HAL Id: hal-00866559
https://hal.archives-ouvertes.fr/hal-00866559
Preprint submitted on 2 Oct 2013HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
La thermo-ionisation en spectrométrie de masse
isotopique
Wulfran Barthelemy
To cite this version:
Centre Européen
de Recherche et d’Enseignement Géosciences de l’Environnement (CEREGE)
Europôle Méditerranéen de l’Arbois CNRS UMR 7330 Aix-Marseille Université 13545 Aix-en-Provence cedex 4
La thermo-ionisation
en spectrométrie de masse isotopique
Wulfran Barthélemy
INTRODUCTION
Dans le cadre de la formation permanente du CNRS, le colloque annuel d'instrumentation scientifique « Isotrace » organisé depuis quelques années par la communauté des chercheurs en géochimie isotopique réunit régulièrement des membres de plusieurs laboratoires français de géosciences. Ces rencontres inter-laboratoires de chercheurs et de personnels techniques sont l’occasion de faire le point sur l’évolution des techniques employées en spectrométrie de masse isotopique.
Ces réunions ont été l'occasion de proposer la rédaction d’un document de synthèse rassemblant les informations disponibles sur les méthodes de dépôt sur filament des échantillons analysés par la technique TIMS (thermal ionisation mass spectrometry). Dans ce type d’instrument, les éléments à analyser sont dissous et déposés sur des filaments de métal qui sont introduits dans la source d’ions. L’un des points faibles de la technique TIMS est la mauvaise efficacité de l’émission des ions. Les méthodes couramment utilisées sont empiriques et les comportements émissifs diffèrent fortement entre les éléments.
Plusieurs laboratoires intéressés ont accepté de nous communiquer leurs protocoles de dépôts, pour une mise en commun du savoir-faire courant. Ce document reporte l'expérience des laboratoires de géochimie associés au CNRS, à laquelle s'ajoutent des informations diffusées par l'Isotope Geology Laboratory de Boise (Idaho, USA) et par le Lamont-Doherty Earth
Observatory de Columbia (New-York, USA). L'objectif est de transmettre l'expérience acquise et de mieux cerner les processus physico-chimiques ou de proposer des améliorations.
Le présent document contient des informations sur des éléments couramment analysés avec la technique TIMS : strontium, néodyme, plomb, uranium, thorium. Il pourra être mis à jour et complété par d’autres éléments plus « exotiques ». Sont également proposées des discussions sur le comportement des ions au cours de la thermo-émission, ainsi que quelques éléments bibliographiques.
GENERALITES
La thermo-ionisation, ou ionisation de surface, est la technique employée pour produire des faisceaux d’ions dans les sources des spectromètres de masse TIMS utilisés pour l’analyse isotopique. Elle consiste à faire évaporer et ioniser par chauffage l’élément chimique à
analyser, provenant d’un échantillon solide ou liquide déposé sur un ruban de métal. l’échantillon est préalablement purifié en salle de chimie, puis déposé avec une microseringue sur le filament métalllique, séché puis installé dans la source de l’instrument. Après mise en pompage, le ruban est parcouru sous vide par un courant électrique qui provoque son échauffement par effet Joule. L’échantillon chauffé s’évapore et s’ionise, le plus souvent positivement par la perte d’un électron, pour former un faisceau d’ions qui est accéléré par un champ électrostatique ; ce faisceau est ensuite analysé dans le spectromètre de masse par une séparation en masse donnant plusieurs faisceaux détectés par des collecteurs d’ions. Les résultats sont présentés sous forme de rapports isotopiques, donnés par les intensités relatives mesurées des différents faisceaux.
Avant d’être déposés, les échantillons naturels sont préalablement dissous et purifiés en laboratoire de chimie. La séparation élémentaire consiste à éliminer tous les éléments, sauf celui dont on veut connaître la composition isotopique et qui sera présent sur le filament. Cette étape s’impose car la présence d’autres éléments a tendance à gêner l’émission. En fin de préparation chimique, l’élément se trouve habituellement dissous dans un acide ayant servi lors de la purification.
La précision des résultats des analyses dépend en particulier de l’intensité du faisceau d’ions obtenu et de sa durée de vie. Elle est donc fonction de la quantité de l’échantillon disponible, mais aussi de l’efficacité de la thermo-ionisation. Il arrive en effet qu’une grande proportion d’atomes s’évaporent sans perdre d’électron, ce qui les empêche de réagir aux forces électromagnétiques et donc d’être anlysés. Ce mauvais taux ionisation est l’une des limites essentielles de la technique. Selon les conditions d’analyse, la qualité de l‘émission peut être très différente. Elle dépend notamment de la nature de l’élément à mesurer, de sa quantité, de sa pureté, d’un additif éventuel (activateur), du matériau qui constitue le ruban, de sa température et de la géométrie du dépôt et de la source.
Une approche théorique est fournie par l’équation de Saha-Langmuir, qui tente d’évaluer le taux d’ionisation. Lorsque des atomes interagissent avec la surface d’un matériau porté à haute température, une fraction d’entre eux sont réémis sous forme d’ions positifs, et une autre sous forme d’atomes neutres. L’équation donne le taux d’ionisation, c’est-à-dire à chaque instant et pour une température T, à l’équilibre thermique, la proportion de particules réémises sous forme d’ions et d’atomes neutres :
ni / no = A exp ((W–I) / k T) Taux d’ionisation
(équation de Saha-Langmuir)
Dans cette expression, A est un coefficient dépendant de l’élément ; W est la fonction de travail de sortie du filament, c’est-à-dire l’énergie qu’il faut fournir à un électron pour qu’il puisse quitter la surface d’un matériau (en eV) ; I est l’énergie de première ionisation, c’est-à-dire celle nécessaire pour ioniser un atome de l’échantillon (en eV) ; k est la constante de Boltzmann et T la température (en K).
D’un point de vue physique, une compétition s’instaure entre l’atome émis et le support pour retenir les électrons. Pour qu’un atome de l’échantillon quitte le ruban métallique en abandonnant un électron, il faut que l’affinité du support pour les électrons soit plus forte que
la sienne. Autrement dit, la fonction de travail de sortie du ruban (W) devrait donc être plus haute que l’énergie qu’il faut donner à l’atome émis (I) pour l’ioniser. Or, on est le plus souvent dans le cas contraire, ce qui peut expliquer l’efficacité souvent mauvaise de la thermo-ionisation.
En effet, dans la plupart des situations, le terme (W-I) étant négatif, le taux d’ionisation augmente donc comme exp (-1/T), donc croît avec la température. Mais il sera d’autant plus élevé que l’écart W–I sera faible en valeur absolue. Pour avoir de bonne efficacité d’ionisation, on aurait donc intérêt à ce que la fonction de travail du ruban soit élevée, et que l’énergie d’ionisation du dépôt soit faible.
L’équation de Saha-Langmuir, qui est tirée d’une loi d’action de masse, donne une idée théorique de l’efficacité d’ionisation pour un élément, mais elle est difficile à utiliser concrètement. En effet elle ne tient pas compte de certains phénomènes comme les propriétés volatiles de chaque espèce ou même d’éventuels processus chimiques supplémentaires au niveau du filament (Delmore, 1991).
La nécessité d’obtenir de hautes précisions de mesure a conduit à rechercher les meilleures conditions possibles d’émission en termes d’intensité, de durabilité et de stabilité des faisceaux d’ions. De nombreuses expériences ont été menées dans différents laboratoires pour tenter d’améliorer l’ionisation.
Le choix du métal constituant le ruban doit être nécessairement celui d’un matériau ayant une fonction de travail élevée et supportant les hautes températures. Les métaux couramment utilisés sont donc ceux qui combinent haute fonction de travail et point de fusion élevé : le tungstène (4,53 eV – 3370 °C), le rhénium (5,1 eV – 3180 °C) et le tantale (4,19 eV – 2890 °C). De leur côté, les éléments les plus faciles à analyser sont préférentiellement ceux qui ont une énergie d’ionisation basse : radium (5,28 eV), néodyme (5,51 eV), samarium (5,6 eV), strontium (5,69 eV).
On définit un rendement d’ionisation par le rapport entre les quantités de matière ionisées et déposées sur le filament. Ce rendement (à ne pas confondre avec le taux d’ionisation) indique la proportion des atomes introduits dans la source qui ont été effectivement détectés tout au long de l’émission. Il est assez facile à estimer concrètement en fin d’analyse, en faisant le bilan des espèces par intégration approximative des signaux et par calcul de la masse de l’échantillon déposée. Le rendement d’ionisation peut varier de plusieurs ordres de grandeur, selon les éléments et les conditions d’analyse.
Curieusement, l’expérience a montré que pour des quantités d’échantillon importantes, le rendement était souvent peu intéressant, tandis qu’il l’est davantage avec de faibles teneurs : l’efficacité diminue avec la quantité. Pour compenser cette perte, il a été mis au point une technique utilisant un filament double ou triple : le dépôt est fait sur un filament, d’où il s’évapore pour frapper un autre filament plus chaud où s’opère l’ionisation (Ingram et Chupka, 1952). Ainsi les deux phénomènes d’évaporation et d’ionisation peuvent être contrôlés séparément. La technique du filament unique reste utilisée pour des dépôts de l’ordre du nanogramme ou moins, tandis que celle du filament multiple est pertinente à partir de plusieurs dizaines ou centaines de nanogrammes.
