants d’histones ... 36
1.2.3 Régulation de la dynamique de la chromatine ... 38
1.2.3.1
Les enzymes catalysant les modifications post-‐traductionnelles des histones .... 38
1.2.3.2
Les complexes de remodelage de la chromatine ATP-‐dépendants ... 42
1.2.3.3
Les chaperons d’histone ... 45
1.2.4 Activation de la transcription dans un contexte chromatinien ... 46
1.3.1 Niveaux supérieurs de l’organisation chromatinienne ... 48
1.3.1.1
La chromatine interphasique est organisée de façon hiérarchique ... 48
1.3.1.2
Étude de l’organisation tridimensionnelle du génome ... 52
1.3.2 Les protéines impliquées dans le repliement du génome ... 56
1.3.2.1
La protéine architecturale CTCF ... 56
1.3.2.2
Le complexe cohésine ... 59
1.3.2.3
Autres protéines architecturales ... 66
1.3.3 Lien entre l’organisation 3D du génome et l’activité transcriptionnelle ... 69
1.3.3.1
L’inactivation du chromosome X ... 70
1.3.3.2
Colocalisation de gènes corégulés dans le noyau ... 71
1.3.3.3
La mutation de protéines architecturales ou la perturbation de leur liaison à l’ADN mènent à une dérégulation de la transcription ... 73
1.4
Objectifs de travail ... 75
CHAPITRE 2 : Connected gene communities underlie transcriptional changes in Cornelia de Lange Syndrome ... 77
2.1
Avant-‐propos ... 78
2.2
Résumé ... 79
2.3
Abstract ... 80
2.4
Introduction ... 81
2.5
Materials and methods ... 83
2.5.1 Gene expression datasets ... 83
2.5.2 Cell culture ... 83
2.5.3 ChIP-‐Seq ... 83
2.5.4 Overlap with genomic features ... 84
2.5.5 Annotation of interaction points ... 84
2.5.6 Identification of connected gene communities ... 85
2.5.7 Enrichment within connected gene communities ... 86
2.5.8 Coherency within a connected gene community ... 86
2.5.9 Proximity of NIPBL-‐ and SMC1A-‐occupied nodes to CdLS-‐misregulated genes ... 86
2.5.10 Data availability ... 87
2.6
Results ... 87
2.6.1 Variable occupancy of NIPBL and cohesin at CdLS-‐deregulated genes ... 87
2.6.2 NIPBL and cohesin are constituents of noncoding regulatory regions within connected gene communities ... 88
2.6.3 NIPBL and cohesin are central to connected gene communities ... 89
2.6.4 Gene communities connect deregulated genes in CdLS ... 90
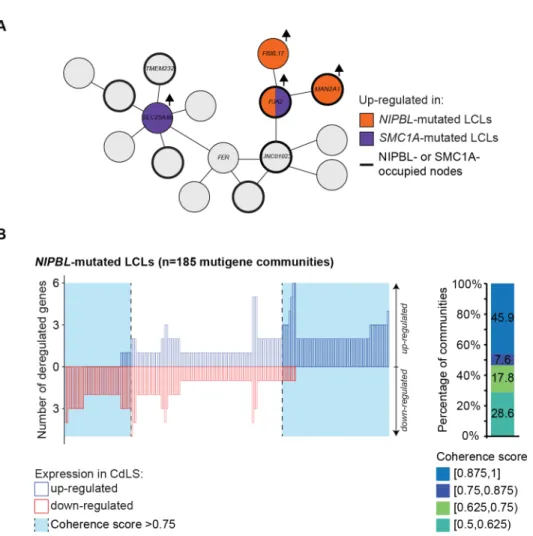
2.6.5 NIPBL mutations lead to coordinated gene expression changes within communities .... 91
2.7
Discussion ... 92
2.8
Figures ... 95
2.9
Supplementary figures ... 103
2.10
Supplementary tables ... 108
CHAPITRE 3 : NIPBL et cohésine régulent des étapes précoces de la transcription dans les cellules souches embryonnaires de souris ... 109
3.1
Avant-‐propos ... 109
3.2
Résumé ... 110
3.3
Introduction ... 111
3.4.1 La perte de fonction du complexe cohésine affecte les propriétés clés des ESCs ... 114
3.4.2 NIPBL et cohésine régulent différentiellement la machinerie transcriptionnelle ... 115
3.4.3 La perte de fonction de NIPBL, cohésine et Médiateur affecte l’enrichissement de l’ARN Pol II au niveau des TSSs ... 117
3.4.4 NIPBL et cohésine régulent la transcription à différentes étapes ... 118
3.5
Discussion ... 120
3.6
Figures ... 124
3.7
Matériel et méthodes ... 128
3.7.1 Culture cellulaire ... 128
3.7.2 Perte de fonction par transduction lentivirale ... 128
3.7.3 Immunoprécipitation d’extraits nucléaires ... 129
3.7.4 ChIP-‐qPCR et ChIP-‐Seq ... 129
3.7.5 Extraction d’ARN et RT-‐qPCR ... 129
3.7.6 Analyses bio-‐informatiques ... 130
3.7.7 Fractionnement cellulaire ... 130
3.7.8 Extraction des protéines totales ... 131
3.7.9 Liste des anticorps utilisés ... 131
3.7.10 Différenciation à l’acide rétinoïque ... 132
3.7.11 Coloration de l'activité de la phosphatase alcaline ... 132
CHAPITRE 4 : NIPBL et cohésine régulent la réponse transcriptionnelle des récepteurs nucléaires ... 133
4.1
Avant propos ... 133
4.2
Résumé ... 134
4.3
Introduction ... 135
4.4
Résultats ... 136
4.4.1 NIPBL régule la réponse transcriptionnelle associée au récepteur aux estrogènes ... 136
4.4.2 NIPBL et le complexe cohésine sont recrutés en réponse à une stimulation aux glucocorticoïdes ... 138
4.4.3 NIPBL et le complexe cohésine régulent l’expression des gènes de réponse aux glucocorticoïdes ... 139
4.4.4 La perte de fonction de TBLR1 affecte la dynamique d’interaction entre NIPBL et GR 140
4.5
Discussion ... 141
4.6
Figures ... 144
4.7
Matériel et méthodes ... 147
4.7.1 Culture cellulaire et traitements hormonaux ... 147
4.7.2 Perte de fonction par transduction lentivirale ... 148
4.7.3 Extraction d’ARN, RT-‐qPCR et RNA-‐Seq ... 148
4.7.4 ChIP-‐Seq ... 149
4.7.5 Analyses bioinformatiques ... 149
4.7.6 Fractionnement cellulaire et immunoprécipitation ... 149
4.7.7 Liste des anticorps utilisés ... 150
CHAPITRE 5 : Discussion ... 151
5.1
L’analyse de l’architecture tridimensionnelle du génome permet de mieux comprendre des défauts de régulation transcriptionnelle ... 151
5.2
Qu’est-‐ce qu’une communauté de gènes ? ... 154
5.3
Deux mécanismes différents mènent au repliement du génome ... 157
5.5
Contribution de l’architecture chromatinienne contrôlée par le complexe cohésine à la régulation de la transcription ... 161
Bibliographie ... 165
Annexes ... 196
Liste des tableaux
Table 2.S1. Uniform gene symbols for genes misregulated in NIPBL-mutated CdLS
probands.
