by Bigyan R. Bista
B.S., Biology; B.S., Mathematics Saint Peter’s College, 2007
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY JUNE 2016
© 2016 Massachusetts Institute of Technology All Rights Reserved
Signature of Author:………. Department of Biology May 20, 2016 Certified By:………. Richard O. Hynes Daniel K. Ludwig Professor for Cancer Research Thesis Supervisor Accepted By: ……….
Michael Hemann Associate Professor of Biology Co-Chair, Biology Graduate Committee
by Bigyan R. Bista
Submitted to the Department of Biology on May 20, 2016, In Partial Fulfillment of the Requirements for the Degree
Of Doctor of Philosophy in Biology
ABSTRACT
Adhesion-GPCRs, a novel family of G protein-coupled receptors (GPCRs), are characterized by
an extended extracellular region linked to a seven-pass transmembrane moiety via GPCR proteolytic site (GPS)-containing stalk region known as GAIN domain. The name adhesion refers to the presence of functional domains in the extracellular region that commonly mediate cell-cell and cell-matrix interactions in various contexts. Recently, many genome-scale analyses of genetic alterations across diverse cancer types have revealed significant alterations (copy number and mutational) in adhesion-GPCRs, yet no comprehensive examination of their roles in cancer biology exists. Through a systematic screening for all adhesion-GPCRs by RT-qPCR in murine mammary carcinoma cell lines with varying metastatic abilities as well as tumor samples of different grades, I have identified several candidate genes with possible roles in breast cancer progression and metastasis. Based on these analyses and cross-referencing with the published gene expression data on human breast cancer cell lines and patient samples, I chose two candidate genes, CELSR2 and GPR126, for more detailed investigation. To elucidate their functions in cancer biology, I investigated the effects of their perturbations using RNAi (loss-of-function) methods both in vitro and in vivo. The results from my work reveal that loss of CELSR2 affects neither tumor growth nor lung metastasis in a xenograft mouse model of breast cancer, despite enhancing invadopodial activity in vitro. I also show that highly metastatic breast cancer and melanoma cells have elevated levels of GPR126, and confirm the significance of this result by revealing (a) reduction in pulmonary metastasis without affecting primary tumor growth in a spontaneous metastasis model of breast cancer, and (b) reduction in lung metastasis in three different experimental metastasis models of breast cancer and melanoma, upon shRNA-mediated knockdown of GPR126. After probing the different steps in the metastatic cascade to investigate how GPR126 promotes metastasis, I demonstrate that GPR126 specifically affects extravasation, most likely through its engagement with type IV collagen in the sub-endothelial basement membrane. Thus, the work described in this thesis contributes to our overall understanding of the perplexing problem of cancer metastasis via identification of novel regulators of distinct steps along the ominous path of malignant cells from primary sites to distant organs.
Thesis Supervisor: Richard O. Hynes
I dedicate this thesis to
my parents,
Jhapindra Bista
and
Bimala Bista
,
my sister,
Bibechana Bista
,
my wife and soulmate,
Elina Pradhan
,
Title Page ……….1
Abstract ………...…………... 2
Dedication ………. 3
Table of Contents ……… 4
Chapter 1: Introduction ……… 6
Chapter 2: Screening Approach to Uncover Novel Adhesion-GPCRs Involved in Breast Cancer Progression and Metastasis ...………... 39
Chapter 3: Investigation of the Role of CELSR2 in Breast Cancer Growth and Metastasis ………. 58
Chapter 4: GPR126 Enhances Extravasation and Metastasis of Breast Cancer and Melanoma Cells ..……… 96
Chapter 5: Discussion and Future Work ……… 133
Materials & Methods ………..………...…. 147
Acknowledgments ……….. 160
CHAPTER 1.
Introduction
A preamble to the investigation of an intriguing surface receptor family
There is no dearth of reasons to be terrified, mortified, petrified, stupefied by metastasis. What originated from the Greek word methistanai in the 16th century as rhetorical term for ‘rapid transition from one point to another’ is now best known in the context of cancer biology to describe the ominous march of malignant cells from the primary tumor to distant secondary sites in a patient’s body almost always choreographing a fatal conclusion. The journey from the primary site to distant organ for tumor cells is, however, a perilous one, with many challenges to surmount. The metastatic cascade is known to involve multiple steps, including local invasion, intravasation into blood or lymphatic vasculature, survival in circulation and systemic dissemination, arrest in capillary beds and extravasation into secondary organs, and finally outgrowth after varying degrees of latency to form clinically relevant lesions (Labelle and Hynes, 2012a; Nguyen et al., 2009).
The conclusions that most circulating tumor cells (CTCs) succumb to stresses and insults along the way, and that metastatic colonization represents one of the most restrictive bottlenecks are rooted in clinical evidence. First, the numbers of CTCs in blood from cancer patients greatly outnumber the overt metastases that develop (Nagrath et al., 2007); second, disseminated tumor cells (DTCs) that survive after infiltrating secondary sites can be found in bone marrow of breast cancer patients for years, and yet only about half of these patients develop overt metastases (Braun et al., 2005). Finally, more than half a dozen studies on pulmonary metastases in breast cancer and soft tissue sarcoma patients, and liver metastases in uveal melanoma patients report that, in >75% of cases,
human malignant tumors were already disseminated when the primary tumors were detected but stayed dormant for many years (Breur, 1966; Collins et al., 1956; Eskelin et al., 2000; von Fournier et al., 1980; Rööser et al., 1987; Tubiana et al., 1975). The inefficiency notwithstanding, even small tumors can shed millions of cancer cells and a few cells with the appropriate genetic and epigenetic (in the broadest sense) makeup to make it past the metastatic cascade could deal a fatal blow (Butler and Gullino, 1975). The fact that metastases often affect vital internal organs and are systemically disseminated with potential for latent growth poses vexing problems in cancer treatment since surgical resection and adjuvant therapies mostly restrict tumor growth at the primary site and have had limited success in treating metastatic diseases. These clinical realities emphasize the need to understand the mechanisms underlying metastasis, which are also interesting in their own right.
At the time the work described in this thesis began, much progress had been made in the field to gain fundamental insights into the molecular bases for metastasis. Several gene expression studies had revealed that the metastatic potential of human tumors was encoded in the bulk of a primary tumor (Paik et al., 2004; Ramaswamy et al., 2003; Sørlie et al., 2001; van ’t Veer et al., 2002) and challenged the notion that metastases arise from rare, variant cells within the primary tumor, whereas other studies had demonstrated alterations in expression of specific genes in metastatic cells and supported the idea of organ-specific metastatic tropism (Clark et al., 2000; Kang et al., 2003; Minn et al., 2005; Xu et al., 2006). It was prudently pointed out that the two ideas need not be incompatible (Hynes, 2003): the shared gene expression pattern between cells derived
from metastases and from their corresponding primary tumors could represent global predisposition to complete some, but not all, steps of the metastatic cascade; this allows fully competent variants to arise through accumulation of additional alterations in the expression of a limited number of genes such that the overall resemblance to the primary tumor was maintained (Hynes, 2003; Kang et al., 2003). Moreover, this model also highlighted the potential roles of non-neoplastic cells, collectively a ‘stromal response’, in clonal evolution of metastatic cells and their spread. Notably, among the most prominently overexpressed genes in various organ-specific metastatic signatures were those encoding cell-surface and secreted proteins, several of which were causally linked to interactions with, or modifications of, the host tissue environment (Bos et al., 2009; Kang et al., 2003; Minn et al., 2005). We were interested in identifying novel mediators of the metastatic cascade, and an emerging family of G-protein-coupled receptors (GPCRs), called adhesion-GPCRs, intrigued us for many reasons as will be explained in the subsequent sections. Work from our lab had recently demonstrated the first genetic evidence for involvement of a member of this family in tumor growth and metastasis (Xu et al., 2006), and we decided to examine closely the entire family in the context of tumorigenesis and metastasis. During the period of this thesis work, much additional data emerged that conformed with this initial hypothesis even as my work was further implicating adhesion-GPCRs in cancer. I will review the development of this deepening understanding of this intriguing family of receptors in the following sections.