Dans tous les cas, les meilleurs résultats obtenus avec les éléments les plus facilement ionisables (Ra, Re, Os …) ne dépassent jamais 30% (Birck, 2001). L’existence de ce plafond
reflète vraisemblablement la transmission de l’instrument, qui multipliée par le rendement d’ionisation proprement dit, donne l’efficacité globale des ions détectés. On peut penser que la transmission instrumentale est de l’ordre de 30%, et que les rendements d’ionisation proprement dits des éléments les plus émissifs sont proches de 100 %.
Elément Fonction de travail W (en eV) Energie de 1ère ionisation I (en eV) Température de fusion (°C) Température d’ébullition (°C) Cd Pb Hf Th Zr Ca U Sr Sm Nd Lu Ra Rb Pt Ir Re W Ta Ti 5,77 5,7 5,1 4,53 4,19 4,1 8,99 7,42 7 6,95 6,84 6,11 6,08 5,69 5,6 5,51 5,42 5,28 4,18 321 327 2200 1750 1852 839 1132 769 1074 1024 1663 700 39 1772 2410 3180 3410 2996 1660 765 1755 5200 4790 4375 1484 3818 1384 1794 3074 3395 1140 696 3800 4130 5627 5660 6000 3287
Tableau I : Propriétés physiques de quelques éléments. Dans la partie basse du tableau sont figurés entre autres les métaux constitutifs des filaments.
STRONTIUM
Généralités et historique de la méthodologie
Le strontium est un des éléments les plus faciles à analyser en TIMS, surtout depuis qu’une technique de préparation spécifique avec activateur a été mise au point avec succès. Le dépôt se fait sur simple filament de tungstène ou de rhénium. La méthode désormais classique consiste à lui ajouter une solution d’activateur chimique à base d’oxyde de tantale, mise au point par J.-L. Birck à l’IPG de Paris (Birck, 1986).
L’origine de l’élaboration de cet activateur remonte aux années 1950, lorsqu’il fut constaté que les oxydes métalliques avaient des fonctions de travail plus hautes (donc meilleures) que les métaux purs. Pour cette raison on commença à déposer les échantillons de strontium sur des filaments de tantale oxydés en surface. Cette méthode, qui fut utilisée par le laboratoire CALTECH en Californie pour analyser les échantillons lunaires, avait ses limites. En effet, à moins de 50 ng de strontium, l’émission était capricieuse et difficile à contrôler, et le rendement obtenu s’avérait très décevant. De plus, les filaments de tantale avaient le défaut de se rompre fréquemment sous l’effet de la chaleur. Pourtant la température d’analyse ne dépassait pas les 1500°C, bien en dessous du point de fusion de ce métal (3000°C). Une solution provisoire consista à alimenter le filament en courant alternatif, au moyen d’une alimentation à découpage spécialement adaptée mais particulièrement onéreuse. Signalons par ailleurs qu’à San Diego, de la poudre de tantale posée sur des filaments de rhénium a été employée à titre expérimental.
Une nouvelle investigation a été menée à l’IPG de Paris, où l’on opta pour des filaments en tungstène ou en rhénium, bien plus résistants à la chaleur, sur lesquels on déposait un activateur chimique à base d’oxyde de tantale. L’idée était de rétablir la présence du tantale, et donc de séparer la fonction chauffage (filament) de la fonction ionisation (fonction de travail), la présence du tantale oxydé faisant augmenter la fonction de travail. Expérimenté sur du tungstène, le dépôt était effectué en emprisonnant le strontium dans l’oxyde de tantale. La technique a donné de très bons résultats, en particulier pour le rendement des petites quantités qui a pu dépasser les 5%.
La composition de l’activateur inclut plusieurs ingrédients (voir annexe). Pour le réaliser, on fait un mélange à partir d’un sel d’oxyde de tantale (Ta2O5) qui doit être pur à au moins
99,5%. L’acide fluorhydrique (HF) permet de le dissoudre. L’acide orthophosphorique (H3PO4) sert de colle, par le fait qu’il empêche l’oxydation du tungstène pendant le chauffage
du dépôt à l’air libre. En effet le tungstène s’oxyde au contact de l’air, et cet oxyde s’évapore à seulement 100°C sous vide, tendant à détacher le dépôt du filament avant qu’il n’atteigne les 1200-1400°C nécessaires à l’analyse. Enfin, l’acide nitrique (HNO3) est un indicateur visuel
qui devient blanc lorsque l’oxyde de tantale s’est bien cristallisé après chauffage du filament en fin de dépôt.
L’IPG de Paris a décrit en détail la méthode qu’il utilise pour effectuer les dépôts proprement dits à l’aide de l’activateur. Avec une microseringue et un cathéter, 0,5 microlitres d’activateur sont prélevés et déposés lentement (en moins de 5 minutes) sur un filament de tungstène porté à 1,2 A pour accélérer son séchage entre chaque couche (pour un filament de 25 microns d’épaisseur). Le courant est ensuite poussé jusqu’à 2A, sans atteindre la température d’évaporation de l’acide phosphorique. L’échantillon de strontium quant à lui est dissous dans 0,5 µl d’acide nitrique concentré, puis déposé lentement sur la couche d’activateur de tantale. Le filament est alors chauffé précautionneusement au rouge sombre. L’acide phosphorique fume, et le dépôt blanchit. Le tungstène s’oxyde en devenant jaune, puis bleu, puis violet foncé. Dès que le dépôt devient blanchâtre, on abaisse rapidement la température. Le filament ne doit pas être laissé plus de cinq minutes au rouge sombre. On réduit alors le courant jusqu’à 1,2 A, pour déposer encore 0,2 µl de tantale par très petites gouttes (qui ont tendance à s’étaler) ; enfin on chauffe à nouveau pendant une seconde au rouge sombre.
Dans le cas d’un standard, il est possible aussi de mélanger les premiers 0,5 µl de l’activateur avec la solution de standard, et de les déposer ensemble. Après quoi on porte alors le filament au rouge sombre avant de déposer la seconde couche d’activateur.
Les autres laboratoires effectuent leurs dépôts avec des méthodes très proches de celle de l’IPG. La plupart font leurs dépôts par couches successives « en sandwich », c’est-à-dire que la couche de strontium est déposée entre deux couches d’activateur ; d’autres font un mélange activateur-échantillon ; au laboratoire américain IGL de Boise (Idaho), on ne dépose qu’une seule couche de tantale sous le strontium. On a aussi avantage à éviter l’étalement des gouttes sur le ruban. C’est ce que font le LMTG de Toulouse et le Lamont Observatory, qui utilisent des « barrières de cathéter » consistant à coller sur le filament deux bandes adhésives transversales qui limitent l’étalement du dépôt.
Si le ruban de métal est en rhénium, la procédure est la même à la différence que les courants de chauffe sont plus faibles à cause de la différence de résistivité électrique. Avec ce métal, les dépôts se font à seulement 0,9 A, et ils sont portés au rouge à 1,8 A.
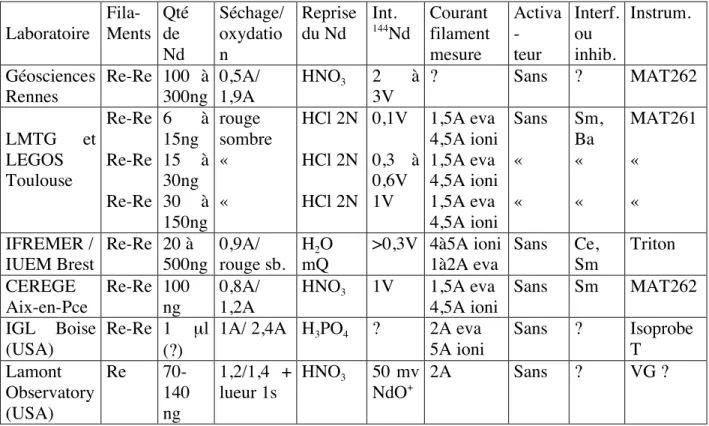
Le tableau suivant résume quelques informations recueillies auprès de plusieurs laboratoires. Notons que les différents modèles d’instruments employés n’utilisent pas tous la même longueur de filament, ce qui explique également les différences entre les courants de chauffe.
Laboratoire Fila- ment Qté de Sr Séchage/ oxydation Acide de dilution du Sr Int. 88Sr Courant de mesure Activat. TaF Interfé- rences ou inhib. Instru- ment IPG Paris W Re 10-500 ng 50-500 ng 1,2A rge fcé 0,9/1,8A HNO3 conc. HNO3 conc. 2-3V 10V 3-3,2A 2,1-2,6A oui oui Rb;occas. Ca, K Rb;occas. Ca, K VG ou MAT262 Géosciences Rennes W 50-300 ng
4,9A ? 3,5V ? oui ? MAT262
LMTG/LEGOS Toulouse W + cath ? 1,5A puis ? H3PO4+ HCl
IFREMER / IUEM Brest W 500 ng 1,1A/ 4,6A H2O 4V 4,2 à 4,6A
oui Ca, K, Rb Triton
CEREGE Aix-en-Pce Re 100 ng 0,8A/ rge1,8A HNO3 1 à 5V
3A oui Rb rarem. MAT262
IGL Boise (USA)
Re ? 2,5A H3PO4 3-4V 3-3,2A 1couche
infér. ? Isoprobe T Lamont Observatory (USA) W + Parf 100 ng 1,1A puis lueur 20s HNO3 4V ? "Sr Loader" ? VG ?