Table 2.S2. Uniform gene symbols for genes misregulated in SMC1A-mutated CdLS
probands.
Table 2.S3. List of publicly available datasets used throughout the manuscript. Table 2.S4. Proportion of genes with a NIPBL- or SMC1A-occupied region. Table 2.S5. Connected gene communities in GM12878 cells.
Table 2.S6. Nature of NIPBL and SMC1A occupied regions in mutated cells.
Liste des figures
CHAPITRE 1
Figure 1.1. Les régions régulatrices de la transcription. Figure 1.2. Les éléments composant le promoteur minimal.
Figure 1.3. Représentation schématique du mécanisme d’action des séquences régulatrices distales.
Figure 1.4. La machinerie transcriptionnelle eucaryote. Figure 1.5. Les différentes formes du complexe Médiateur. Figure 1.6. Les étapes de la transcription par l’ARN Pol II.
Figure 1.7. Patrons de phosphorylation du CTD de l’ARN Pol II au cours de la transcription.
Figure 1.8. Établissement et relâche de la pause de l’ARN Pol II.
Figure 1.9. Les complexes contenant P-TEFb.
Figure 1.10. Terminaison de la transcription dépendante du signal de polyadénylation. Figure 1.11. Les différents niveaux de compaction de la chromatine.
Figure 1.12. Le nucléosome.
Figure 1.13. Les modifications post-traductionnelles des histones. Figure 1.14. Exemples de rôles de la méthylation.
Figure 1.15. Les enzymes régulant la dynamique des principales méthylations de lysine
des histones H3 et H4.
Figure 1.16. Le remodelage ATP-dépendant de la chromatine.
Figure 1.17. La chromatine interphasique est organisée de façon hiérarchique. Figure 1.18. Les techniques d’étude de la conformation des chromosomes. Figure 1.19. La protéine architecturale CTCF.
Figure 1.20. Le complexe cohésine.
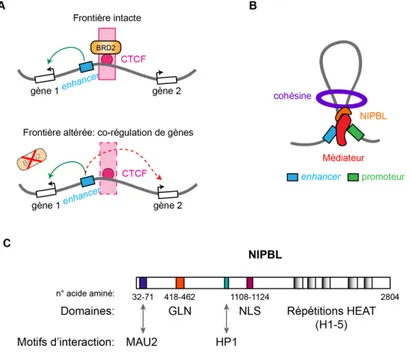
Figure 1.21. Dynamique du complexe cohésine au cours du cycle cellulaire. Figure 1.22. Modèle de l’ARN Pol II comme médiateur de l’extrusion de boucles. Figure 1.23. BRD2, Médiateur et NIPBL sont des protéines architecturales. Figure 1.24. Représentation schématique d’une communauté de gènes.
CHAPITRE 2
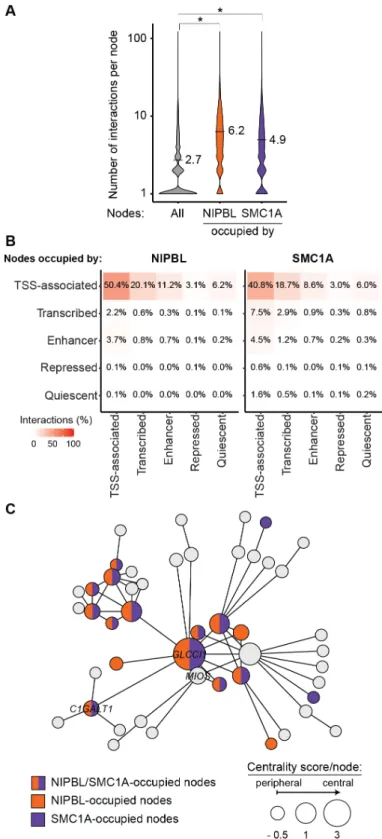
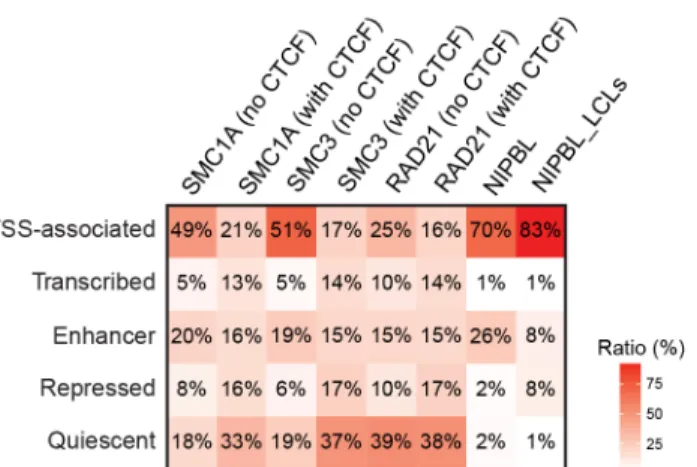
Figure 2.1. NIPBL and cohesin occupy a fraction of CdLS-deregulated genes.