Seven-‐transmembrane (7TM) receptor superfamily
7TM receptors, also known as G-protein-coupled receptors (GPCRs), constitute the largest group of plasma membrane receptors that mediate a wide variety of physiological responses to hormones, neurotransmitters and environmental stimulants in mammals and regulate such crucial sensory modalities as olfaction, gustation and vision. More than 800 genes encoding these receptors have been identified in the human genome and approximately 40% of all clinically approved drugs in the market are known to target them. While the exceptional nature of 7TM receptors - their diversity, ubiquity, versatility and pharmacological importance - is widely recognized today, the concept of a functional cell surface receptor was met with skepticism well into the 60s and 70s (Lefkowitz, 2004) despite having originated more than 100 years ago in Paul Ehrlich’s ‘side-chains’ and John Newport Langley’s ‘receptive substance’ theories (Maehle, 2009). Only with the advent of novel biochemical and molecular biological tools and techniques in the last 40 years, namely radioligand binding, receptor purification and reconstitution, and receptor cloning, were researchers able to elucidate molecular understanding and functional significance of prototypical 7TM receptors such as β2-adrenoreceptor and
rhodopsin (Lefkowitz, 2004). Moreover, identification of the handful of prototypical receptors allowed subsequent discovery of the astonishingly large superfamily of 7TM receptors via sequence-based searches against genomes of many species (Fredriksson et al., 2003a, 2003b).
7TM receptors are characterized by the presence of seven transmembrane α-helical segments of relatively high hydrophobicity that are connected by alternating extracellular
and intracellular loop regions. The N-terminus of the receptor lies extracellularly which in combination with the extracellular loops forms a ligand recognition site and also modulates ligand accessibility. The C-terminal region is intracellular and interacts with cytosolic G-proteins and myriad non-G-protein effectors such as G protein-coupled receptor kinases (GRKs), arrestins, multidomain scaffolding proteins and accessory/chaperone molecules to transmit signals (Brady and Limbird, 2002). The versatility of 7TM receptors is also reflected in the diversity of extracellular stimuli that they recognize, including ions, photons, organic odorants, amines, neurotransmitters, chemokines, hormones, peptides, lipids, nucleotides, nucleosides etc. (Lagerström and Schiöth, 2008). The classical role of a GPCR is to couple ligand binding and consequent conformational changes to activation of heterotrimeric G-proteins (Gαβγ) and modulation
of downstream effector proteins to initiate different physiological and pathological processes (Rosenbaum et al., 2009). The G proteins can be divided into several families such as Gs, Gi, Gq and G12/13 based on the functions of the α-subunit. Upon nucleotide
exchange of GDP for GTP, dissociation of the tightly associated Gα and Gβγ follows and
each of which can then regulate distinct signaling cascades. It should be noted that G proteins represent just one out of a multitude of mechanisms of signal transduction, and interaction with G proteins has not been demonstrated for most receptors in the superfamily; as such, ‘7TM receptors’ is considered functionally more appropriate term for the superfamily than the more common ‘GPCRs’. Since canonical GPCR biology has been extensively reviewed in the literature (Lefkowitz, 2007; Oldham and Hamm, 2008), we will turn our focus to the novel family of adhesion-GPCRs.
Classification of the 7TM receptors
Several groups have utilized the diversity in the primary structures of 7TM receptors to classify the members of this superfamily. A sequence-based fingerprint of the seven characteristic hydrophobic domains was first used as a diagnostic tool in identifying all sequences belonging to the 7TM superfamily from OWL composite sequence database (Attwood and Findlay, 1993, 1994). This method increased the number of known 7TM receptors from 240 to 393, and distinguished ‘clans’ of pheromone, cAMP (3’-5’-cyclic adenosine monophosphate) and secretin-like receptors from the prototypical rhodopsin-like family. Kolakowski developed a database of sequences and known mutations in 7TM receptors in vertebrates as well as invertebrates and used the criterion of G-protein binding as well as parsimony analyses to group the receptors into the well known A-F classification system (Kolakowski, 1994). A similar approach was used in combination with structural and ligand-binding criteria to divide the receptors into families 1-5 (Bockaert and Pin, 1999). The large sequence differences between mammalian and invertebrate 7TM receptors, however, made these classification systems cumbersome in human-specific studies.
The publication of the reference sequences of the human genome at the turn of the 21st century was a momentous occasion that spurred advances in comparative genomics and provided unprecedented insights into human evolution, physiology and disease processes (Lander et al., 2001; Venter et al., 2001). The field of 7TM receptor biology was among the direct beneficiaries of the effort that resulted in identification of 802 (known and predicted) human 7TM receptors, including 342 functional non-olfactory receptors, and
their phylogenetic classification into Glutamate, Rhodopsin, Adhesion, Frizzled/Taste2 and Secretin families, termed the ‘GRAFS’ classification system (Fredriksson et al., 2003b). The five families, besides having structural differences in the 7TM regions, vary greatly in their extracellular domains. The Rhodopsin family is the most-studied family with more than 670 members of which many are amine-, peptide- and purine-binding receptors and the largest cluster is formed by olfactory receptors. Most Rhodopsin family receptors have short N-termini and are among the most important drug targets: the histamine receptors, the serotonin receptors, the adrenoreceptors, the muscarinic receptors, the prostanoid receptors and the cannabinoid receptors. The Glutamate family comprises 22 human proteins including metabotropic glutamate receptors, gamma-aminobutyric acid (GABA) receptors, the calcium-sensing receptor, taste receptors and several orphan receptors. Generally, the N-terminal region of these receptors is folded into two lobes separated by a cavity for ligand binding. The Frizzled/Taste2 family consists of frizzled receptors, smoothened receptor, and taste receptors that show weak similarity to the frizzled receptors. The frizzled receptors are best known for mediating Wnt signaling during development. Their N-termini are 200-320 amino acids long with conserved cysteine-rich regions for efficient binding of the curled and twisted Wnt glycoproteins. The Secretin family contains long peptide-binding receptors such as the calcitonin receptor, the glucagon receptor and the parathyroid hormone receptor. The N-termini are 60-80 amino acids long and contain several cysteine bridges that form ligand-binding pockets. The fifth family, the Adhesion family, is the second largest family and has striking structural features that distinguish these receptors from other 7TM receptor
The Adhesion G Protein-‐Coupled Receptors (Adhesion-‐GPCRs)
Discovery and evolutionary history
The Adhesion family consists of 33 receptors in humans and has been further subdivided into nine distinct sub-families (I-IX) (Bjarnadóttir et al., 2004) based on phylogenetic similarity in their 7TM domains and this classification is also bolstered by the molecular signature of the N-termini (Figure 1). The first widely studied adhesion-GPCR was the murine macrophage-specific antigen recognized by the monoclonal antibody F4/80 (Austyn and Gordon, 1981) which was itself not recognized as a 7TM receptor at the time of discovery; nonetheless, it served as an excellent marker for exploring the reticuloendothelial system in the 80s and led to identification of various murine macrophage subpopulations such as microglia, Kupffer cells and Langerhans cells etc. (Yona et al., 2008). F4/80 receptor would be ultimately linked to induction of CD8+ regulatory T cells in peripheral tolerance (Lin et al., 2005). The human ortholog of F4/80, EMR1, was found to be restricted to eosinophils, while another related protein CD97 turned out to be leukocyte activation antigen. Together, these discoveries pointed to a special class of 7TM receptors that had tandemly arranged EGF-like modules in their extended extracellular regions and played key roles in immunology (Baud et al., 1995; Gray et al., 1996; Hamann et al., 1995).