Tableau II : Méthodes de dépôt du strontium utilisées dans différents laboratoires. L’activateur est composé de Ta2O5 , HF, H3PO4 et HNO3.
Annexe : détail de la préparation chimique de l’activateur
Préparation de l’activateur d'oxyde de tantale :
La recette suivante permet de préparer 100 ml d’activateur en solution, pouvant servir à analyser le strontium, le potassium ou le rubidium. Elle nécessite les réactifs suivants :
- 2 g de chlorure de tantale TaCl5 ;
- 1,2 ml d’acide fluorhydrique HF concentré à 50% ;
- 1,2 ml d’acide orthophosphorique H3PO4 concentré à 85% ;
- 20 ml d’acide nitrique HNO3 concentré à 70 % ;
- 80 ml d’eau distillée ou dé-ionisée.
Le mode opératoire est le suivant :
- peser 2 g (+/- 10%) de chlorure de tantale TaCl5 dans une bouteille de 125 ml en téflon PFE
propre et sèche ;
- ajouter 30 ml d‘eau. Le TaCl5 est hydrolysé pour former un solide blanc (Ta2O5) ;
- ajouter les acides fluorhydrique (HF), orthophosphorique (H3PO4) et nitrique (HNO3) ;
- le solide blanc en suspension doit se dissoudre immédiatement, ou au plus tard dans les deux à trois heures ;
- compléter à 100 ml en versant encore 50 ml d’eau.
La solution obtenue contient alors près de 1 g de tantale pour 100 ml de solution. Sa concentration n’a pas besoin d’être très précise (+/- 10 % suffisent).
Remarques : pour la réaliser, on a intérêt à utiliser un chlorure de tantale TaCl5 de la meilleure
qualité possible. Un réactif qui convient bien est celui vendu par Aldrich sous la référence 21863-4, pur à 99,9 %. Il donne au final un blanc de dépôt en strontium de 1 ou 2 pg.
Si ce niveau de pureté n’est pas suffisant, le tantale peut être traité comme suit : entre l’hydrolyse du TaCl5 et l’ajout du HF, la poudre blanche obtenue à ce stade (Ta2O5 + eau)
peut être centrifugée. Le surnageant sera éliminé, et la poudre lavée à nouveau plusieurs fois de suite avec de l’acide chlorhydrique (1N à 3N) ou nitrique (1N à 8N). Le tantale ne se
dissout pas. Après ces lavages successifs, on poursuit la procédure ci-dessus (ajout des acides, etc.). Le volume d’eau à verser à la fin est alors de 80 ml. Le blanc de strontium doit alors être inférieur à 0,5 pg.
Si l’on souhaite obtenir un blanc de dépôt d’un niveau encore plus bas, l’acide phosphorique peut être purifié par dilution à 8-9 %, puis passé en colonne échangeuse de cations (par exemple, une colonne AG 50 X 8 à 2 ml fera l’affaire, prénettoyée avec 20 ml d’HCl 6N, puis avec deux fois 10 ml d’eau). On procède ainsi : diluer 1,2 ml de H3PO4 dans 10 ml d’eau, et
charger le tout dans la colonne. Collecter l’effluent. Ajouter 5 ml d’eau dans la colonne. Verser les deux effluents dans la bouteille de 125 ml contenant le tantale. A la fin on ajoutera seulement 65 ml d’eau.
NEODYME
Généralités et historique de la méthodologie
Le néodyme émis par thermo-ionisation a le défaut de sortir préférentiellement sous une forme oxydée NdO+, au détriment de sa forme atomique Nd+. Il serait donc tentant d’analyser
le néodyme sous la forme NdO+, mais cette méthode pose un problème d’interprétation, dû à
la présence des isotopes de l’oxygène qui compliquent le dépouillement des mesures. Pour cette raison, le néodyme est mesuré le plus souvent sous sa forme atomique. Des activateurs à base de gel de silice ou d’acide phosphorique ont été essayés, mais ils tendent à favoriser encore davantage l’émission de la forme oxydée NdO+ (Thirlwall, 1991). Par conséquent,
aucun activateur ne semble adéquat pour le néodyme. Il est donc habituellement déposé directement en solution sur double ou triple filament, et analysé sous forme Nd+.
Des essais ont cependant été effectués pour analyser du néodyme en faible quantité (de l’ordre du ng), situation où l’analyse de la forme NdO+ est alors la seule possible. L’analyse est
pertinente à condition de la corriger des rapports de l’oxygène (Lugmair et Marti, 1977). Il est dans ce cas impératif d’obtenir un dépôt très pur en néodyme, sinon d’autres éléments de masses proches interfèrent également, tels le cérium (Ce) et le praséodyme (Pr). La chimie d’extraction utilise à cette fin un acide alpha-hydroxybutyrique, (Griselin et coll., 2001). Les dépôts peuvent être faits sur rhénium avec un gel de silice, ou sur tungstène avec un activateur de tantale.
Wakaki et coll. (2007) rapportent également des mesures de néodyme en quantités très faibles mais sous forme non oxydée (de 0,1 à 5ng). Ils utilisent une méthode d’évaporation totale incluant une normalisation du fractionnement de masse. Le standard JNdi-1 dissous dans de l’acide nitrique est déposé en triple filament, avec ajout d’acide phosphorique. La précision des résultats est de 140 ppm (2 SD), avec un rendement d’ionisation de 1,3%.
Auteur Méthode Quantité Rendement Lugmair 1977 NdO+ et acide
hydroxybutyrique
Quelques ng ?
Griselin 2001 NdO+ et HPLC Quelques ng ?
Wakaki 2007 Evap. totale, HNO3 +
H3PO4
0,1 à 5 ng Env. 1,3 %
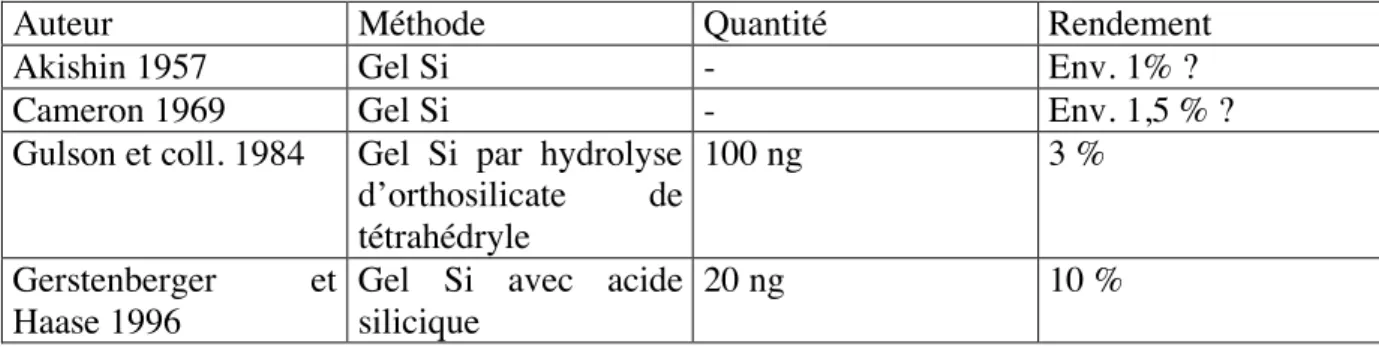
Tableau III : Techniques de dépôt du néodyme et rendements d’ionisation obtenus, d’après la bibliographie.
Méthodes pratiquées dans différents laboratoires
Les laboratoires recensés qui analysent le néodyme pratiquent apparemment tous la même méthode, à quelques détails près. Quelles que soient les quantités d’échantillon disponibles, les dépôts sont invariablement faits sur des filaments multiples de rhénium, sans emploi d’activateur. Les mesures se font sur des ions monoatomiques, non combinés avec l’oxygène. Les valeurs des paramètres et la nature des solvants diffèrent en fonction des conditions locales.