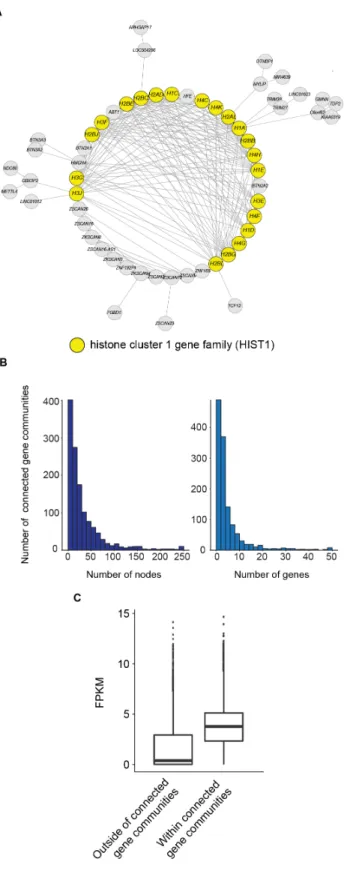
Figure 2.2. Active noncoding regulatory regions are occupied by NIPBL and cohesin within connected gene communities.
Figure 2.3. Nodes occupied by NIPBL and cohesin create more interactions.
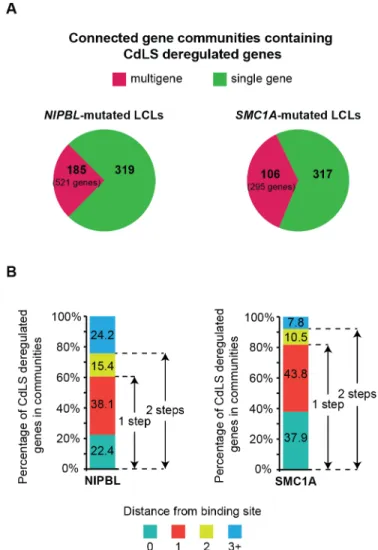
Figure 2.4. Deregulated genes in CdLS are within reach of NIPBL- and cohesin-
occupied regions.
Figure 2.5. Coordinated deregulation of gene expression is associated with NIPBL
mutations.
Figure 2.S1. Comparison of the genome-wide occupancy between NIPBL and cohesin
subunits ChIP-Seq datasets.
Figure 2.S2. Characterization of connected gene communities in normal lymphoblastoid
cells.
Figure 2.S3. Identification of the central nodes within connected gene communities. Figure 2.S4. Promoter Capture Hi-C-defined connected gene communities identify
CHAPITRE 3
Figure 3.1. Les pertes de fonction de NIPBL et SMC1A affectent différentiellement l’état cellulaire des ESCs.
Figure 3.2. NIPBL et SMC1A régulent différentiellement la machinerie transcriptionnelle.
Figure 3.3. La perte de fonction de NIPBL, cohésine et Médiateur affecte la liaison de l’ARN Pol II au niveau génomique.
Figure 3.4. Effet de la perte de fonction de NIPBL et SMC1A au niveau des promoteurs
liés par NIPBL et cohésine.
CHAPITRE 4
Figure 4.1. NIPBL est impliqué dans la régulation de la réponse au récepteur aux estrogènes.
Figure 4.2. NIPBL et cohésine sont recrutés aux sites occupés par GR en réponse à la dexamethasone.
Figure 4.3. NIPBL et cohésine régulent l’expression des gènes de réponse aux glucocorticoïdes.
Figure 4.4. La perte de fonction de TBLR1 affecte l’interaction entre GR et NIPBL.
Abréviations
2i Deux inhibiteurs de kinases
3C Chromosome conformation capture
3D Tridimensionnel
4C Chromosome conformation capture-on-chip 5C Carbon copy chromosome conformation capture 5hmC 5-hydroxymethylcytosine
5mC 5-methylcytosine
6mA N6-methyladenine
AD Activation domain
ADN Acide désoxyribonucléique
AF9 ALL1-fused gene from chromosome 9 AP Alkaline phosphatase
APC/C Anaphase promoting complex/cyclosome AR Androgen receptor
ARN Acide ribonucléique ARNlnc Long ARN non codant
ARNm ARN messager
ARNr ARN ribosomique
ARNsn Petit ARN nucléaire (sn : small nuclear) ARNsno Petit ARN nucléolaire (sno : small nucleolar) ARNt ARN de transfert
Asf1 Anti-silencing function 1 ATM Ataxia telangiectasia mutated
ATR Ataxia telangiectasia and RAD3-related BAF BRG1-associated factor
BRD4 Bromodomain-containing 4 BRE TFIIB recognition element BRG1 Brm-related gene 1
BRM Brahma homologue
C-terminal Carboxy-terminal
C. elegans Caenorhabditis elegans
CBS Cockayne syndrome B
CDK1 Cyclin-dependent kinase 1 CdLS Cornelia de Lange syndrome CenH3 Centromeric histone variant H3 CENP-A Centromere protein A
CFIA Cleavage factor IA
CHD Chromodomain-helicase-DNA binding
ChIP-Seq Chromatin immunoprecipitation coupled with massively parallel DNA sequencing
CoAT Cohesin acetyltransferase CpG Cytosine-phospho-guanine
CPSF Cleavage and polyadenylation specificty factor
CstF Cleavage stimulatory factor CTCF CCCTC-binding factor CTD Carboxy-terminal domain
DBD DNA-binding domain
DCE Downstream core element
Dex Dexamethasone
DNA-PK DNA-dependent protein kinase DNMT DNA methyltransferase
DOT1L DOT1-like
DPE Downstream promoter element
DRB 5,6-dichloro-1-b-D-ribofuranosyl-benzimidazole DSIF DRB-sensitivity inducing factor
ELL Eleven nineteen lysine rich leukemia ENL Eleven-nineteen leukemia
ER Estrogen receptor
eRNAs ARNs associés aux enhancers ESCs Embryonic stem cells
EtOH Éthanol
FISH Fluorescent in situ hybridization
GC Glucocorticoïdes
GFP Green fluorescent protein GNAT Gcn5 N-acteyltransferase GR Glucocorticoid receptor GRO-Seq Global run-on sequencing GTF General transcription factor
H Histone
HAT Histone acetyltransférase HDAC Histone déacetylase
HDM Histone démethylase
HEAT Huntingtin-elongation-A subunit-TOR
HEXIM Hexamethylene bisacetamide inducible protein Hi-C High throughput 3C
HIPMap High-throughput imaging position mapping HMT Histone methytransférase
HP1 Heterochromatin protein 1
HSA Helicase-SANT
ICR Imprinting control region IKK-α Iκb kinase-α
INO80 Inositol requiring 80 Inr Élément initiateur
IP Immunoprécipitation
ISWI Imitation switch
kb Kilo bases
KDM Lysine demethylase