Around the same time, researchers studying the action of black widow spider toxin, α-latrotoxin, on neurons discovered a different 7TM receptor that also featured a long extracellular domain and contained a combination of hormone-binding, lectin-like and olfactomedin-like modules and named it latrophilin (Krasnoperov et al., 1997; Lelianova
et al., 1997). It was quickly realized that both the EMR-related proteins and latrophilin were peculiar 7TM receptors with sequence homology in the 7TM regions, and were eventually grouped together as adhesion-GPCRs after the extent of the family was realized (Fredriksson et al., 2003b). The family derives its name from the presence of protein domains commonly associated with cell-cell and cell-matrix interactions in the elaborate N-terminal region that is fused to the 7TM domain. As alluded to earlier, it was not until the human genome project was completed that all adhesion-GPCRs were discovered and this helped in identifying orthologs across diverse species. Other than EMR2 and EMR4, every human adhesion-GPCR has 1-to-1 correspondence with the mouse genome (Bjarnadóttir et al., 2004, 2006; Fredriksson et al., 2003a).
Despite being one of the most recently discovered human gene families, adhesion-GPCRs are of ancient origin. Not only are they found in mammals and higher vertebrates, but their homologs have also been observed in urochordates and cephalochordates such as sea squirts and Amphioxus and invertebrates such as worms, sea urchins and fruitflies (Nordström et al., 2008; Usui et al., 1999; Whittaker et al., 2006). Putative adhesion-GPCRs have also been reported in single-celled protozoans, including Tetrahymena thermophilia, Dictyostelium discoideum and Monosiga brevicollis (Yona et al., 2008), indicating (i) an ancient association of the modular extracellular regions with 7TM moieties, (ii) potential exogenous ligands, (iii) non-adhesive roles, very likely in innate immunity, beyond the presumed roles in multi-cellular evolution, and (iv) protein evolution by molecular tinkering that gave rise to the complex structure. It has been posited that the adhesion-GPCRs evolved some 1275 million years ago before the split of
unikonts from the common eukaryotic ancestor, most likely from the cAMP receptor (Krishnan et al., 2012; Langenhan et al., 2013). Curiously, while the latrophilin and flamingo homologs have been observed in primitive organisms, the EMR1-4 cluster is only found in higher vertebrates hinting at the continuity of evolutionarily conserved functions and simultaneous rise of novel functions for the family to execute (Kwakkenbos et al., 2006).
Gene and protein structure
All adhesion-GPCRs are characterized by their complex genomic and protein structures. The chimaeric receptors are encoded by multiexonic genes and some of the closely related subfamily members such as the EMRs occupy distinct clusters in the human genome suggesting diversification through exon shuffling, and gene duplication, deletion and conversion events (Kwakkenbos et al., 2006). The multi-exon structure also permits further receptor diversity via tissue-specific or developmentally regulated alternative splicing (McMillan et al., 2002; Shiratsuchi et al., 1997) that can generate receptors with different combination of extracellular modules (Hamann et al., 1996), dominant-negative receptors (Davies et al., 2007), and putative soluble and non-cleavable isoforms, e.g. GPR124 (Vallon and Essler, 2006).
Adhesion-GPCRs possess many unique features in their protein structures that distinguish them from the rest of the 7TM receptor families: (i) as mentioned before, their extracellular regions are among the longest among all 7TM receptors, and contain some combination of fourteen distinct functional domains that are generally not found in other 7TM receptors (Bjarnadóttir et al., 2007), (ii) the long N-termini of most
adhesion-GPCRs have relatively high percentage (~20%) of serine and threonine residues creating many putative O- and N-glycosylation sites with functional relevance (Bjarnadóttir et al., 2007), (iii) the majority are orphan receptors and neither their ligand nor physiological functions are clearly known, (iv) none of the ligands for deorphanized adhesion-GPCRs bind within the 7TM bundle unlike other 7TM receptors (Boucard et al., 2012; Luo et al., 2011; Prömel et al., 2012; Silva et al., 2011; Xu et al., 2006), and (v) all adhesion-GPCRs, with the exception of GPR123, contain a ~40 residue GPCR-proteolysis site (GPS) domain in the extracellular region. The GPS domain is, in turn, an integral part of a much larger ~320 residue domain called GPCR-Autoproteolysis INducing (GAIN) immediately distal to the first TM helix; the GAIN domain mediates autoproteolytic cleavage within the GPS domain (Araç et al., 2012).
The GAIN domain has been found to be a highly evolutionarily conserved feature in adhesion-GPCRs. Structure-based BLAST (Basic Local Alignment Search Tool) searches against the genome of Disctyostelium discoideum have revealed GAIN domains, and the single-celled ciliate, Tetrhymena thermophilia, is now known to encode more than eight GAIN domains (Prömel et al., 2013). However, the GAIN domains are not found in the adhesion-GPCR precursors of these unicellular organisms, and rather exist in the N-terminal region of other transmembrane proteins (McGuffin et al., 2000; Pei et al., 2007). Intriguingly, all PKD (polycystic kidney disease) proteins in humans, thought to function as mechanosensors in renal tubes, also contain GAIN domains despite bearing little other similarity to adhesion-GPCRs. Within adhesion-GPCRs themselves, the GAIN domains between BAI3 and Latrophilin-1 share low sequence identity (~24%) but
have highly similar domain structure (Prömel et al., 2013). Together, these results suggest that the 3D structure might dictate the functional utility of GAIN domains that seems to have found important use throughout evolution, spreading itself across diverse phyla.
