Conformational Dynamics Control Catalysis in Disparate Systems:
Structural Insights from DNA Repair and Antibiotic Biosynthetic Enzymes
by
Jeremy Wayne Setser
OF TECHNOLOGYJUN 3
0
2014
B.S., Chemistry (2008)1
The University of Akron
LIBRARIES
Submitted to the Department of Chemistryin Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2014
C 2014 Massachusetts Institute of Technology. All rights reserved.
Signature of Author ...
Signature redacted
Department of Chemistry May 9,2014
Signature redacted,.
C ertified by ... .Catherine L. Drennan Professor of Chemistry and Biology Howard Hughes Medical Institute Investigator and Professor Thesis Supervisor
A ccepted by ...
Signature redacted
This doctoral thesis has been examined by a
Committee of the Department of Chemistry as follows:
Signature redacted
Assistk1't Professor Elizabeth M. Nolan Committee Chairman Pfizer-Laubach Career Development Assistant Professor of Chemistry
Signature redacted
Professor Catherine L. Drennan Research Supervisor Professor of Chemistry and Biology Howard Hughes Medical Institute Investigator and Professor
Signature redacted
Professor Leona D. SamsonCommittee Member Professor of Biological Engineering and Biology Uncas and Helen Whitaker Professor American Cancer Society Research Professor
Conformational Dynamics Control Catalysis in Disparate Systems: Structural Insights from DNA Repair and Antibiotic Biosynthetic Enzymes
by
Jeremy Wayne Setser
Submitted to the Department of Chemistry on May 9, 2014 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in
Biological Chemistry
ABSTRACT
Chemical reactions allow biological systems to function. The majority of these biochemical reactions occur due to the work of protein catalysts known as enzymes. These biocatalysts are often thought of as pre-formed, static 'locks' that bind, and subsequently transform, their substrate molecule 'keys'. However, scientists are increasingly finding that dynamic movements of enzymes are a crucial aspect of catalysis. One such example of a system that relies on conformational flexibility is the human DNA repair protein alkyladenine DNA glycosylase (AAG). To efficiently repair DNA, AAG must search the million-fold excess of unmodified DNA bases to find a handful of DNA lesions. Such a search can be facilitated
by the ability of glycosylases, like AAG, to interact with DNA using two affinities: a lower-affinity
interaction in a searching process, and a higher-affinity interaction for catalytic repair. We have captured crystallographic snapshots of AAG bound to DNA in both high- and lower-affinity states. These depictions reveal several significant and unexpected protein structural rearrangements, providing molecular insight into the DNA-searching process adopted by AAG. By combining these new insights with existing biochemical and structural data, we are able to relate AAG to the big picture question of how DNA binding proteins find their binding sites in the vast expanse of the genome.
In another study, a member of a biosynthetic pathway for antibiotic natural products, called kutznerides, was shown to be dependent on conformational changes. The enzyme in question, KtzI, uses a bound flavin cofactor, reducing equivalents from NADPH, and molecular oxygen to install a hydroxyl group on the side-chain nitrogen of the amino acid L-ornithine, which is subsequently incorporated into the kutzneride scaffold. KtzI was structurally characterized after being subjected to various chemical and environmental factors, capturing the enzyme in several states along its catalytic trajectory. These states suggest that a novel conformational change of both the protein backbone and the flavin moiety must take place in order to complete the enzymatic cycle of KtzI. This drastic rearrangement was also shown to be chemically interchangeable in the protein crystal, suggesting that these dynamic motions are catalytically relevant.
Thesis Supervisor: Catherine L. Drennan
Title: Professor of Chemistry and Biology
ACKNOWLEDGEMENTS
I am indebted to so many people for so many things.
First, I would like to say thank you to Cathy for accepting me into your group, and believing I could succeed. Joining a completely foreign research area was daunting enough to me; I cannot imagine having the amount of trust needed to believe I could learn from the ground up. It really has been an amazing experience over the past six years. I would also like to thank Liz Nolan, Leona Samson, and Sarah O'Connor for their scientific insights and guidance during this time. A large part of my graduate experience was built by the support of my fellow lab members, to whom I owe many thanks.