KDM1A Lysine specific demethylase 1A KMT Lysine methyltransferase LARP7 La-related protein 7 LBD Ligand binding domain LCR Locus control region LIF Leukemia inhibitory factor LSD1 Lysine specific demethylase 1
m7G 7-methylguanosine
MBD Methyl-cpg-binding domain MBT Malignant brain tumor
MePCE Methyl phosphate capping enzyme
miARN Micro ARN
MPT Modification post-traductionnelle
MRD21 Autosomal dominant mental retardation 21 MSK Mitogen- and stress-activated kinase MTE Motif ten element
MYST MOZ/Morf, Ybf2, Sas2 et Tip60 N-terminal Amino-terminal
NAD Nicotinamide adenine dinucleotide Nap1 Nucleosome assembly protein 1 NASP Nuclear autoantigenic sperm protein NCoR Nuclear receptor corepressor
NDR Nucleosome-depleted region NE Nuclear extract
NELF Negative elongation factor complex NF-kB Nuclear factor-k B
NIPBL Nipped-B-like protein
NURD Nucleosome remodeling and deacetylase P-TEFb Positive transcription elongation factor b
pb Paire de bases
PBAF Polybromo BRG1-associated factor PDS5 Precocious dissociation of sisters PET Paired-end tags
PHD Plant homeodomain
PIC Pre-initiation complex PLK1 Polo-like kinase 1
Pol Polymérase
PP2A Protein phosphatase 2A
qPCR Quantitative polymerase chain reaction RA Retinoic acid
RBS Roberts syndrome
RITS RNA-induced transcriptional silencing complex
RN Récepteur nucléaire
RNA-Seq RNA sequencing
RPB1 RNA polymerase II subunit B1 RSK2 Ribosomal S6 kinase
S. cerevisiae Saccharomyces cerevisiae S. pombe Schizosaccharomyces pombe SA Stromal antigen
SAM S-adenosylmethionine
Scc1 Sister chromatid cohesion protein 1 SEC Super elongation complex
SET Suppressor of variegation, Enhancer of zeste, Trithorax
SGO1 Shugoshine 1
shRNA Short haipin RNA SLIDE SANT-like ISWI
SMC Structural maintenance of chromosomes
SMRT Silencing mediator for retinoid and thyroid hormone receptors snRNP Small nuclear ribonucleoprotein
SPA Signal poly(A)
Srb Suppressor of RNA polymerase B SV40 Simian virus 40
SWI/SNF Switch/surcrose nonfermenting TAD Topologicaly associated domain TAF TBP-associated factors
TBP TATA-box binding protein TC Territoires chromosomiques TCR Transcription coupled repair TFBS Transcription factor binding site TFII Transcription factor II
TR Traveling ratio
TSS Transcription start site TTS Transcription termination site
Ubx Ultrabithorax
WAPL Wings apart-like protein homologue WB Western blot
Xist X inactive specific transcript XRN2 Exoribonuclease 2
YY1 Yin yang 1
ZNF143 Zinc finger protein 143
Épigraphe
“Begin at the beginning, and go on till you come to the end: then stop.”Remerciements
En premier lieu, je souhaite remercier mon directeur de recherche, le Dr Steve Bilodeau, pour m’avoir accueilli au sein de son équipe en tant que première étudiante du laboratoire. Ce défi, bien que souvent difficile à relever, s’est révélé être l’expérience la plus formatrice de mon apprentissage académique. Au-delà de la culture scientifique acquise au cours de mon doctorat, mon passage au sein du laboratoire m’aura également permis de développer mon sens de l’initiative ainsi que mon autonomie. Je ressors grandie de cette expérience.
Je tiens également à remercier les membres de mon jury, le Dr Amine Nourani, le Dr Samer Hussein et le Dr Nicolas Gévry pour avoir accepté d’évaluer ce travail.
Mon doctorat aura été une période remplie de belles rencontres et d’apprentissages de tout genre et je n’en serais pas venue à bout sans le soutien de nombreuses personnes. Merci à l’ensemble des membres présents et passés du laboratoire Bilodeau. Claire, Gaëlle, Fabien, Michèle, j’ai appris de précieuses leçons de vie à vos côtés, et pour ça je vous remercie. Un grand merci tout particulier à Maxime et Éric qui ont été de véritables anges gardiens. Je vous suis très reconnaissante pour tout ce que vous avez fait pour me permettre de finir mon doctorat. Merci pour vos encouragements et pour votre soutien. Maxime, merci d’être un lab manager hors pair et pour tous les moments passés à plaisanter ! Éric, merci pour ta précieuse contribution scientifique et surtout pour les GIFs de chats ! Je remercie également les étudiants d’été qui sont passés au fil des ans, particulièrement Béatrice et Thierry que j’ai eu le plaisir d’encadrer. Votre enthousiasme est toujours une bouffée d’air frais !