Par exemple, au CEREGE d’Aix-en-Provence, on dépose de manière très classique 100 ng ou plus, tandis qu’un courant de 0,8 A circule dans le filament afin d’acccélérer son séchage. A la fin du dépôt le courant est porté à environ 1,2 A, pour faire oxyder le ruban de métal en le chauffant au rouge sombre. L’analyse proprement dite se fait après un chauffage progressif des deux filaments en 15 ou 30 minutes à 4,5 ou 5 A (filament d’ionisation) et 1,5 ou 2 A (filament d’évaporation). On vérifiera qu’il n’y a pas d’éléments isobariques qui risquent d’interférer avec le néodyme, comme le samarium. Le cas du samarium illustre une manière de s’en affranchir. Cet élément qui possède des isotopes aux masses 144, 148, 149, 150, peut être détecté par la visualisation de son isotope 147, sachant qu’il n’existe pas de néodyme à cette masse. Si tel est le cas, l’interférence est considérée comme négligeable en dessous de 300 coups/seconde de samarium 147 environ. Au-delà, une correction est possible en soustrayant aux signaux de néodyme, les pics parasites recalculés à partir du samarium 147. D’autres éléments peuvent interférer, tels le cérium et le baryum, pour lesquels une démarche similaire peut être menée.
A l’IFREMER de Brest, où l’on analyse des échantillons à partir de 20 ng, le néodyme prêt à être déposé est repris avec de l’eau ultrapure, puis chargé sur un filament de rhénium raffiné par zone. Le séchage du dépôt est effectué à l’aide d’un courant de 0.5 à 1 A. Pour améliorer sa fixation sur le filament, on porte ensuite celui-ci rapidement au rouge sombre avant de l’éteindre immédiatement.
Le LEGOS et le LMTG de Toulouse peuvent descendre jusqu’à des quantités de 6 ng, grâce au protocole suivant : après séparation chimique, les échantillons amenés à sec sont repris avec 1,5 à 2 µl d'acide chlorhydrique à 2 mol/l, pour être déposés sur des filaments de rhénium dégazés entre deux barrières de cathéter fondu à 1,2 A. Ces barrières évitent l’étalement du dépôt et facilitent la mesure, surtout lorsqu'il s'agit de très petites quantités. On dépose entre 6 et 150 ng de néodyme, toujours sur double filament. Pendant que le dépôt est effectué, le ruban métallique est parcouru par un courant de 0,8 A qui favorise un séchage rapide. A la fin il est chauffé brièvement au rouge pour permettre une meilleure adhésion de l’échantillon. Au moment de l’analyse, les filaments sont portés au maximum à 4,5 A pour le filament ionisant et à 1,5 A pour l'évaporant. On vérifie qu'il n'y ait pas de samarium ; s’il est présent, ce qui reste exceptionnel, l'échantillon n'est pas mesuré mais récupéré et soumis à une nouvelle purification.
Les chercheurs du Lamont Observatory analysent le néodyme sous sa forme oxydée (NdO+).
Ils injectent dans la source un flux de gaz O2 et mesurent les pics autour de la masse 160,
c'est-à-dire 144Nd16O, en contrôlant la pression qui ne doit pas excéder 10-6 mbar, avec un
Laboratoire Fila- Ments Qté de Nd Séchage/ oxydatio n Reprise du Nd Int. 144Nd Courant filament mesure Activa - teur Interf. ou inhib. Instrum. Géosciences Rennes Re-Re 100 à 300ng 0,5A/ 1,9A HNO3 2 à 3V ? Sans ? MAT262 LMTG et LEGOS Toulouse Re-Re Re-Re Re-Re 6 à 15ng 15 à 30ng 30 à 150ng rouge sombre « « HCl 2N HCl 2N HCl 2N 0,1V 0,3 à 0,6V 1V 1,5A eva 4,5A ioni 1,5A eva 4,5A ioni 1,5A eva 4,5A ioni Sans « « Sm, Ba « « MAT261 « « IFREMER / IUEM Brest Re-Re 20 à 500ng 0,9A/ rouge sb. H2O mQ >0,3V 4à5A ioni 1à2A eva Sans Ce, Sm Triton CEREGE Aix-en-Pce Re-Re 100 ng 0,8A/ 1,2A
HNO3 1V 1,5A eva
4,5A ioni Sans Sm MAT262 IGL Boise (USA) Re-Re 1 µl (?)
1A/ 2,4A H3PO4 ? 2A eva
5A ioni Sans ? Isoprobe T Lamont Observatory (USA) Re 70-140 ng 1,2/1,4 + lueur 1s HNO3 50 mv NdO+ 2A Sans ? VG ?
PLOMB
Généralités et historique de la méthodologie
L’énergie de première ionisation du plomb est une des plus élevées (7,42 eV), ce qui rend a
priori difficile son analyse en TIMS. Cependant, une méthode empirique a fait ses preuves, qui consiste à mélanger l’échantillon de plomb avec un gel de silice. Le principe est de former un verre qui permet de faire émettre le plomb à basse température. En fait la nature du processus qui facilite l’ionisation est mal comprise (Delmore, 1991). On ne sait pas si le rendement est amélioré grâce à un phénomène électrochimique ou grâce à une limitation physique de la volatilité du plomb.
La méthode de préparation la plus ancienne de ce gel a été mise au point par Akishin et coll. (1957), puis améliorée par Cameron et coll. (1969). En 1984, Gulson et coll. ont décrit une méthode de préparation de particules de gel par hydrolyse d’orthosilicate de tétraéthyle. Ces auteurs insistent sur la taille réduite et homogène des particules (moins de 0,2 um) qui ne doivent pas floculer. Ils chargent par électrodépôt quelques centaines de ng de plomb dissous dans de l’acide phosphorique. L’efficacité de l’ionisation est supérieure à 3% pour 100 ng.
Une nouvelle préparation de gel de silice a été mise au point par Gerstenberger et Haase en 1996. Elle contient de l’acide phosphorique et de l’acide silicique en solution colloïdale. La nouveauté essentielle réside dans le choix de l’acide silicique utilisé. Elle a donné des rendements d’ionisation de 10 %, alors que les méthodes classiques ne dépassaient pas 1 ou 1,5 %.
La précision des mesures isotopiques du plomb dépend entre autres de l’absence de contaminations organiques. Celles-ci apparaissent habituellement à basse température. Au-delà de 1000°C elles deviennent négligeables (Doucelance et Manhès, 2001).
Auteur Méthode Quantité Rendement
Akishin 1957 Gel Si - Env. 1% ?
Cameron 1969 Gel Si - Env. 1,5 % ? Gulson et coll. 1984 Gel Si par hydrolyse
d’orthosilicate de tétrahédryle
100 ng 3 %
Gerstenberger et Haase 1996
Gel Si avec acide silicique
20 ng 10 %
Tableau V : Techniques de dépôt du plomb et rendements d’ionisation obtenus, d’après la bibliographie.
Après séparation élémentaire, les dépôts de plomb se font généralement sur des filaments de rhénium simples. Un échantillon peut contenir environ 100 ng de plomb ou plus pour être mesurable sur cage de Faraday.
Dans la méthode pratiquée au CEREGE, le plomb est d’abord dissous dans de l’acide phosphorique. Le gel de silice est déposé dans le becher sur la goutte d’échantillon, puis l’ensemble est repris plusieurs fois avec la seringue. Le mélange est déposé sur un filament de rhénium porté à 0,9 A. Le courant dans le filament est ensuite augmenté très lentement jusqu’à 1,5 A, par paliers de 0,2 A qui durent trois minutes. Au maximum du courant, on doit voir s’échapper la fumée de l’acide phosphorique. Le verre se forme sous vide dans la source pendant le chauffage de l’échantillon. L’analyse a lieu autour de 1100°C.
Le LMTG de Toulouse utilise la même méthode, l’échantillon étant déposé tandis qu’un courant de 0,8 A circule dans le filament, poussé ensuite progressivement jusqu’à 2 A. La température d’analyse contrôlée au pyromètre optique est voisine de 1300 °C lorsque le filament émet un signal pouvant atteindre 5 volts de plomb 208.
Le laboratoire américain de Boise (Idaho) décrit une pratique très semblable. Le gel de silice est mélangé à l’acide phosphorique avant d’être ajouté à l’échantillon de plomb, que l’on laisse se dissoudre pendant une minute. L’ensemble est déposé sur le filament resté froid. Celui-ci est ensuite chauffé par un courant de 1,5 A. Après séchage, on fait croître le courant par paliers de 0,2 A qui durent une minute, pour atteindre environ 2,5 A. L’échantillon devient gris ou noir vers 2,2 A, puis se met à fumer vers 2,5 A en devenant gris clair. Il faut alors le refroidir immédiatement avant qu’il ne tourne au blanc, car il risque de se détacher du filament.
Le Lamont Isolab dépose le plomb puis sous 1A ajoute le gel de silice, puis de l'acide phosphorique. Le dépôt est séché entièrement à 1,3A, puis chauffé davantage jusqu'à légère incandescence et enfin refroidi.
Laboratoire Fila- men t Qté de Pb Séchage/ oxydatio n Dilutio n du Pb Int. 208Pb Couran t/ Temp. mesure Activate ur Instrum. IPG Paris Re 5 à 20ng ? ? ? ? Si gel VG ou MAT262 LMTG/LEGOS Toulouse
Re ? 0,8/2A ? 5 V ? Si gel MAT261
IFREMER / IUEM Brest Re >50 ng 1A / rge ou vap.ac H3PO4 variable variabl e Si gel Triton CEREGE Aix-en-Pce Re 100 ng
0,9/1,5 A Si gel 2 V 1,5 A Si gel MAT262
IGL Boise (USA) Re ? 1,5/2,5 A Si gel- H3PO4 ? ? Si gel Isoprobe T
URANIUM
Généralités et historique de la méthodologie
Le rendement d’ionisation de l’uranium est plutôt faible et dépasse rarement les quelques pour cent. Ces mauvaises conditions sont un facteur limitant pour la précision des mesures, notamment lorsque les échantillons sont peu abondants.