Despite the presence of GPS domains in all adhesion-GPCRs, not every receptor is predicted to be cleaved due to absence of a consensus catalytic triad; in fact, GPR111, GPR115 and CELSR1 have been experimentally shown to be non-cleavable adhesion-GPCRs (Formstone, 2010; Prömel et al., 2012), and EMR1, GPR123, GPR124, GPR125 and CELSR3 have been predicted to be uncleaved as well (Langenhan et al., 2013). In the majority of adhesion-GPCRs that undergo autoproteolysis, the reaction occurs in the endoplasmic reticulum (ER) in the early secretory pathway and is dependent on the GPS motif; however, it is the GAIN domain and not GPS motif which is the minimal structural unit necessary and sufficient for autoproteolysis (Araç et al., 2012). Crucially, even though the autoproteolytic reaction produces an extracellular N-terminal fragment and C-terminal fragment, their dissociation is usually prevented due to the strong hydrogen bonds and hydrophobic interactions maintained by the cleaved β-strand within the GAIN domain. Thus, much like the PKD proteins, the mature adhesion-GPCRs undergo heterodimerization upon autoproteolysis at the GPS site and a high fraction of both protein family members exist as homogeneric dimers at the plasma membrane (Araç et al., 2012; Krasnoperov et al., 1997; Qian et al., 2002). The importance of cleavage at the GPS is not entirely understood. While some groups have reported impaired processing and membrane trafficking of proteins that have mutations in the GPS (Jin et al., 2007; Krasnoperov et al., 1997), it is unclear whether such impairment is due to lack of cleavage or to an effect of the mutations on the overall domain structure. In fact, studies
involving mutations in Latrophilin-1 and BAI3, which impaired autoproteolysis but did not affect protein folding and surface localization, suggest that GPS/GAIN domain might have key roles in signaling beyond mediating proteolysis alone (Araç et al., 2012).
To avoid discrepancies and confusion in receptor terminology for an already complex protein family, the Adhesion GPCR Consortium (http://www.adhesiongpcr.org/) recommends following the topology and cleavage-based lexicon as depicted in figure 2. Briefly, like other 7TM receptors, adhesion-GPCRs can be divided into an extracellular domain (ECD), a 7TM domain, and an intracellular domain (ICD) based on receptor topology. Given the autoproteolysis and retained association events, adhesion-GPCRs can also be divided into an N-terminal fragment (NTF) and a C-terminal fragment (CTF). While the NTF contains all extracellular protein domains and much of the cleaved GAIN domain, the CTF contains the C-terminal piece of the cleaved GAIN domain, a short linker region, the 7 TM bundle and the ICD. The consortium, in concert with Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC) and International Union of Basic and Clinical Pharmacology (IUPHAR), has recently also recommended using a revised nomenclature for the Adhesion family (Hamann et al., 2015) as shown in Table 1. For simplicity and ease of reference to the published literature, this thesis work will mostly use the old names; the readers are kindly requested to refer to the table to become acquainted with new names.
Binding partners, signaling and functional roles in development and physiology
As predicted from the domain architecture, most of the interacting partners that have been discovered for a rather small subset of deorphanized adhesion-GPCRs over the last two decades are cellular and extracellular matrix (ECM) ligands, and each of these few receptors has been associated with multiple ligands. The nature of the ligands and promiscuity of the receptors stand in sharp contrast to the other 7TM families that bind specific short peptides or small molecules (Langenhan et al., 2013). Prominent examples are: (i) CD97 on inflammatory cells can bind CD55 (decay-accelerating factor), the first adhesion-GPCR ligand identified, via their EGF-like domains to modulate leukocyte activation, migration and adhesion (Hamann et al., 1996, 1998; Lin et al., 2001), chondroitin sulfate to mediate leukocyte-B cell interaction (Kwakkenbos et al., 2005; Stacey et al., 2003), and α5β1 and αvβ3 integrin counterreceptors on endothelial cells to enhance neovascularization in tumors and endothelial invasion (Wang et al., 2005), (ii) GPR56 NTF can bind tissue transglutaminase (TG2), a calcium-dependent ECM-modifying enzyme, to inhibit melanoma progression and metastasis (Xu et al., 2006), and collagen III to regulate neuronal migration during cortical development (Luo et al., 2011), (iii) Latrophilins (1-3) bind single-span transmembrane molecules such as teneurins (O’Sullivan et al., 2012; Prömel et al., 2012; Silva et al., 2011), FLRTs (fibronectin leucine-rich transmembrane proteins) (O’Sullivan et al., 2012) and neurexins (Boucard et al., 2012) to mediate axon guidance, trans-synaptic adhesion and neuronal connectivity, and synapse function and maintenance, (iv) BAI1 on macrophages binds phosphatidylserine to enable both the recognition and internalization of apoptotic cells (Park et al., 2007) while BAI3 interacts with C1q-like molecules in modulating synapse
formation (Bolliger et al., 2011), and (v) CELSR family members exhibit cadherin-mediated hemophilic adhesions to establish and transmit planar cell polarity in various developmental processes, such as neural tube closure (Nishimura et al., 2012), somitogenesis (Formstone and Mason, 2005) and organ morphogenesis (Cortijo et al., 2012; Yates et al., 2010), and also influence axon guidance, dendritic morphogenesis and neuronal migration (Tissir et al., 2010).
The Adhesion family-specific unique features are not limited to the remarkable extracellular region; receptors belonging to the BAI and CELSR subfamilies, for example, have relatively long intracellular domains that suggest potential for complex intracellular interactions. Indeed, the intracellular domain of BAI1 is known to mediate phagocytosis of dying cells and Gram-negative bacteria by forming a complex with ELMO (engulfment and cell motility) and Dock180 (dedicator of cytokinesis) proteins which activates the Rac signaling pathway (Das et al., 2011; Park et al., 2007). Additionally, the intracellular tail also can interact with Par3/Tiam1 complex, independent of the ELMO/Dock/Rac pathway, to localize it to synaptic sites for synapse formation (Duman et al., 2013). CELSR1 has been shown to direct actomyosin-dependent planar-polarized contraction to initiate neural tube closure during chick embryogenesis via intracellular interaction with Frizzled/DAAM1 and PDZ-RhoGEF to activate Rho kinase (Nishimura et al., 2012). Furthermore, GPR97 was linked to Rho kinase activation without any apparent involvement of G proteins in migration of lymphatic endothelial cells (Valtcheva et al., 2013). Lastly, β-arrestin-mediated signaling has also emerged as a possibility for GPR56, GPR97, BAI1 and BAI3 (Paavola and Hall,
2012; Southern et al., 2013; Stephenson et al., 2013), thereby significantly increasing the signaling repertoire, both G protein-dependent and independent, downstream of the cellular context sensed by adhesion-GPCRs.
In spite of the identification of the various interaction partners mentioned above, whether adhesion-GPCRs serve purely adhesive functions or are capable of activating G-protein-mediated signaling has been difficult to study, mainly due to incomplete knowledge of expression patterns across and within species, notoriously low endogenous expression in cells and tissues when detected, and lack of reliable reagents such as antibodies and convenient signaling readouts. Several recent studies, however, suggest that most, if not all, adhesion-GPCRs, might actually couple to and signal via G proteins. Teneurin-2 binding to Latrophilin-1 leads to intracellular Ca++ release (Silva et al., 2011); collagen III binding to GPR56 activates RhoA signaling which is abrogated by dominant negative Gα13 (Luo et al., 2011); myelination defects in zebrafish gpr126 mutants are rescued by
forskolin-induced cAMP stimulation indicative of Gαs coupling (Monk et al., 2009); and
in vitro assays where different members of the G protein and Adhesion family were overexpressed in HEK293 cells in a combinatorial fashion revealed that many adhesion-GPCRs including EMR2, GPR110, GPR115, BAI1, GPR97, GPR114 and GPR126 can induce intracellular accumulation of inositol phosphate or stimulation of cAMP, implying signaling via G protein-coupling (Gupte et al., 2012).