Mes remerciements seraient incomplets si je ne mentionnais pas les membres du laboratoire voisin. Laurence, Jonathan et Alexia, en particulier, m’ont accueilli à mes débuts et ont grandement contribué à mon intégration à Québec. Merci pour les sorties et de m’avoir occupé quand je n’avais pas encore de manips.
Je souhaite également exprimer ma gratitude à Sylvie Dodin et au réseau REAGIR ainsi qu’au comité étudiant du centre de recherche pour m’avoir donné la possibilité de m’investir dans divers projets. Ces activités ont été extrêmement enrichissantes et ont considérablement contribué à ma formation.
Mon passage à Québec n’aurait jamais été le même sans les amitiés qui se sont forgées au cours des années. Je tiens à chaleureusement remercier Claire, Manu et Alexandra
dîner ont toujours été LE moment de décompression de prédilection ! Merci pour ces moments de folie passagère ! Un grand merci tout particulier à mes PPFs d’amour, Alice, Carole et Lauriane, votre soutien au cours de mon parcours m’a aidé à traverser ces années de doctorat. Merci pour votre confiance, votre écoute et votre bienveillance. Je souhaite également remercier JC et Laurent d’avoir répondu présents lorsque j’avais besoin de leur aide et pour les discussions scientifiques et philosophiques de l’un, et les bons petits plats de l’autre. Merci à tous, sans oublier Alice, Solenn, Gaëlle et Mélanie, pour les innombrables soirées passées ensemble, vous êtes trop canons !
Enfin, à mon cher époux, merci d’être venu bravé le froid Québécois avec moi, merci pour ta patience à toute épreuve, et merci pour ton soutien et tes judicieux conseils. Merci d’être toi, my best husband !
Je souhaite dédier cette thèse à mes parents, Said et Fadela, qui ont toujours eu une confiance indéfectible en moi, même dans mes plus grands moments de doute. Sans vous, ce travail n’aurait pas été possible.
Bonne lecture !
Avant-propos
Les résultats présentés dans cette thèse sont le résultat de cinq années d’études au sein du laboratoire du Dr Steve Bilodeau. Au cours de mon doctorat, j’ai participé à plusieurs projets dont l’un a abouti à la réalisation d’un article. De plus, j’ai contribué à des collaborations avec d’autres laboratoires du Centre de recherche sur le cancer de l’Université Laval.
Un des projets sur lesquels j’ai travaillé, qui fait l’objet du Chapitre 2, montre que les gènes dérégulés dans le syndrome de Cornelia de Lange sont préférentiellement organisés au sein de communautés de gènes où ils sont en contact dans l’espace chromosomique tridimensionnel avec des régions du génome occupées par NIPBL et cohésine. Ce projet a donné lieu à une publication dans le journal Genetics (Imène Boudaoud, Éric Fournier, Audrey Baguette, Maxime Vallée, Fabien C. Lamaze, Arnaud Droit and Steve Bilodeau. Connected Gene Communities Underlie Transcriptional Changes in Cornelia de Lange Syndrome. Genetics. vol. 207 no. 1 139-151 (2017)).
Au sein de mon laboratoire d’accueil, j’ai également eu l’occasion de mener deux autres projets de recherche visant à caractériser le rôle de NIPBL et du complexe cohésine dans la régulation de la transcription. Les travaux présentés dans le Chapitre 3 visent à identifier la ou les étape(s) de la transcription régulée(s) par NIPBL et cohésine dans les cellules souches embryonnaires de souris. Dans le Chapitre 4, nous décrivons une étude cherchant à caractériser le rôle de ces facteurs dans la régulation de la réponse transcriptionnelle contrôlée par les récepteurs nucléaires.
Durant mon doctorat, j’ai également eu l’opportunité de collaborer sur deux projets de recherche différents portant sur le cancer de la prostate. En collaboration avec l’équipe du Dr Vincent Fradet, nous avons entrepris d’établir un profil épigénétique de cellules normales de patients atteints ou non d’un cancer de la prostate plus ou moins avancé dans le but d’identifier un nouveau biomarqueur du microenvironnement prostatique. De plus, j’ai participé à un projet portant sur la caractérisation d’un partenaire du récepteur aux androgènes en collaboration avec les équipes des Drs Nicolas Bisson et Frédéric Pouliot.
CHAPITRE 1 : Introduction
Un des grands défis de la biologie moléculaire moderne est de comprendre comment se crée la diversité fonctionnelle des types cellulaires eucaryotes à partir d’un patrimoine génétique commun. En effet, en utilisant le même génome, chaque cellule d’un organisme pluricellulaire acquiert des fonctions spécifiques lui permettant de répondre et de s’adapter à divers stimuli internes et externes. Ces fonctions sont définies par la coordination spatio- temporelle de programmes d’expression, c’est-à-dire l’expression d’une sous-population de gènes, spécifiques à chaque type et état cellulaire.
Le mécanisme de sélection des gènes à exprimer dans chaque cellule est la résultante de nombreux niveaux de régulation. En effet, la mise en place d’un programme d'expression donné passe par la régulation de l’activité de l’ARN polymérase II et de la liaison de facteurs de transcription et de cofacteurs au niveau de séquences régulatrices, par le contrôle de la compaction de la chromatine et enfin par la coordination du repliement des chromosomes dans le noyau.
Au cours des dernières années, un grand nombre de recherches faisant appel aux méthodes les plus modernes en biologie cellulaire et moléculaire, en particulier la génomique et la transcriptomique, ont permis de mettre en lumière divers mécanismes de régulation impliqués dans les différents niveaux de contrôle de l’expression génique.
1.1 Régulation de la transcription par l’ARN polymérase II
La transcription est le processus ubiquitaire permettant la biosynthèse de l’ARN, une copie de l’information encodée par la molécule d’ADN. Chez les eucaryotes, ce phénomène est accompli par trois différents types d’ARN polymérases (Pol). Ces polymérases ADN- dépendantes varient par le type et le nombre de sous-unités qu’elles contiennent ainsi que par la classe de gènes qu’elles transcrivent1 :
- L’ARN Pol I transcrit les gènes de classe I codant pour les ARN ribosomiques (ARNr) 18S, 28S et 5.8S.