En fonction de la quantité d’uranium disponible dans un échantillon, on choisit habituellement entre deux types de dépôt. Les échantillons en faible quantité (quelques nanogrammes) sont déposés sur simple filament de rhénium. On ajoute éventuellement un agent chimique facilitant l’ionisation (voir ci-dessous). Les échantillons en quantités supérieures à quelques dizaines de nanogrammes sont déposés sur double ou triple filament de rhénium, sans activateur.
Diverses investigations ont été menées pour tenter d’améliorer ce rendement, notamment pour les échantillons de faibles teneurs déposés sur simple filament. En réalité une fraction de l’uranium est toujours ionisée sous forme d’oxydes : U0+, U0
2
+… (Studier et coll. 1962).
Même sous ultra-vide, une proportion importante du dépôt n’échappe pas à cette oxydation. A tel point que certains laboratoires ont choisi d’analyser l’uranium sous une forme oxydée. L’uranium ainsi mesuré a donné un rendement d’ionisation de 0,5 % (Yokoyama et coll. 2001). L’inconvénient majeur de ce type d’analyse est qu’il multiplie le nombre d’espèces chimiques sur le spectre de masses, nécessitant de faire une correction des résultats par les rapports isotopiques de l’oxygène.
A l’exception de cette méthode rarement employée, l’analyse de l’uranium sous forme monoatomique (U+) est largement préférée dans la plupart des laboratoires. On cherche donc
plutôt à limiter l’oxydation des ions d’uranium émis. Pour cela une méthode consiste à rajouter à l’échantillon un agent réducteur. Un élément relativement efficace dans cette fonction est le carbone, qui a été testé dans cette fonction sous plusieurs formes. Sous sa forme atomique dans un réseau de cristal (graphite), le carbone est quatre fois réducteur puisqu’il est tétravalent. En outre, cet effet se double d’une influence sur la fonction de travail du filament de rhénium. En effet, la présence de graphite en couches sur le filament de rhénium fait baisser sa fonction de travail de 0,9 eV. Cela dit, les effets de l’ajout de carbone sont complexes. A la différence de la forme graphitique, si le carbone est en « solution solide » dans le rhénium (c’est-à-dire mélangé au métal de manière désordonnée ou amorphe), il accroît au contraire de 0,25 eV la fonction de travail du substrat. Une troisième forme, le carbure de rhénium (cristal régulier) semble l’accroître de 0,4 eV (Wayne et coll. 2002).
Afin de réaliser une solution solide de carbone dans le métal, des expériences ont été menées pendant l’étape du dégazage des filaments, mettant en œuvre une introduction de benzène dans l’enceinte sous vide pour faire diffuser du carbone dans les filaments incandescents (Studier et coll. 1962, Pallmer et Gordon 1980, Warnecke 2002). Ces investigations difficiles à mettre en œuvre sont apparemment restées à l’état expérimental.
Une autre approche a consisté à déposer simplement sur le ruban de métal une solution aqueuse de graphite, mélangée à l’échantillon d’uranium ou bien déposée en « sandwich » avant et après lui (Goldstein et Stirling, 2003 ; Warnecke 2002). Cette pratique facile et rapide est devenue la plus courante.
Cependant l’ajout de carbone n’a pas été la seule voie explorée, et l’utilisation d’autres substances comme activateurs potentiels a été expérimentée. Il semblerait par exemple que l’émission soit améliorée par un ajout préalable d’iridium métallique en poudre sur le filament simple de rhénium. La poudre d’iridium est fondue sur le filament au cours du dégazage de celui-ci (Lugmair, cité par Rubin, 2001).
Une autre méthode empirique se fonde sur la réalisation d’un verre autour de l’échantillon. L’activateur à base de gel de silice mis au point pour le plomb, peut aussi être avantageusement utilisé pour l’uranium (Gerstenberger et Haase 1997). Cette préparation est déposée avec l’échantillon sur le filament, et le verre se forme au moment du chauffage sous vide précédant l’analyse. Le processus théorique qui permet d’améliorer par ce biais le rendement d’ionisation reste mal compris.
En théorie, si les rendements d’ionisation sont faibles, c’est parce que la majorité des atomes quittent le dépôt sous forme neutre, et ne sont donc pas analysables. Ceci est dû au fait que l’énergie d’évaporation de l’uranium est bien plus basse que son énergie d’ionisation. Une grande proportion de l’échantillon déposé est ainsi perdue.
A partir de ce raisonnement, des tentatives ont été faites pour retarder l’émission des atomes d’uranium, afin que l’évaporation se produise à peu près en même temps que l’ionisation. La proportion d’atomes ionisés serait donc accrue. A cette fin, la diffusion contrôlée consiste à enfermer (ou encapsuler) le dépôt d’uranium sous une couche de métal qui doit freiner son évaporation. Lors du préchauffage, les atomes à analyser diffusent lentement à travers la couche de métal et ne sortent qu’à une température où ils peuvent être ionisés. Les métaux qui ont été employés avec succès pour réaliser cette couche superficielle sont essentiellement le platine et le rhénium, dont les fonctions de travail et les températures de fusion sont relativement élevées.
Il existe plusieurs manières de réaliser cette couche de métal. Parmi celles-ci, l’électrodéposition est une électrolyse provoquée sur le filament au moment du dépôt (Rokop et coll. 1982 ; Perrin et coll. 1985 ; Ramebäck et coll. 2002 ; Jones 2003). Un bain électrolytique est réalisé sur le filament, matérialisé par une grosse goutte dans une cuvette limitée par du ruban adhésif. Deux microélectrodes sont immergées dans le bain. Le dépôt sur le filament de l’échantillon en solution se fait en même temps que celui du métal. Le rendement d’ionisation atteint péniblement les 0,015 % (Rokop et coll., 1982). Le platine semble plus efficace que le rhénium. Cette méthode est en outre difficile à mettre en œuvre.
Une autre façon de recouvrir le dépôt d’une couche métallique est la projection par chauffage sous vide, à partir d’une source de métal très proche (Rec et coll., 1974). Cette source n’est autre qu’un second ruban de métal, approché à 1 ou 2 mm du premier et chauffé pour émettre des atomes qui rencontrent la surface à recouvrir restée froide. Le filament de la source est chauffé progressivement par un courant alternatif. Le rhénium semble avoir donné d’excellents résultats : pour des quantités d’uranium de l’ordre du picogramme, le rendement d’ionisation obtenu pour l’uranium étant multiplié par 4 au moins. Quoique l’épaisseur de la
couche métallique soit difficile à contrôler, la méthode est relativement simple à mettre en oeuvre.
La thèse de Kraiem (2007) fait une bonne synthèse de toutes ces investigations. Elle a étudié la thermo-ionisation de l’uranium en testant les différentes méthodes, et en analysant systématiquement aux rayons X les produits présents sur les filaments à différentes étapes du chauffage. Deux voies sont explorées : l’augmentation de la fonction de travail du substrat, et la réduction chimique des espèces présentes pour limiter l’oxydation de l’uranium. Pour la première, le filament de rhénium est recouvert d’un film métallique à haute fonction de travail (or, platine) sur lequel l’uranium est déposé. La deuxième voie a été l’occasion de tester plusieurs activateurs contenant du carbone sous diverses formes : Collodion (solution de nitrocellulose mélangée à de l’alcool et à de l’éther), film de graphite réalisé par projection, et Aquadag (suspension de graphite colloïdal). Après plusieurs essais incluant la combinaison des deux voies, il est nettement apparu que le principal facteur d’augmentation du rendement d’ionisation de l’uranium n’était pas la présece d’un film métallique à haute fonction de travail, mais précisément l’ajout de carbone. En l’absence de carbone, quel que soit le support, la quasi-totalité de l’uranium s’évapore sous forme d’oxydes UO2
+ et UO+. En
revanche, le carbone favorise l’émission d’ions U+, améliorant ainsi le rendement
d’ionisation. On constate également dans ce cas la formation d’un carbure d’uranium à la surface du filament. En outre, la nature de la source de carbone a des effets sur les conditions d’analyse. La suspension de graphite Aquadag apporte une pollution en uranium 238 ; elle ne peut pas être utilisé dans des quantités très reproductibles, et permet difficilement l’obtention d’un dépôt ponctuel. La source de carbone déposée sous forme de film graphitique provoque elle aussi un étalement du dépôt. Le Collodion semble être la source de carbone la mieux adaptée, car il permet la formation d’un dépôt d’uranium ponctuel en formant un microfilm transparent fixant l’uranium au centre du filament.
Auteur Méthode Quantité Rendement Studier et coll. 1962 Carbone par dégazage
avec benzène
1 ng à 10 ug -
Rec et coll. 1974 Projection Re sur Re simple
2 pg Env. 10 % ?