Implications in tumor biology
Given their roles as key regulatory elements in a broad range of physiological processes, it is reasonable to expect that aberrant expression and activity of GPCRs, G proteins and other components of GPCR-signaling pathways could contribute in carcinogenesis. Indeed, activating hotspot mutations in genes for G protein α subunits, particularly GNAS, GNAQ and GNA11, are frequently seen in and have been causally linked to cancer progression (Dhanasekaran et al., 1995; Dorsam and Gutkind, 2007); however, the discovery from a systematic analysis of somatic mutations in cancer genomes (Kan et al., 2010) that GPCRs themselves are mutated in approximately 20% of all cancers surprised many (O’Hayre et al., 2013) and revealed that such mutations are widespread across tumors arising in large intestine, skin, ovary, prostate, breast, thyroid, central nervous system, lung, stomach, haematopoietic and lymphoid tissue among others. The most exciting finding from this survey for us was that the highest frequency of alterations among GPCRs was observed in the coding sequences of the members of the most poorly understood family of GPCRs, the Adhesion family.
Despite lack of information on functional significance of the somatic mutations, we had good reasons to hypothesize that adhesion-GPCRs might be playing crucial roles in tumorigenesis and metastasis:
1. The best-known physiological roles for adhesion-GPCRs involve key processes in embryonic development and immunology - including but not limited to stemness, EMT and collective-cell migration behaviors, planar cell polarity signaling in epitheliua, cell differentiation, leukocyte activation,
adhesion and migration - all of which have been linked to cancer biology (Lim and Thiery, 2012; Thiery, 2003; Trinchieri, 2012; de Visser et al., 2006).
2. The N-terminal domain architecture seems poised to mediate various cell-cell and cell-matrix interactions, features that regulate virtually every aspect of tumor growth, and metastatic dissemination and colonization (Labelle and Hynes, 2012b; Lu et al., 2012; Nguyen et al., 2009).
3. Many adhesion-GPCRs are known to have restricted expression patterns in specific cell types or tissues such as leukocytes, smooth muscle cells, ependymal epithelial cells and brain (Stacey et al., 2000; Yona et al., 2008). Both normal and aberrant activity of such receptors might affect the metastatic proclivity of tumor cells at the primary site as well as their growth at the secondary site.
4. The potential for combinatorial engagement of the diverse protein folds in the N-terminal region with extracellular ligands and the myriad signaling pathways that can be activated in response by the C-terminal region could impart molecular rheostat-like traits that can modulate the dynamic behavior and unique abilities required of cancer cells as they navigate the difficult path from primary tumor to distant sites.
This thesis seeks to explore the putative roles of adhesion-GPCRs in cancer progression and metastasis in a systematic and comprehensive manner, not only to uncover novel determinants of tumorigenesis and metastasis but also to address the lack of knowledge of physiologically and pathologically important functions in human biology for this
fascinating family of surface receptors. The subsequent chapters are structured in the following manner:
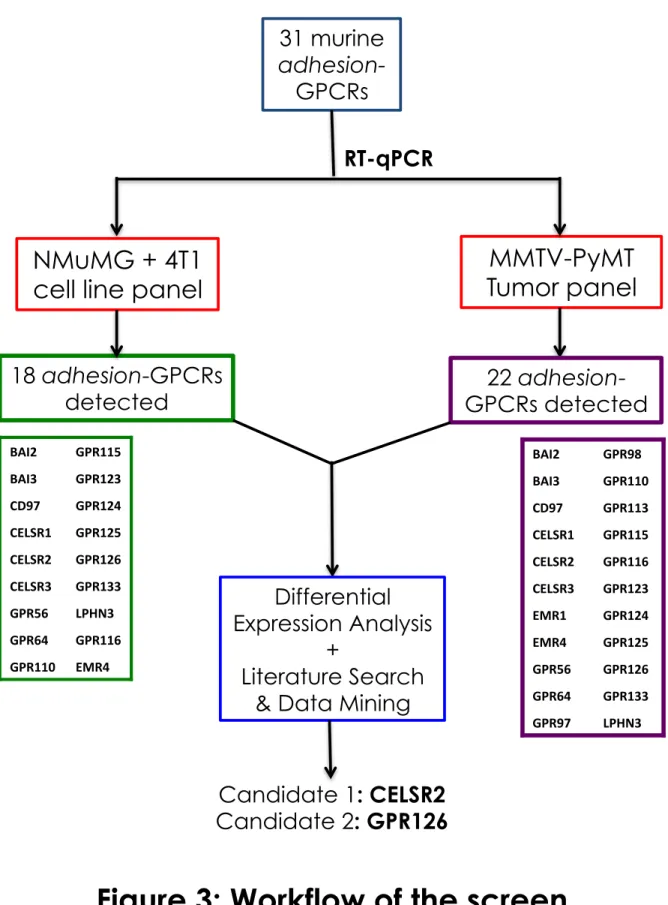
• In Chapter 2, I will describe the screening method used to identify candidate adhesion-GPCRs for their putative roles in tumorigenesis and metastasis. I will also provide an overview of my survey of published literature and data mining that implicates the Adhesion family in cancer progression and metastasis, and validates the choice of candidate genes taken forward for further investigation.
• In Chapter 3, I will provide an overview of the CELSR family within adhesion-GPCRs, of which one member – CELSR2 – was significantly downregulated in highly metastatic breast cancer cells. This will be followed by a discussion about our attempts to test the putative tumor-suppressor role of CELSR2 in both experimental and spontaneous metastasis mouse models.
• In Chapter 4, I will provide an overview of the GPR126 protein, a candidate metastasis enhancer identified in our screen. This will be followed by a description of our experimental results that demonstrate GPR126 as an enhancer of metastasis and implicate the protein in extravasation in in vivo models of breast cancer as well as melanoma.
• In Chapter 5, I will summarize the work described in this thesis, the outstanding questions that came out of the studies, and the potential avenues for further
REFERENCES
Araç, D., Boucard, A.A., Bolliger, M.F., Nguyen, J., Soltis, S.M., Südhof, T.C., and Brunger, A.T. (2012). A novel evolutionarily conserved domain of cell-‐adhesion GPCRs mediates autoproteolysis. EMBO J. 31, 1364–1378.
Attwood, T.K., and Findlay, J.B. (1993). Design of a discriminating fingerprint for G-‐ protein-‐coupled receptors. Protein Eng. 6, 167–176.
Attwood, T.K., and Findlay, J.B. (1994). Fingerprinting G-‐protein-‐coupled receptors. Protein Eng. 7, 195–203.
Austyn, J.M., and Gordon, S. (1981). F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 11, 805–815.
Baud, V., Chissoe, S.L., Viegas-‐Péquignot, E., Diriong, S., N’Guyen, V.C., Roe, B.A., and Lipinski, M. (1995). EMR1, an unusual member in the family of hormone receptors with seven transmembrane segments. Genomics 26, 334–344.
Bjarnadóttir, T.K., Fredriksson, R., Höglund, P.J., Gloriam, D.E., Lagerström, M.C., and Schiöth, H.B. (2004). The human and mouse repertoire of the adhesion family of G-‐ protein-‐coupled receptors. Genomics 84, 23–33.
Bjarnadóttir, T.K., Gloriam, D.E., Hellstrand, S.H., Kristiansson, H., Fredriksson, R., and Schiöth, H.B. (2006). Comprehensive repertoire and phylogenetic analysis of the G protein-‐coupled receptors in human and mouse. Genomics 88, 263–273.