- L’ARN Pol II catalyse la transcription des gènes de classe II à l’origine des ARNs messagers (ARNm) ainsi que certains ARNs non codants comme les micro-ARN (miARN), les petits ARN nucléaires (ARNsn) et nucléolaires (ARNsno) et les longs ARN non codants (ARNlnc).
- L’ARN Pol III transcrit les gènes de classe III codant pour les ARNs de transfert (ARNt) ainsi que l’ARNr 5S.
Nous nous concentrerons dans cette partie sur la transcription par l’ARN polymérase II des gènes codant pour des protéines.
1.1.1 Les éléments régulateurs de la transcription
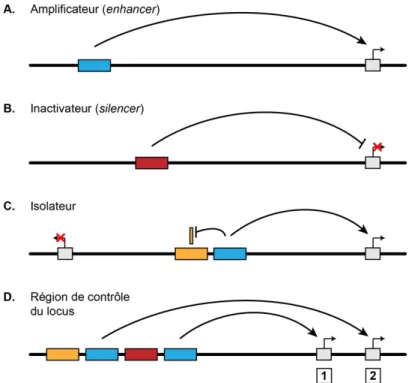
Chez les eucaryotes, un grand nombre de séquences régulatrices, auxquelles s’associe la machinerie moléculaire contrôlant la transcription, permettent d’établir les patrons d’expression uniques des gènes. Il existe deux grands types de régions régulatrices agissant en cis : le promoteur minimal associé à des éléments régulateurs proximaux, et des éléments régulateurs distaux qui peuvent être des séquences amplificatrices (enhancers), inactivatrices (silencers), isolatrices ou des régions de contrôle du locus (LCR, pour locus control region) (Figure 1.1).
Figure 1.1: Les régions régulatrices de la transcription.2
Les séquences régulatrices de la transcription sont de deux grands types : le promoteur et les régions régulatrices distales. Le promoteur, composé d’un promoteur minimal associé à des éléments régulateurs proximaux, s’étend en général sur moins de 1000 paires de bases (1 kb). Les régions régulatrices distales, qui incluent des séquences amplificatrices (enhancers), inactivatrices (silencers), isolatrices et des régions de contrôle du locus (LCR), peuvent être situées à de très grandes distances du promoteur qu’elles régulent. Les éléments régulateurs distaux peuvent entrer en contact avec le promoteur par l’intermédiaire de boucles d’ADN.
1.1.1.1 Le promoteur
La région promotrice des gènes eucaryotes possède une organisation complexe où le contenu et la disposition des éléments composant le promoteur sont variables. On
distingue néanmoins deux grandes régions : le promoteur minimal et le promoteur proximal.
Le promoteur minimal est la région en amont du gène qui est utilisée comme site d’ancrage de la machinerie transcriptionnelle de base pour permettre l’assemblage du complexe de pré-initiation (PIC pour pre-initiation complex) de la transcription. Il définit la position du site d’initiation (TSS pour transcription start site) ainsi que la direction de la transcription. Il comprend, par définition, la séquence localisée de part et d’autre du TSS et est constitué d’une combinaison variable d’une ou de plusieurs séquences conservées au sein de cette région3,4 (Figure 1.2).
Figure 1.2: Les éléments composant le promoteur minimal.2
Les promoteurs minimaux des métazoaires sont constitués d’une combinaison variable d’éléments aux séquences conservées qui peuvent inclure une boîte TATA, un élément initiateur (Inr) ainsi des éléments DPE (downstream promoter element), BRE (TFIIB recognition element), DCE (downstream core element) et MTE (motif ten element). Leur position relative au TSS (représenté par une flèche) ainsi que les facteurs de transcription qui se lient à ces éléments sont indiqués. Le DCE est représenté séparément uniquement pour faciliter son illustration.
La boîte TATA, localisée en amont du TSS, est le premier élément du promoteur minimal à avoir été décrit5. Sa séquence consensus, TATAA, permet la liaison de la protéine TBP
(TATA-box binding protein) faisant partie du PIC. Les boîtes TATA sont souvent associées à des promoteurs forts tissus-spécifiques et ne sont retrouvées que dans une minorité des promoteurs eucaryotes (5 à 20%)6–8. Un autre élément important du promoteur minimal est l’élément initiateur (Inr) riche en pyrimidine localisé sur le TSS. L’élément Inr et la boite TATA sont les deux seuls éléments du promoteur minimal à être capables de recruter le PIC et d’initier la transcription7.
Les autres éléments pouvant être associés au promoteur minimal sont les suivants: - L’élément DPE (downstream promoter element), se situant en aval du TSS, est
un site de reconnaissance de TFIID, mais ne peut agir indépendamment d’un élément Inr6.
- L’élément de reconnaissance de TFIIB, le BRE (TFIIB recognition element), localisé en amont de la boite TATA. Le BRE peut activer ou réprimer la transcription11.
- L’élément DCE (downstream core element) en aval du TSS.
- L’élément MTE (motif ten element), également situé en aval du TSS.
TFIID et TFIIB sont deux facteurs généraux de la transcription (voir section 1.1.2.1). De plus, il est intéressant de noter qu’à l’exception du BRE, tous les éléments du promoteur minimal sont des sites d’interaction du complexe TFIID (Figure 1.2)11,12.
Le promoteur minimal contrôle la transcription basale des gènes. Cependant, des éléments régulateurs proximaux peuvent s’y associer afin de moduler cette régulation. Le promoteur proximal est alors défini comme la région immédiatement en amont, et s’étendant sur quelques centaines de paires de bases, du promoteur minimal. Il contient par exemple la boîte CAAT ainsi que la boite GC et possède généralement plusieurs sites de liaison pour des activateurs7,11.