Rokop et coll. 1982 Electrodéposition Re sur Re simple
10 ng 0,015%
CEREGE 2006 (n. publ.)
Re triple sans activateur 1,5 ug 0,16 %
Yokoyama et coll. 2001 Acide silicique et H3PO4 pour UO2 + simple Re 10 à 100ng 0,5 % Ramebäck 2002 Electrodéposition Pt sur Re simple - -
Kraiem 2007 Graphite ou Collodion sur Re, avec ou sans Au ou Pt
0,1 à 1 ng 0,028 % à 0,069 %
Tableau VII : Techniques de dépôt de l’uranium et rendements d’ionisation obtenus, d’après la bibliographie.
Méthodes pratiquées dans différents laboratoires
Les analyses d’uranium se font en général sur filaments de rhénium multiples pour les grandes quantités, et simples pour les quantités faibles. Dans ce dernier cas un activateur est souvent usité, graphite ou gel de silice.
L’IPG de Paris emploie le gel de silice, avec la même méthode que pour le plomb, sachant qu’elle est pertinente pour des quantités d’uranium comprises entre 5 et 20 nanogrammes. L’uranium est analysé sous forme d’oxyde, les masses à mesurer étant 265 (233UO
2), 267
(235UO
2) et 270 ( 238UO
2). Au moment de l’analyse, le filament est porté à une tempérture
comprise entre 900 et 1350 °C, par étapes de 50 degrés qui durent cinq minutes. Le fractionnement de masse obtenu est de 1,5.10-3 par écart de masse (Manhès, comm. pers.).
Au CEREGE d’Aix-en-Provence, les échantillons d’uranium en faible quantité sont mesurés sous la forme non oxydée. Après purification chimique, l’uranium dissous dans de l’acide nitrique est déposé sur simple filament de rhénium, « en sandwich » entre deux couches d’une solution colloïdale de graphite. Notons que la « solution » de graphite, hétérogène, est passée aux ultra-sons avant chaque usage. Au-delà de plusieurs dizaines de nanogrammes d’uranium, le dépôt se fait sur triple filament de rhénium et sans activateur. Dans les deux cas, pendant la réalisation du dépôt on fait circuler 0,8 A dans le filament pour accélérer son séchage. Un soin particulier est pris pour réaliser un dépôt le plus ponctuel et centré possible. A la fin du dépôt on fait croître le courant jusqu’à 1,2 A pour provoquer l’évaporation de l’acide, puis on le fait redescendre lentement jusqu’à zéro. Une fois le dépôt installé dans la source, un préchauffage automatique précédant l’analyse génère en une demi-heure 4,5 A en simple filament, ou 4,5 / 1,8 A en triple filament.
Initialement les dépôts étaient effectués sur filaments latéraux de tantale, mais ce métal n’est plus utilisé à cause de sa trop grande inertie thermique, causant un long temps de réaction des faisceaux à une augmentation de courant. L’emploi du rhénium est devenu systématique, étant plus prompt à répondre à la chauffe. Globalement, les rendements d’ionisation pour l’uranium obtenus au CEREGE ne dépassent pas 0,16 %.
L' Isotope Geology Laboratory américain de Boise (Idaho) décrit une méthode identique pour l’uranium et pour le thorium : un microlitre d’acide chlorhydrique concentré à 3 mol/l est ajouté à l’échantillon pour le dissoudre ; puis deux microlitres de graphite colloïdal sont déposés au centre du ruban de rhénium. La solution de graphite utilisée contient 7 volumes d’eau dé-ionisée pour un volume de graphite « Ted Pella ». L’échantillon est ensuite déposé sur la solution de graphite. Le courant est alors porté à 1,5 A pour le séchage, puis augmenté jusqu’à 2,5 A par paliers de 0,1 A qui durent 15 secondes. Le filament est refroidi dès qu’il commence à devenir lumineux.
Le même laboratoire cite également une autre méthode, utilisable pour l’uranium et pour le plomb, qui consiste à analyser l’uranium sous forme d’oxyde (UO2
+
), en l’enfermant dans une capsule de verre formée avec un gel de silice exactement comme pour le plomb (voir plus haut).
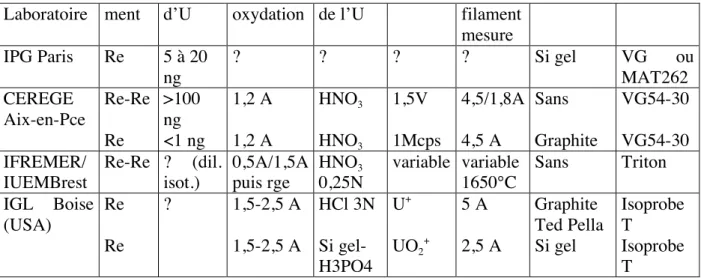
Laboratoire ment d’U oxydation de l’U filament mesure IPG Paris Re 5 à 20 ng ? ? ? ? Si gel VG ou MAT262 CEREGE Aix-en-Pce Re-Re Re >100 ng <1 ng 1,2 A 1,2 A HNO3 HNO3 1,5V 1Mcps 4,5/1,8A 4,5 A Sans Graphite VG54-30 VG54-30 IFREMER/ IUEMBrest Re-Re ? (dil. isot.) 0,5A/1,5A puis rge HNO3 0,25N variable variable 1650°C Sans Triton IGL Boise (USA) Re Re ? 1,5-2,5 A 1,5-2,5 A HCl 3N Si gel- H3PO4 U+ UO2 + 5 A 2,5 A Graphite Ted Pella Si gel Isoprobe T Isoprobe T
THORIUM
Généralités et historique de la méthodologie
l’analyse isotopique du thorium, très utile en particulier pour la datation uranium-thorium, montre des rendements d’ionisation particulièrement faibles, ce qui constitue le principal facteur limitant pour la précision, surtout lorsqu’il se trouve en petite quantité. La recherche de solutions pour tenter d’améliorer les rendements d’ionisation a suscité des investigations relativement poussées.
Après séparation chimique élémentaire, les échantillons de thorium sont généralement déposés sur filaments simples pour les petites quantités, ou triples pour les quantités supérieures.
Dans les conditions du simple filament, le rendement d’ionisation du thorium est d’autant plus faible que la quantité d’échantillon est importante. Les rendements obtenus sont inversement corrélés aux quantités déposées (Edwards 1988). Ce facteur passe de 0,1 % à 0,001 %, pour des dépôts chargés entre 40 pg et 400 ng respectivement.
Comme pour l’uranium, il est courant d’utiliser le graphite comme activateur pour tenter de réhausser le rendement d’ionisation. Là aussi, le processus physico-chimique qui permet cette amélioration reste mal compris. Plus encore que pour l’uranium, des expériences ont été réalisées avec le graphite afin de mieux maîtriser les conditions de la thermo-ionisation. Pour le thorium on s’est intéressé notamment à l’influence possible des différentes phases du mélange carbone-métal sur la fonction de travail du filament. En théorie, au moment du chauffage dans la source, le carbone déposé sur le filament est logiquement présent sous l’une des trois formes suivantes : soit il forme une simple pellicule de graphite superficielle, soit il pénètre dans le ruban incandescent pour former une solution solide amorphe (désordonnée) de carbone dans le métal, soit il forme un cristal régulier avec le métal, c’est-à-dire un carbure. L’une des questions essentielles était de savoir si le graphite pouvait éventuellement changer de phase et se mélanger avec le métal.
Le graphite utilisé sur des filaments de tungstène formerait un carbure de tungstène, d’après les résultats des essais réalisés à l’IPG de Paris (Manhès, comm. pers.). Dans le cas du rhénium cependant, la présence dans les filaments de la phase carbure de rhénium n’a pas été démontrée. La documentation fournit les diagrammes de phase binaires du mélange rhénium-carbone, qui indiquent que le rhénium ne peut former un carbure que s’il est soumis à des pressions extrêmement fortes (Elliott 1965 ; Jones 2003). En basse pression une solution solide amorphe de carbone dans le rhénium peut exister, mais il n’est pas certain que dans les conditions de l’ultra-vide un cristal de carbure de rhénium puisse se former.
L’un des meilleurs rendements d’ionisation obtenus pour le thorium est celui d’Esat (1995), qui annonce 4 à 6 %. Il s’agit de 100 à 300 pg d’une uraninite déposés sans séparation chimique U/Th, et analysés par la méthode de l’intégration de charge. Ses filaments de rhénium (qui présentent un creux en forme de V, ou dimple filaments) sont dégazés une
première fois à 1500°C, puis une seconde fois à 2000°C avec une couche de graphite pour former le carbure. Ils sont stockés sous ultra-vide en attendant leur utilisation, afin d’éviter l’oxydation. Au moment du dépôt les échantillons de thorium sont déposés entre deux nouvelles couches de graphite étalées sur le filament. Esat explique son excellent résultat par le soin apporté pour empêcher l’oxydation du rhénium, et par la bonne géométrie de l’instrument.