Bjarnadóttir, T.K., Fredriksson, R., and Schiöth, H.B. (2007). The adhesion GPCRs: a unique family of G protein-‐coupled receptors with important roles in both central and peripheral tissues. Cell. Mol. Life Sci. CMLS 64, 2104–2119.
Bockaert, J., and Pin, J.P. (1999). Molecular tinkering of G protein-‐coupled receptors: an evolutionary success. EMBO J. 18, 1723–1729.
Bolliger, M.F., Martinelli, D.C., and Südhof, T.C. (2011). The cell-‐adhesion G protein-‐ coupled receptor BAI3 is a high-‐affinity receptor for C1q-‐like proteins. Proc. Natl. Acad. Sci. U. S. A. 108, 2534–2539.
Bos, P.D., Zhang, X.H.-‐F., Nadal, C., Shu, W., Gomis, R.R., Nguyen, D.X., Minn, A.J., van de Vijver, M.J., Gerald, W.L., Foekens, J.A., et al. (2009). Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009.
Boucard, A.A., Ko, J., and Südhof, T.C. (2012). High Affinity Neurexin Binding to Cell Adhesion G-‐protein-‐coupled Receptor CIRL1/Latrophilin-‐1 Produces an
Brady, A.E., and Limbird, L.E. (2002). G protein-‐coupled receptor interacting proteins: emerging roles in localization and signal transduction. Cell. Signal. 14, 297–309.
Braun, S., Vogl, F.D., Naume, B., Janni, W., Osborne, M.P., Coombes, R.C., Schlimok, G., Diel, I.J., Gerber, B., Gebauer, G., et al. (2005). A Pooled Analysis of Bone Marrow Micrometastasis in Breast Cancer. N. Engl. J. Med. 353, 793–802.
Breur, K. (1966). Growth rate and radiosensitivity of human tumours. II. Radiosensitivity of human tumours. Eur. J. Cancer 2, 173–188.
Butler, T.P., and Gullino, P.M. (1975). Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 35, 512–516.
Clark, E.A., Golub, T.R., Lander, E.S., and Hynes, R.O. (2000). Genomic analysis of metastasis reveals an essential role for RhoC : Article : Nature. Nature 406, 532–535. Collins, V.P., Loeffler, R.K., and Tivey, H. (1956). Observations on growth rates of human tumors. Am. J. Roentgenol. Radium Ther. Nucl. Med. 76, 988–1000. Cortijo, C., Gouzi, M., Tissir, F., and Grapin-‐Botton, A. (2012). Planar Cell Polarity Controls Pancreatic Beta Cell Differentiation and Glucose Homeostasis. Cell Rep. 2, 1593–1606.
Das, S., Owen, K.A., Ly, K.T., Park, D., Black, S.G., Wilson, J.M., Sifri, C.D., Ravichandran, K.S., Ernst, P.B., and Casanova, J.E. (2011). Brain angiogenesis inhibitor 1 (BAI1) is a pattern recognition receptor that mediates macrophage binding and engulfment of Gram-‐negative bacteria. Proc. Natl. Acad. Sci. U. S. A. 108, 2136–2141.
Davies, J.Q., Chang, G.-‐W., Yona, S., Gordon, S., Stacey, M., and Lin, H.-‐H. (2007). The Role of Receptor Oligomerization in Modulating the Expression and Function of Leukocyte Adhesion-‐G Protein-‐coupled Receptors. J. Biol. Chem. 282, 27343–27353. Dhanasekaran, N., Heasley, L.E., and Johnson, G.L. (1995). G protein-‐coupled
receptor systems involved in cell growth and oncogenesis. Endocr. Rev. 16, 259– 270.
Dorsam, R.T., and Gutkind, J.S. (2007). G-‐protein-‐coupled receptors and cancer. Nat. Rev. Cancer 7, 79–94.
Duman, J.G., Tzeng, C.P., Tu, Y.-‐K., Munjal, T., Schwechter, B., Ho, T.S.-‐Y., and Tolias, K.F. (2013). The Adhesion-‐GPCR BAI1 Regulates Synaptogenesis by Controlling the Recruitment of the Par3/Tiam1 Polarity Complex to Synaptic Sites. J. Neurosci. 33, 6964–6978.
Eskelin, S., Pyrhönen, S., Summanen, P., Hahka-‐Kemppinen, M., and Kivelä, T. (2000). Tumor doubling times in metastatic malignant melanoma of the uvea: tumor
progression before and after treatment. Ophthalmology 107, 1443–1449.
Formstone, C.J. (2010). 7TM-‐Cadherins: developmental roles and future challenges. Adv. Exp. Med. Biol. 706, 14–36.
von Fournier, D., Weber, E., Hoeffken, W., Bauer, M., Kubli, F., and Barth, V. (1980). Growth rate of 147 mammary carcinomas. Cancer 45, 2198–2207.
Fredriksson, R., Gloriam, D.E.I., Höglund, P.J., Lagerström, M.C., and Schiöth, H.B. (2003a). There exist at least 30 human G-‐protein-‐coupled receptors with long Ser/Thr-‐rich N-‐termini. Biochem. Biophys. Res. Commun. 301, 725–734.
Fredriksson, R., Lagerström, M.C., Lundin, L.-‐G., and Schiöth, H.B. (2003b). The G-‐ protein-‐coupled receptors in the human genome form five main families.
Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272.
Gray, J.X., Haino, M., Roth, M.J., Maguire, J.E., Jensen, P.N., Yarme, A., Stetler-‐ Stevenson, M.A., Siebenlist, U., and Kelly, K. (1996). CD97 is a processed, seven-‐ transmembrane, heterodimeric receptor associated with inflammation. J. Immunol. Baltim. Md 1950 157, 5438–5447.
Gupte, J., Swaminath, G., Danao, J., Tian, H., Li, Y., and Wu, X. (2012). Signaling property study of adhesion G-‐protein-‐coupled receptors. FEBS Lett. 586, 1214– 1219.
Hamann, J., Eichler, W., Hamann, D., Kerstens, H.M., Poddighe, P.J., Hoovers, J.M., Hartmann, E., Strauss, M., and van Lier, R.A. (1995). Expression cloning and
chromosomal mapping of the leukocyte activation antigen CD97, a new seven-‐span transmembrane molecule of the secretion receptor superfamily with an unusual extracellular domain. J. Immunol. Baltim. Md 1950 155, 1942–1950.
Hamann, J., Vogel, B., van Schijndel, G.M., and van Lier, R.A. (1996). The seven-‐span transmembrane receptor CD97 has a cellular ligand (CD55, DAF). J. Exp. Med. 184, 1185–1189.
Hamann, J., Stortelers, C., Kiss-‐Toth, E., Vogel, B., Eichler, W., and van Lier, R.A.W. (1998). Characterization of the CD55 (DAF)-‐binding site on the seven-‐span transmembrane receptor CD97. Eur. J. Immunol. 28, 1701–1707.
Hamann, J., Aust, G., Araç, D., Engel, F.B., Formstone, C., Fredriksson, R., Hall, R.A., Harty, B.L., Kirchhoff, C., Knapp, B., et al. (2015). International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-‐coupled receptors. Pharmacol. Rev. 67, 338–367.
Hynes, R.O. (2003). Metastatic potential: generic predisposition of the primary tumor or rare, metastatic variants-‐or both? Cell 113, 821–823.