Enfin, des études effectuées à l’échelle du génome ont montré que la majorité des gènes possèdent plusieurs promoteurs au sein desquels sont retrouvés différents TSS dont l’utilisation différentielle permet une diversification du transcriptome7. Ainsi, la complexité
de l’organisation des régions promotrices permet un contrôle combinatoire de la régulation des promoteurs, ce qui augmente de façon exponentielle le nombre potentiel de patrons d’expression uniques qui peuvent être créés.
1.1.1.2 Les séquences amplificatrices distales ou enhancers
Les enhancers (Figure 1.3.A) ont d’abord été définis comme des régions du génome étant capables d’activer la transcription dans une construction plasmidique. En effet, ces éléments ont été caractérisés en premier lieu, et sont souvent encore fonctionnellement spécifiés ainsi, en utilisant des essais de gènes rapporteurs dans des cellules en culture13.
La première description d’un enhancer dans un tel système remonte au début des années 1980 lorsqu’il a été observé qu’une région du génome du virus simien 40 (SV40) pouvait augmenter la transcription d’un gène humain14. Le premier enhancer humain, quant à lui, a été découvert dans le locus de la chaîne lourde des gènes d'immunoglobuline15.
Au cours des trente dernières années, de nombreuses études ont permis d’identifier les caractéristiques clés de ces régions régulatrices. Premièrement, les enhancers exercent leur fonction indépendamment de leur localisation ou orientation par rapport à leur(s) promoteur(s) cible(s). De plus, les enhancers peuvent agir à de très grandes distances et maintiennent leur fonction indépendamment de la séquence d’ADN (par exemple : dans un système rapporteur hétérologue). Enfin, les enhancers sont modulaires, permettant ainsi à un unique promoteur d’être sous la régulation de plusieurs éléments enhancers à différents moments, dans différents tissus, ou encore en réponse à différents stimuli13,16.
Ces séquences régulatrices sont généralement composées de sites de liaison pour des facteurs de transcription groupés en clusters et fonctionnant de manière coopérative afin de rehausser la transcription. Les enhancers peuvent être liés par les mêmes activateurs que les promoteurs proximaux, mais ils sont plus éloignés du promoteur minimal. À titre d’exemple, les enhancers peuvent être situés à plusieurs centaines de kilobases en amont d’un promoteur, en aval d’un promoteur, ou même au-delà de la partie 3’ du gène17. Pour permettre la mise en place d’une régulation sur de grandes distances, il est communément admis que des boucles d’ADN rapprochent les enhancers de leurs promoteurs cibles (discuté dans la partie 1.3). De plus, il a été montré que des enhancers peuvent être localement regroupés dans certaines régions du génome afin de former des super- enhancers qui régulent des gènes clés de l’identité cellulaire. Ces régions diffèrent des enhancers classiques par leur taille, la densité des facteurs de transcription tissus- spécifiques qui s’y lient ainsi que leur capacité à activer la transcription18,19.
Parmi les éléments distaux qui régulent la transcription, les enhancers montrent la plus grande diversité et spécificité cellulaire et jouent ainsi un rôle primordial dans la régulation spécifique de l’expression des gènes. De nombreuses études ont donc été entreprises dans le but de mieux comprendre la fonction et les mécanismes de régulation des enhancers. Il a par exemple été décrit que l’ARN Pol II est présente au niveau des régions enhancers où elle permet la transcription d’une nouvelle classe d’ARNs non-codants, les ARNs associés aux enhancers ou eRNAs20,21. Bien que la fonction exacte des eRNAs soit encore mal comprise, ces observations suggèrent que les enhancers peuvent fonctionner comme des unités transcriptionnelles22. De plus, il a été proposé que l’assemblage du PIC
pourrait avoir lieu au niveau de certains enhancers afin de permettre un contrôle plus précis du timing de l’activation transcriptionnelle23. Ce mécanisme serait plus commun
Figure 1.3: Représentation schématique du mécanisme d’action des séquences régulatrices distales.2
A. et B. Les enhancers activent la transcription alors les silencers la répriment. C. Les
séquences isolatrices forment une frontière qui permet de bloquer l’effet d’autres éléments régulateurs. D. Les régions de contrôle du locus (LCR) sont composées de plusieurs séquences régulatrices permettant de contrôler l’expression spatio-temporelle d’un groupe de gènes.
1.1.1.3 Les autres éléments régulateurs distaux
À l’opposé fonctionnel des enhancers, on retrouve des séquences inactivatrices ou silencers (Figure 1.3.B). Ces derniers sont des éléments possédant des sites de liaison pour des facteurs ayant un effet répressif sur l’expression des gènes24. Ils peuvent faire
partie d’un promoteur proximal, d’un enhancer, ou constituer un module de régulation distal indépendant. Les silencers partagent de nombreuses propriétés avec les enhancers. En effet, ils agissent généralement de manière indépendante de la distance ou de leur orientation par rapport au promoteur qu’ils régulent. De plus, ils peuvent être situés à une grande distance de leur gène cible, dans un intron, ou dans la région 3’UTR. Par ailleurs, les silencers peuvent agir de façon coopérative et synergique25,26 et ont la capacité
d’interagir avec des promoteurs par l’intermédiaire de boucles d’ADN27,28. Enfin, ils
Les isolateurs, aussi connus sous le nom d’éléments frontière, permettent d’isoler des séquences données de l’activité transcriptionnelle environnante (Figure 1.3.C). Ils limitent l’action d’éléments régulateurs à des régions bien définies et partitionnent ainsi le génome en différents domaines d’expression. Les isolateurs sont de deux types : les isolateurs bloquant la communication enhancer-promoteur et ceux empêchant la dispersion de la chromatine répressive29. Plusieurs modèles ont été proposés pour expliquer les mécanismes par lesquels les isolateurs exercent leur activité de blocage de la communication enhancer-promoteur. L’isolateur pourrait par exemple recruter la machinerie transcriptionnelle, l’empêchant ainsi de se lier au promoteur. Il pourrait également agir comme barrière physique ou encore former des boucles entre isolateurs ou avec d’autres séquences d’ADN, interférant ainsi avec la fonction des enhancers. L’activité de limitation de la dispersion de la chromatine répressive pourrait quant à elle passer par le recrutement de complexes protéiques maintenant la chromatine ouverte30.