Le choix du type de graphite utilisé semble avoir son importance. En comparant plusieurs graphites de provenances différentes, Xia et coll. (2000) observent des différences significatives dans les conditions d’émission. Apparemment ce n’est pas tant la taille ni la pureté des particules de graphite qui fait la différence, que la qualité du réseau cristallin qui doit être la plus parfaite possible. Le carbone pur donne un résultat désastreux, et l’ionisation se fait d’autant mieux que le graphite n’en contient pas. Leurs résultats obtenus avec du bore et du chlore en ions composés positifs, pourraient avoir une incidence sur le choix du graphite à utiliser pour l’ionisation d’autres éléments.
Le carbone a également été testé sous forme de diamant. Une couche de poudre de diamant recouvrant un dépôt de thorium réalisé sur filament simple de rhénium, a été essayée sur un spectromètre VG 354. Cette expérience faite sur des quantités de thorium de l’ordre de 70 ng, a donné des faisceaux assez peu stables et intenses (Chabaux, cité par Claude-Ivanaj, 1997).
Une autre série d’expériences qui a été menée consistait à mélanger le graphite avec de l’iridium métallique en poudre : le rendement atteignait 0,08 % pour 50 à 100 ng de thorium (Rubin, 2001). Par ailleurs, 1 mg d’iridium posé sur un filament de rhénium simple en V, et fondu lors du dégazage, a servi de support au graphite et au thorium. L’intensité et la stabilité des faisceaux y ont gagné, l’efficacité de l’ionisation atteignant 0,1 %.
Quelques analyses de thorium ont été faites sur des filaments simples de tungstène, pour éviter de polluer en rhénium un instrument servant par ailleurs à analyser ce métal (Manhès, thèse). Le thorium dissous dans de l’acide sulfurique était mélangé à la solution colloïdale de graphite et déposé en même temps. Ensuite le courant dans le filament était porté à 1,8 A pour laisser s’évaporer l’acide.
Des mesures de thorium ont aussi été faites sur des filaments doubles de tungstène sur un spectromètre MAT262-RPQ (Claude-Ivanaj, 1997). La fonction de travail du tungstène (4,19 eV) étant plus faible que celle du rhénium (4,53 eV), l’utilisation du tungtène a fait perdre plus d’un ordre de grandeur au rendement d’ionisation.
En revanche, C. Claude-Ivanaj a effectué des essais préliminaires pour réaliser une « diffusion contrôlée ». Une pellicule de tungstène a été réalisée sur le dépôt, grâce à un préchauffage du filament d’ionisation pendant une heure à 5 A, ce qui donnait une projection de tungstène équivalant à une intensité de 5 mV, tandis que celui d’évaporation restait froid. Ce dernier était ensuite chauffé à 2 A pour l’analyse proprement dite (en double filament), et pouvait alors produire un faisceau de thorium durable. Cette expérience de métallisation du dépôt a donné un rendement un peu meilleur qu’en l’absence de pellicule. Il a aussi été remarqué que l’émission du thorium sous forme d’oxyde (Th0+) était d’autant plus importante que le
filament du dépôt était peu chauffé, à l’inverse de celui d’ionisation. Les rendements d’ionisation ainsi obtenus étaient compris entre 0,006 % et 0,03 %, pour des quantités déposées de 200 ng à 1 ug de thorium.
Des tests de métallisation du dépôt par projection ont aussi été faits au CEREGE, cette fois avec du rhénium. L’effet obtenu a été de décaler considérablement vers le haut le courant de chauffe auquel le thorium émettait, mais sans améliorer de manière perceptible le rendement d’ionisation.
Signalons enfin une méthode très différente, testée apparemment avec succès pour l’analyse du thorium en grande quantité : celle de la source en creuset. L’échantillon est enfermé dans un long réceptacle en tungstène qui est chauffé par bombardement d’électrons. Cette technique donne de très bons rendements d’ionisation, jusqu’à 5,2 % pour le thorium (Kirchner 1990 ; Duan et al 1997 ; Wayne et coll. 2002) mais nécessite de modifier la géométrie de la source.
Auteur Méthode Quantité Rendement Edwards 1988 Simple Re + graphite 40 pg à 400 ng 0,001 à 0,1 % Goldstein 1989 Triple Re 200 ng 0,04 % McDermott 1992 Double Re+graphite 23 ng 0,05 % Palacz 1991 Triple Re 760 ng - Chabaux 1993 Simple Re+diamant 76 ng - Esat 1995 Simple Re + graphite 100 à 300 pg 4 à 6 % Claude-Ivanaj 1997 Double Re +
préchauffage
200 ng à 1 ug 0,006 à 0,03 %
Kirchner 1990 Creuset - 5,2 %
Delanghe 2002 Simple Re + graphite 250 pg 0,22 % Rubin 2001 Simple Re+iridium 50 ng (?) 0,1 %
Table IX : Techniques de dépôt du thorium et rendements d’ionisation obtenus, d’après la bibliographie (notamment Claude-Ivanaj 1997 modifié).
Méthodes pratiquées dans différents laboratoires
Effectués sur du rhénium ou du tungstène, les dépôts de thorium se font habituellement en filament simple avec du graphite pour les quantités de l’ordre du ng, et en filament multiple à partir de quelques dizaines de ng. Les méthodes pratiquées en routine montrent peu de différences dans l’utilisation du graphite.
L’IPG de Paris dépose ses échantillons de thorium sur des filaments simples de tungstène avec graphite. Après évaporation dans 5 µl d’acide sulfurique concentré à 0,05 mol/l, la fraction thorium de l’échantillon est reprise à chaud dans 2 µl d’eau et déposée sur le ruban métallique. Une suspension de graphite (2 µl à 5 mg/ml) est alors ajoutée à la goutte déposée. Le filament est chauffé à 1 A pendant une minute pour que l’eau s’évapore, puis monté et maintenu un moment à 1,8 A pour que l’acide s’évapore.
L’analyse proprement dite se fait à une température de l’ordre de 1450°C. Tandis que le courant dans le filament est maintenu constant dans ces conditions pendant une dizaine de
minutes, une réaction a lieu entre le carbone et le tungstène (carburation ?). Ce phénomène provoque sans doute une variation de résistance du filament, entraînant un accroissement de température de l’ordre de 150°C. Après quoi le courant de chauffe est augmenté manuellement, pour générer une température proche de 1900-1950°C, à laquelle la prise de données est effectuée.
La méthode utilisée au LMTG et au LEGOS de Toulouse, mise au point par M. Roy-Barman et L. Coppola, est décrite en détail par C. Venchiarutti. Après purification chimique, l’échantillon de thorium est évaporé jusqu’à son séchage presque complet, avant d’être déposé sur un filament de rhénium préalablement dégazé à 4,5 A pendant 40 minutes. Le dépôt de 1,5 à 2 ng de thorium se fait sur un filament simple. L’échantillon est dissous dans 1 µl d’eau ultrapure avec une pipette réglée sur 1µl. Puis il est mélangé à 1µl d’une suspension de graphite dans un cathéter monté sur une seringue. Pour ce faire on prélève d’abord 1 µl de graphite, on laisse entrer un peu d’air et on préleve le µl d’échantillon. Le mélange se fait à l’extrémité du cathéter, dans une goutte qu’il faut aspirer et refouler plusieurs fois de suite pour l’homogénéiser. Le mélange est déposé en plusieurs fois sur un filament chauffé à 1 A, un séchage presque complet ayant lieu après chaque couche. Le dépôt observé à la loupe binoculaire doit être concentré et légèrement bombé. Enfin il est chauffé très rapidement au rouge puis refroidi.
Le graphite permet d’améliorer et de rendre plus progressive l’émission du thorium. Le rendement est d’autant meilleur qu’il y a davantage de graphite ; cependant il faut savoir que le graphite rejette de 1 à 2 pg de thorium 232. Le chauffage du filament ne doit pas nécessairement être long. La mesure se fait sous un courant de 3 à 4 A.
A Toulouse également, J. Schmeleff (comm. pers.) préfère quant à lui déposer ses échantillons de thorium avec l'activateur de tantale de J.-L. Birck conçu pour le strontium. Le profil du faisceau obtenu présente deux plateaux dont le second plus élevé dure environ 40 minutes.
Au CEREGE d’Aix-en-Provence, les échantillons riches en thorium sont déposés sur triple filament de rhénium sans activateur. Pour les échantillons à faible teneur, on utilise la méthode du simple filament avec graphite dont voici le détail : après un premier dégazage « classique » (4 A pendant 30 minutes), le ruban de rhénium est recouvert d’une première couche de graphite colloïdal (environ 1 µg), pour être de nouveau dégazé sous vide avec un courant de 5,2 A durant 30 minutes. Cette opération préalable vise à chasser l’oxygène du métal, et à incorporer du carbone dans le filament pour tenter de former un carbure de rhénium (Esat, 1995). Grâce à cette étape préliminaire, le rendement d’ionisation a pu être décuplé (Delanghe, 2002). Enfin l’échantillon de thorium est déposé « en sandwich », avant et après l’ajout de deux nouvelles couches de graphite (environ 1 µg) sur toute la surface du ruban de métal. Le courant est maintenu à 0,8 A pendant la réalisation du dépôt, puis il est finalement porté à 1,3 A durant quelques secondes. Une variante consistant à mélanger le graphite et le thorium dans la goutte à l’extrémité du cathéter a également été essayée, mais elle n’a pas donné de différence significative dans le comportement des faisceaux.