Jin, Z., Tietjen, I., Bu, L., Liu-‐Yesucevitz, L., Gaur, S.K., Walsh, C.A., and Piao, X. (2007). Disease-‐associated mutations affect GPR56 protein trafficking and cell surface expression. Hum. Mol. Genet. 16, 1972–1985.
Kan, Z., Jaiswal, B.S., Stinson, J., Janakiraman, V., Bhatt, D., Stern, H.M., Yue, P.,
Haverty, P.M., Bourgon, R., Zheng, J., et al. (2010). Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873.
Kang, Y., Siegel, P.M., Shu, W., Drobnjak, M., Kakonen, S.M., Cordón-‐Cardo, C., Guise, T.A., and Massagué, J. (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549.
Kolakowski, L.F. (1994). GCRDb: a G-‐protein-‐coupled receptor database. Receptors Channels 2, 1–7.
Krasnoperov, V.G., Bittner, M.A., Beavis, R., Kuang, Y., Salnikow, K.V., Chepurny, O.G., Little, A.R., Plotnikov, A.N., Wu, D., Holz, R.W., et al. (1997). alpha-‐Latrotoxin
stimulates exocytosis by the interaction with a neuronal G-‐protein-‐coupled receptor. Neuron 18, 925–937.
Krishnan, A., Almén, M.S., Fredriksson, R., and Schiöth, H.B. (2012). The Origin of GPCRs: Identification of Mammalian like Rhodopsin , Adhesion , Glutamate and Frizzled GPCRs in Fungi. PLOS ONE 7, e29817.
Kwakkenbos, M.J., Pouwels, W., Matmati, M., Stacey, M., Lin, H.-‐H., Gordon, S., Lier, R.A.W. van, and Hamann, J. (2005). Expression of the largest CD97 and EMR2
isoforms on leukocytes facilitates a specific interaction with chondroitin sulfate on B cells. J. Leukoc. Biol. 77, 112–119.
Kwakkenbos, M.J., Matmati, M., Madsen, O., Pouwels, W., Wang, Y., Bontrop, R.E., Heidt, P.J., Hoek, R.M., and Hamann, J. (2006). An unusual mode of concerted evolution of the EGF-‐TM7 receptor chimera EMR2. FASEB J. 20, 2582–2584.
Labelle, M., and Hynes, R.O. (2012a). The Initial Hours of Metastasis: The Importance of Cooperative Host–Tumor Cell Interactions during Hematogenous Dissemination. Cancer Discov. 2, 1091–1099.
Labelle, M., and Hynes, R.O. (2012b). The initial hours of metastasis: the importance of cooperative host-‐tumor cell interactions during hematogenous dissemination. Cancer Discov. 2, 1091–1099.
Lagerström, M.C., and Schiöth, H.B. (2008). Structural diversity of G protein-‐coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 7, 339–357.
Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921.
Langenhan, T., Aust, G., and Hamann, J. (2013). Sticky signaling-‐-‐adhesion class G protein-‐coupled receptors take the stage. Sci. Signal. 6, re3.
Lefkowitz, R.J. (2004). Historical review: a brief history and personal retrospective of seven-‐transmembrane receptors. Trends Pharmacol. Sci. 25, 413–422.
Lefkowitz, R.J. (2007). Seven transmembrane receptors: something old, something new. Acta Physiol. Oxf. Engl. 190, 9–19.
Lelianova, V.G., Davletov, B.A., Sterling, A., Rahman, M.A., Grishin, E.V., Totty, N.F., and Ushkaryov, Y.A. (1997). Alpha-‐latrotoxin receptor, latrophilin, is a novel member of the secretin family of G protein-‐coupled receptors. J. Biol. Chem. 272, 21504–21508.
Lim, J., and Thiery, J.P. (2012). Epithelial-‐mesenchymal transitions: insights from development. Dev. Camb. Engl. 139, 3471–3486.
Lin, H.-‐H., Stacey, M., Saxby, C., Knott, V., Chaudhry, Y., Evans, D., Gordon, S.,
McKnight, A.J., Handford, P., and Lea, S. (2001). Molecular Analysis of the Epidermal Growth Factor-‐like Short Consensus Repeat Domain-‐mediated Protein-‐Protein Interactions DISSECTION OF THE CD97-‐CD55 COMPLEX. J. Biol. Chem. 276, 24160– 24169.
Lin, H.-‐H., Faunce, D.E., Stacey, M., Terajewicz, A., Nakamura, T., Zhang-‐Hoover, J., Kerley, M., Mucenski, M.L., Gordon, S., and Stein-‐Streilein, J. (2005). The macrophage F4/80 receptor is required for the induction of antigen-‐specific efferent regulatory T cells in peripheral tolerance. J. Exp. Med. 201, 1615–1625.
Lu, P., Weaver, V.M., and Werb, Z. (2012). The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 196, 395–406.
Luo, R., Jeong, S.-‐J., Jin, Z., Strokes, N., Li, S., and Piao, X. (2011). G protein-‐coupled receptor 56 and collagen III, a receptor-‐ligand pair, regulates cortical development and lamination. Proc. Natl. Acad. Sci. 108, 12925–12930.
Maehle, A.-‐H. (2009). A binding question: the evolution of the receptor concept. Endeavour 33, 135–140.
McGuffin, L.J., Bryson, K., and Jones, D.T. (2000). The PSIPRED protein structure prediction server. Bioinforma. Oxf. Engl. 16, 404–405.
McMillan, D.R., Kayes-‐Wandover, K.M., Richardson, J.A., and White, P.C. (2002). Very large G protein-‐coupled receptor-‐1, the largest known cell surface protein, is highly expressed in the developing central nervous system. J. Biol. Chem. 277, 785–792. Minn, A.J., Gupta, G.P., Siegel, P.M., Bos, P.D., Shu, W., Giri, D.D., Viale, A., Olshen, A.B., Gerald, W.L., and Massagué, J. (2005). Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524.
Monk, K.R., Naylor, S.G., Glenn, T.D., Mercurio, S., Perlin, J.R., Dominguez, C., Moens, C.B., and Talbot, W.S. (2009). A G Protein–Coupled Receptor Is Essential for
Schwann Cells to Initiate Myelination. Science 325, 1402–1405.
Nagrath, S., Sequist, L.V., Maheswaran, S., Bell, D.W., Irimia, D., Ulkus, L., Smith, M.R., Kwak, E.L., Digumarthy, S., Muzikansky, A., et al. (2007). Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239. Nguyen, D.X., Bos, P.D., and Massagué, J. (2009). Metastasis: from dissemination to organ-‐specific colonization. Nat. Rev. Cancer 9, 274–284.
Nishimura, T., Honda, H., and Takeichi, M. (2012). Planar cell polarity links axes of spatial dynamics in neural-‐tube closure. Cell 149, 1084–1097.
Nordström, K.J.V., Fredriksson, R., and Schiöth, H.B. (2008). The amphioxus (Branchiostoma floridae) genome contains a highly diversified set of G protein-‐ coupled receptors. BMC Evol. Biol. 8, 9.
O’Hayre, M., Vázquez-‐Prado, J., Kufareva, I., Stawiski, E.W., Handel, T.M., Seshagiri, S., and Gutkind, J.S. (2013). The Emerging Mutational Landscape of G-‐proteins and G-‐ protein Coupled Receptors in Cancer. Nat. Rev. Cancer 13, 412–424.