La protéine se liant à des séquences isolatrices la plus étudiée chez les mammifères est la protéine CTCF (CCCTC-binding factor). CTCF joue notamment un rôle au niveau du locus H19-Igf2, l’exemple le mieux caractérisé de locus humain soumis à l’empreinte parentale contenant un élément isolateur24(Figure 1.14.B). Les fonctions de CTCF seront discutées
plus en détail dans la 3ème partie de cette introduction.
Les régions de contrôle du locus (LCR, pour locus control region) sont des groupes d’éléments régulateurs impliqués dans le contrôle d’un locus complet ou d’un ensemble de gènes organisés en cluster (Figure 1.3.D). Les LCRs sont généralement composées de plusieurs éléments agissant en cis, incluant notamment des enhancers, silencers et isolateurs. Ces éléments sont liés par des activateurs (ubiquitaires et tissus-spécifiques), des coactivateurs, des répresseurs et des enzymes modifiant de la chromatine. Chacun de ses composants affecte différentiellement l’expression des gènes, et c’est leur activité collective qui définit fonctionnellement un LCR et confère une expression spatio- temporelle adéquate. La propriété principale des LCRs est néanmoins une forte activité enhancer spécifique31.
L’identification d’un grand nombre de LCRs a révélé que, tout comme les enhancers et les silencers, les LCRs peuvent réguler l’expression des gènes à distance et fonctionner indépendamment de leur position. Bien que les LCRs soient généralement positionnés en amont de leurs gènes cibles, ils peuvent aussi être retrouvés au sein d’un intron, en aval d’un gène ou encore dans l’intron du gène voisin. Une des caractéristiques typiques des
LCRs est la présence de plusieurs sites d’hypersensibilité à la DNase I permettant l’interaction avec la machinerie transcriptionnelle2,32.
1.1.2 La machinerie transcriptionnelle eucaryote
Les éléments cis régulateurs de la transcription contiennent des sites de reconnaissance pour des facteurs de transcription se liant à l’ADN et agissant en trans, soit pour activer ou réprimer la transcription. Les facteurs impliqués dans l’activation de la transcription peuvent être classés en trois groupes : les facteurs généraux de la transcription (GTFs pour General Transcription Factors), les protéines activatrices, et les coactivateurs (Figure 1.4).
Figure 1.4: La machinerie transcriptionnelle eucaryote. 2
L’activation de la transcription par l’ARN Pol II fait intervenir trois groupes de facteurs : les activateurs, les coactivateurs et la machinerie transcriptionnelle de base incluant l’ARN Pol II elle-même associée aux facteurs généraux de la transcription (GTFs) TFIIA, TFIIB, TFIID, TFIIE, TFIIF et TFIIH. La machinerie transcriptionnelle se base s’associe au promoteur afin de former le complexe de pré-initiation de la transcription (PIC). Les activateurs, possédant généralement un domaine de liaison à l’ADN (DBD, pour DNA- binding domain) ainsi qu’un domaine d’activation (AD, pour activation domain), sont liés à des séquences activatrices comme les enhancers et permettent de stimuler la transcription. Les cibles directes et indirectes des activateurs peuvent être multiples.
1.1.2.1 Les facteurs généraux de la transcription
Seule, l’ARN polymérase II n’est pas capable de démarrer la transcription. Ce complexe multi-protéique composé de 12 sous-unités chez les eucaryotes nécessite des protéines auxiliaires interagissant notamment avec l’ADN du promoteur pour initier son activité : ce sont les facteurs généraux de la transcription (GTFs). Ils constituent, avec l’ARN Pol II, la machinerie transcriptionnelle de base nécessaire et suffisante pour initier une transcription basale in vitro33,34. Au cours de l’initiation de la transcription, l’ARN Pol II s’associe aux
GTFs au niveau de la région promotrice des gènes afin de former le complexe de pré- initiation de la transcription (PIC). Ce dernier peut se former de façon séquentielle ou peut être recruté sous forme d’holoenzyme déjà préassemblée. Ces deux modèles seront présentés dans la partie 1.1.3.2.
Les facteurs de transcription interagissant avec l’ARN Pol II (TFII, pour transcription factor II) interviennent à différentes étapes de la transcription. Le complexe TFIID, par exemple, sert de plateforme d’assemblage de la machinerie transcriptionnelle au niveau du promoteur. TFIID est constitué de la protéine TBP, sous-unité principale interagissant avec la boîte TATA lorsqu’elle existe, et de 14 TAFs (TBP-associated factors), des protéines extrêmement conservées de la levure à l’homme. L’interaction entre TBP et la boite TATA est stabilisée par le facteur TFIIA qui interagit directement avec TBP ainsi que des séquences en amont de la boîte TATA. TFIIB quant à lui interagit avec le complexe TBP- ADN et recrute l’ARN Pol II. TFIIF, TFIIE et TFFIIH interagissent avec l’enzyme et assurent sa conversion en une forme compétente pour l’élongation de la transcription (Figure 1.4). En particulier, TFIIH possède plusieurs activités enzymatiques dont une activité hélicase, une activité de réparation de l’ADN ainsi qu’une activité kinase phosphorylant l’ARN Pol II au niveau de son domaine C-terminal (Figure 1.7)35,36.
Bien que les sous-unités de l’ARN Pol II ainsi que les GTFs soient fortement conservés au cours de l’évolution, les séquences des régions promotrices sont variées. Comme mentionné précédemment, le séquençage du génome humain a notamment montré que la majorité des promoteurs ne possède pas de séquence TATA7. Par conséquent la protéine TBP ne peut pas, à elle seule, reconnaître le promoteur, et d’autres fonctions de reconnaissance de l’ADN sont requises37. Cela suggère alors que les complexes d’initiation de la transcription ont des compositions variables et que tous les GTFs ne sont pas requis pour la transcription de l’ensemble des gènes35.