Au cours de l’analyse, on constate souvent sur les échantillons graphités un phénomène d’emballement de l’intensité (émission « en cloche »). La température du filament s’accroît parallèlement, pouvant gagner spontanément plus de 200 °C en quelques minutes. Souvent le faisceau diminue ensuite très rapidement, pour se stabiliser à une intensité réduite.
L’existence presque systématique de ce pic d’émission a été attribuée à plusieurs causes possibles. Soit le carbone diffuse dans le ruban, modifiant sa résistivité électrique : la résistance du métal augmentant, elle provoquerait une élévation de température induisant une production d’ions accrue ; soit le graphite s’associe avec le rhénium pour former un cristal régulier de carbure de rhénium, ayant des propriétés physiques spécifiques ; soit tout simplement la couche de graphite superficielle s’évapore, et ne participe plus à la conduction du courant. Quelle qu’en soit la cause, cet effet d’emballement donnant un pic d’intensité de courte durée est gênant pour la qualité de la mesure. Il semble pouvoir être limité difficilement en prévoyant une durée de préchauffage extrêmement longue (jusqu’à quatre ou cinq heures). Les rendements d’ionisation typiques mesurés sont de 0,009 % en simple filament, et de 0,06 % en double ou triple filament. D. Delanghe a obtenu un résultat optimal de 0,22 % en simple filament.
Le laboratoire de Boise (Idaho) utilise pour le thorium la même technique que pour l’uranium, c’est-à-dire qu’il est déposé sur une seule couche de graphite.
Laboratoire Fila- ment Qté de Th Séchage/ oxydation Dilution Du Th Int. 232Th Courant filament mesure Activateur Instrum.
IPG Paris W ? 1/1,8A H2SO4 ? 1900°C Graphite MAT262
LMTG/ LEGOS Toulouse Re 1,5 à 2ng 1A/rouge Eau mQ 1 µl 105 cps 3 à 4A Graphite 1 µl MAT261 LMTG/LE GOS Toulouse Re ? ? ? ? ? ? Oxyde de tantale MAT261 ? CEREGE Aix-en-Pce Re-Re Re >50ng <50ng 0,8/1,3A 0,8/1,3A HNO3 1 à 2V 1 Mcps 5,1/3A 4,9A Sans Graphite VG54-30 VG54-30 IFREMER/ IUEMBrest Re-Re ? (dil. isot.) 0,5A/1,5A puis rge HNO3 0,25N variable variable 1750°C Sans Triton IGL Boise (USA) Re ? 1,5/2,5A HCl 3N ? 5A Graphite Ted Pella Isoprobe T
Tableau X : Méthodes de dépôt du thorium utilisées dans plusieurs laboratoires.
CHLORE
A côté d'autres méthodes d'analyse de ce gaz (IRMS), il est possible de faire réagir le chlore (isotopes 35Cl et 37Cl) avec du césium (un seul isotope, 133Cs) pour former des ions Cs2Cl
+
(aux masses 301 et 305) pour l'analyser en TIMS. Les avantages de cette forme sont ceux de
l'analyse d'ions lourds positifs, c’est-à-dire qu’ils donnent une faible discrimination en masse. Les instruments peuvent faire les mesures à condition de pouvoir dépasser la masse 300. L'analyse se fait sur filament simple graphité.
Plus précisément, l'analyse du Cs2Cl
+ par TIMS permet de descendre à une précision de +/-
0,2 pour mille avec des quantites de 2 ug par dépôt. Les spectromètres de masse à gaz (IRMS) peuvent aussi analyser le chlore sous une forme CH3Cl
+, mais cela nécessite de grandes
quantités d’échantillon (1 mg), tout en permettant néanmoins d’obtenir 0,06 pour mille de précision.
Banks et coll. ont mesuré le delta 37Cl, qui variait dans une gamme de 15 pmil environ, entre
-8 pmil (eaux de mer profondes) à +7 pmil (mineraux). Ils mesurent les rapports du chlore des inclusions de fluides dans les quartz de certaines roches. Dans ce cas le quartz est broyé, nettoyé à la main puis traité par des résines pour supprimer les ions F- et SO4- - et obtenir une
solution CsCl. Les résines sont des BioRAd AG50-X12 de chez Aldrich.
- deux résines conditionnées sous la forme Ca++ pour supprimer les ions F- ;
- une résine conditionnée sous la forme Ba++ pour éliminer les ions SO4- - ;
- une résine conditionnée sous la forme Cs+ pour convertir les espèces en un composé CsCl.
La solution de CsCl est asséchée sous une lampe à infrarouge, puis redissoute de sorte qu'elle contienne 5 ug de chlore par ul. Le pH est de 2,5 avant la réalisation des dépots. Ceux-ci sont faits pour 10 ug de chaque échantillon sur des filaments de tantale, recouverts par 50 ug de graphite (Ultra F Carbon, Johnson Matthey) mélangé dans de l'éthanol et de l'eau. Il faut maintenir identiques la quantité de chlore et le rapport chlore / graphite déposés, car cela affecte les rapports isotopiques. Le pH doit aussi être le même.
Un signal apparaît à partir de 1,2 A. On le laisse se stabiliser autour de 100 mV pendant 20 minutes au moins, en ajustant le courant. On a pu faire 25 blocs de 12 cycles du rapport Cs2 37Cl+ / Cs2 35Cl+. Pour suivre le fractionnement de masse, on mesurait aussi Cs2 35Cl+ /
Cs+ (masses 301 / 133,25). Le rapport < ou > à 1 était corrélé au rapport 37Cl / 35Cl. On surveille aussi le rapport Cs2 35Cl+ / Cs2 F+ car les ions Cs2 F+ inhibent l'ionisation.
Xiao (2002) fait des analyses du chlore dans l'eau de mer. La preparation chimique pour le TIMS se fait, pareillement, par :
- reaction sur une resine au baryum (sous forme Ba2+) pour eliminer les ions S04 2-. - reaction sur une resine aux ions H+ pour transformer les ions Cl- en HCl.
- reaction sur une resine au Cs pour convertir HCl en CsCl, pret pour l'analyse.
Des étapes supplémentaires peuvent etre ajoutées pour ameliorer la pureté. On supprime par exemple les ions SO4 2- par une résine au Na.
Il analyse le CsCl sur simple filament de tantale ou rhénium avec graphite, avec un VG354 ou un MAT 262. On dépose d'abord la suspension de graphite, puis l'échantillon de CsCl et on laisse sécher le tout sous 1 A pensant 5 min.
Le chauffage se fait à 0,8 A en 10 minutes. Ce sont des ions Cs2+ qui sont émis à partir du CsCl. On peut obtenir 500 mV sous seulement 1 A et à basse temprérature (env. 250°C seulement).
Pendant l'analyse (sur cages) on peut surveiller aussi le faisceau de Cs+ (masse 133) qui sert a titre indicatif a evaluer le fractionnement isotopique (Nakamura 2009). L'analyses se font souvent par commutation de pic en faisant varier le champ magnétique.
Le résultat peut etre reporté en delta Cl 37. Un standard NIST existe, le SRM 975. Xiao et al. ont réalisé un standard "artisanal" d'eau de mer, le ISL354.
BORE
Les isotopes du bore (masses 10 et 11) peuvent être analysés par TIMS, en ions négatifs ou positifs.
Sous la forme d’ions négatifs, le bore est analysé en combinaison avec l’oxygène (BO-) aux masses 42 et 43. A l’inverse, en ions positifs le bore est associé à du césium et à de l’oxygène (Cs2BO2
+), dont les masses sont 308 et 309. Le césium étant monoisotopique, il ne crée pas de
problèmes de combinaisons de masses multiples ; cependant la présence de l’oxygène nécessite une correction.
Les analyses en ions négatifs (BO2
-) permettent d’atteindre une précision voisine de 2 %, alors
qu’en positifs (Cs2BO2
+), on peut gagner un ordre de grandeur et obtenir 0,2 ‰.
L’une des raisons de la différence est due au fractionnement de masse, bien moindre avec les masses élevées qu’avec les masses faibles. En revanche, l’avantage des analyses en ions négatifs des échantillons liquides est qu’elle ne nécessite aucune préparation chimique, car les résultats sont indépendants de la pureté. On dépose l’eau à analyser directement sur le filament. Inversement, la méthode des ions positifs nécessite un procédé de purification assez complexe utilisant des résines échangeuses d’ions.
Il existe un standard de bore qui porte le nom de SRM 951 et dont le rapport absolu 10B / 11B
est de 0,2473 +/- 0,0002. Ce standard sert de référence, et les résultats sont souvent présentés sous forme de delta :
Delta 11B = ( ( ( 11B/10B) échantillon / (11B/10B) SRM951 ) – 1 ) . 103
Historique des méthodes employées
Les premières mesures isotopiques du bore furent faites avec des spectromètres à gaz, selon une méthode qui souffrait d’un effet de mémoire important dû au composé BF3 (Bentley