Oldham, W.M., and Hamm, H.E. (2008). Heterotrimeric G protein activation by G-‐ protein-‐coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71.
O’Sullivan, M.L., de Wit, J., Savas, J.N., Comoletti, D., Otto-‐Hitt, S., Yates III, J.R., and Ghosh, A. (2012). FLRT Proteins Are Endogenous Latrophilin Ligands and Regulate Excitatory Synapse Development. Neuron 73, 903–910.
Paavola, K.J., and Hall, R.A. (2012). Adhesion G protein-‐coupled receptors: signaling, pharmacology, and mechanisms of activation. Mol. Pharmacol. 82, 777–783.
Paik, S., Shak, S., Tang, G., Kim, C., Baker, J., Cronin, M., Baehner, F.L., Walker, M.G., Watson, D., Park, T., et al. (2004). A Multigene Assay to Predict Recurrence of Tamoxifen-‐Treated, Node-‐Negative Breast Cancer. N. Engl. J. Med. 351, 2817–2826. Park, D., Tosello-‐Trampont, A.-‐C., Elliott, M.R., Lu, M., Haney, L.B., Ma, Z., Klibanov, A.L., Mandell, J.W., and Ravichandran, K.S. (2007). BAI1 is an engulfment receptor
for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430– 434.
Pei, J., Kim, B.-‐H., Tang, M., and Grishin, N.V. (2007). PROMALS web server for
accurate multiple protein sequence alignments. Nucleic Acids Res. 35, W649–W652. Prömel, S., Frickenhaus, M., Hughes, S., Mestek, L., Staunton, D., Woollard, A.,
Vakonakis, I., Schöneberg, T., Schnabel, R., Russ, A.P., et al. (2012). The GPS Motif Is a Molecular Switch for Bimodal Activities of Adhesion Class G Protein-‐Coupled
Receptors. Cell Rep. 2, 321–331.
Prömel, S., Langenhan, T., and Araç, D. (2013). Matching structure with function: the GAIN domain of adhesion-‐GPCR and PKD1-‐like proteins. Trends Pharmacol. Sci. 34, 470–478.
Qian, F., Boletta, A., Bhunia, A.K., Xu, H., Liu, L., Ahrabi, A.K., Watnick, T.J., Zhou, F., and Germino, G.G. (2002). Cleavage of polycystin-‐1 requires the receptor for egg jelly domain and is disrupted by human autosomal-‐dominant polycystic kidney disease 1-‐associated mutations. Proc. Natl. Acad. Sci. U. S. A. 99, 16981–16986. Ramaswamy, S., Ross, K.N., Lander, E.S., and Golub, T.R. (2003). A molecular signature of metastasis in primary solid tumors. Nat. Genet. 33, 49–54. Rööser, B., Pettersson, H., and Alvegård, T. (1987). Growth rate of pulmonary metastases from soft tissue sarcoma. Acta Oncol. Stockh. Swed. 26, 189–192. Rosenbaum, D.M., Rasmussen, S.G.F., and Kobilka, B.K. (2009). The structure and function of G-‐protein-‐coupled receptors. Nature 459, 356–363.
Shiratsuchi, T., Nishimori, H., Ichise, H., Nakamura, Y., and Tokino, T. (1997). Cloning and characterization of BAI2 and BAI3, novel genes homologous to brain-‐specific angiogenesis inhibitor 1 (BAI1). Cytogenet. Cell Genet. 79, 103–108.
Silva, J.-‐P., Lelianova, V.G., Ermolyuk, Y.S., Vysokov, N., Hitchen, P.G., Berninghausen, O., Rahman, M.A., Zangrandi, A., Fidalgo, S., Tonevitsky, A.G., et al. (2011). Latrophilin 1 and its endogenous ligand Lasso/teneurin-‐2 form a high-‐affinity transsynaptic receptor pair with signaling capabilities. Proc. Natl. Acad. Sci. 108, 12113–12118. Sørlie, T., Perou, C.M., Tibshirani, R., Aas, T., Geisler, S., Johnsen, H., Hastie, T., Eisen, M.B., Rijn, M. van de, Jeffrey, S.S., et al. (2001). Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. 98, 10869–10874.
Southern, C., Cook, J.M., Neetoo-‐Isseljee, Z., Taylor, D.L., Kettleborough, C.A., Merritt, A., Bassoni, D.L., Raab, W.J., Quinn, E., Wehrman, T.S., et al. (2013). Screening β-‐ arrestin recruitment for the identification of natural ligands for orphan G-‐protein-‐ coupled receptors. J. Biomol. Screen. 18, 599–609.
Stacey, M., Lin, H.H., Gordon, S., and McKnight, A.J. (2000). LNB-‐TM7, a group of seven-‐transmembrane proteins related to family-‐B G-‐protein-‐coupled receptors. Trends Biochem. Sci. 25, 284–289.
Stacey, M., Chang, G.-‐W., Davies, J.Q., Kwakkenbos, M.J., Sanderson, R.D., Hamann, J., Gordon, S., and Lin, H.-‐H. (2003). The epidermal growth factor–like domains of the human EMR2 receptor mediate cell attachment through chondroitin sulfate
glycosaminoglycans. Blood 102, 2916–2924.
Stephenson, J.R., Paavola, K.J., Schaefer, S.A., Kaur, B., Van Meir, E.G., and Hall, R.A. (2013). Brain-‐specific Angiogenesis Inhibitor-‐1 Signaling, Regulation, and
Enrichment in the Postsynaptic Density. J. Biol. Chem. 288, 22248–22256. Thiery, J.P. (2003). Epithelial–mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 15, 740–746.
Tissir, F., Qu, Y., Montcouquiol, M., Zhou, L., Komatsu, K., Shi, D., Fujimori, T., Labeau, J., Tyteca, D., Courtoy, P., et al. (2010). Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat. Neurosci. 13, 700–707. Trinchieri, G. (2012). Cancer and inflammation: an old intuition with rapidly
evolving new concepts. Annu. Rev. Immunol. 30, 677–706.
Tubiana, M., Chauvel, P., Renaud, A., and Malaise, E.P. (1975). [Growth rate and natural history of breast cancer]. Bull. Cancer (Paris) 62, 341–358.
Usui, T., Shima, Y., Shimada, Y., Hirano, S., Burgess, R.W., Schwarz, T.L., Takeichi, M., and Uemura, T. (1999). Flamingo, a seven-‐pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell 98, 585–595.
Vallon, M., and Essler, M. (2006). Proteolytically Processed Soluble Tumor
Endothelial Marker (TEM) 5 Mediates Endothelial Cell Survival during Angiogenesis by Linking Integrin αvβ3 to Glycosaminoglycans. J. Biol. Chem. 281, 34179–34188. Valtcheva, N., Primorac, A., Jurisic, G., Hollmén, M., and Detmar, M. (2013). The orphan adhesion G protein-‐coupled receptor GPR97 regulates migration of lymphatic endothelial cells via the small GTPases RhoA and Cdc42. J. Biol. Chem. 288, 35736–35748.
van ’t Veer, L.J., Dai, H., van de Vijver, M.J., He, Y.D., Hart, A.A.M., Mao, M., Peterse, H.L., van der Kooy, K., Marton, M.J., Witteveen, A.T., et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536.
Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. (2001). The Sequence of the Human Genome. Science 291, 1304–1351.