Graduate school is not easy. Experiments fail almost exclusively. Without the support of the Drennan Lab members, past and present, this would have been a crippling undertaking. Thank you all. To Nozomi Ando - I could say that I would have made it through the final years of graduate school without
my lab BFF, but I would probably be lying. Thank you for spending countless hours listening and laughing with me. Your support and friendship bolstered me when I was at my most disillusioned. To Peter Goldman - You were the first person I talked to in the Drennan Lab, and made me understand that a regular dude could succeed in our lab. This realization was a large part of why I joined in the first place. Even if the utter breakdown of your body ruined our chances at volleyball supremacy, you made our lab a fun place to be. To Marco Jost - I was reluctant to accept the 'hot shot German' who knew everything
about crystallography coming into our lab, but you won me over eventually. I have no problem getting your advice now, because I have come to understand you as the 'hot shot German' who knows everything about everything. To Yan Kung and Christine Phillips Piro - Thank you for teaching me what it meant to be a Drennan Lab member. To Mishtu Dey and Danny Yun - Thank you for your ever-patient guidance.
Ultimately, friends and family are the reason I made it through graduate school. I love you all. To Emily Setser - Even though you will always be Emily Lippert to me, I am excited every time I remember we are married. You have been my most important source of support for over six years. Thank you for being the best thing that has happened to me. I could not have done this without you. I love you clown. To Jeff Setser - Thank you for always being there to provide a brother's love when I needed it most. To Diana Setser - I could not have asked for a better Sister-in-law, and the joy I have observed in your
household always reminds me that things will be OK. To Nola, June, and Oden Setser - Thank you for being constant reminders that the future will be bright and joyful. To my Dad, Jim Setser - Thank you for constantly making sure I was healthy and happy. To Mom and Dad Lippert - Thank you for always treating me as your own Son. To Josh Slaga - We have come a long way since 8th grade. Thank you for
handling all best friend-related duties for the last 15 years. To Alyssa Larson - The free stuff line at Sid Pac may not have offered up any useful items, but it did provide me with a best friend here at MIT. Thank you for being a constant source of support for five years and counting. To all CGSC members past and present - Always remember that we actually make an impact around here. Keep it up. To all my other friends here and elsewhere - Thank you for your support and all the fun times had at TGIF, the Muddy, around Boston, in Ohio, and around the country; It has all been instrumental in getting me to this point.
Finally, this thesis would not have been possible without my Mother..She supported me in every endeavor, in any way that she could...even if this meant letting her 'baby' move 650 miles away from home. My Mother passed away in the middle of graduate school, and the period of time before, during, and after this tragedy was the worst experience of my life. I was extremely lucky to have an advisor as understanding as Cathy, as I spent much of my Mother's last months at her bedside. This time allowed me to support and comfort her in what ways I could, and to spend what would end up being our last quality time together. 'Grateful' does not even begin to express the amount of appreciation I have for my Mother's wonderful influence. 'Sorrow' falls incredibly short of describing what I feel in her absence. Therefore, I would like to dedicate this thesis in honor of the kind, strong, generous, and above all, loving woman that was my Mother.
For Judy Ann Setser. I love you Mom.
Table of Contents
ABSTRACT 3
ACKNOWLEDGEMENTS 4
Chapter 1. The Dynamism of DNA-binding and Flavin-dependent Enzymes
I. SUMMARY 13
I.1I INTRODUCTION 13
1.111 DNA repair by human alkyladenine DNA glycosylase (AAG) 15
I.IV Natural product biosynthesis with FAD-dependent N-hydroxylases 16
FIGURES 19
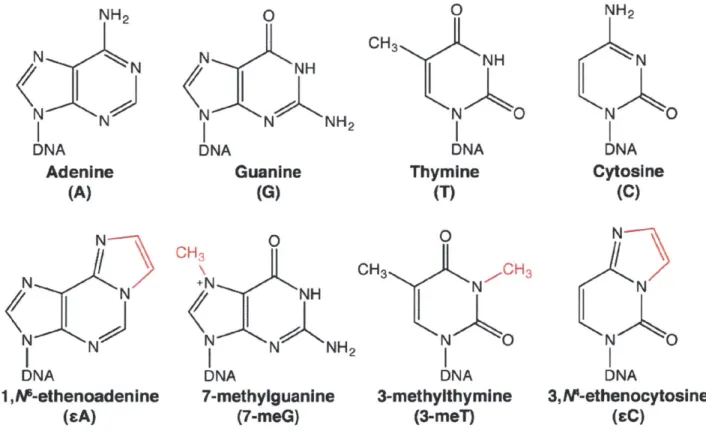
Figure 1.1 The damage of DNA bases.
Figure 1.2 Base excision repair (BER) in humans.
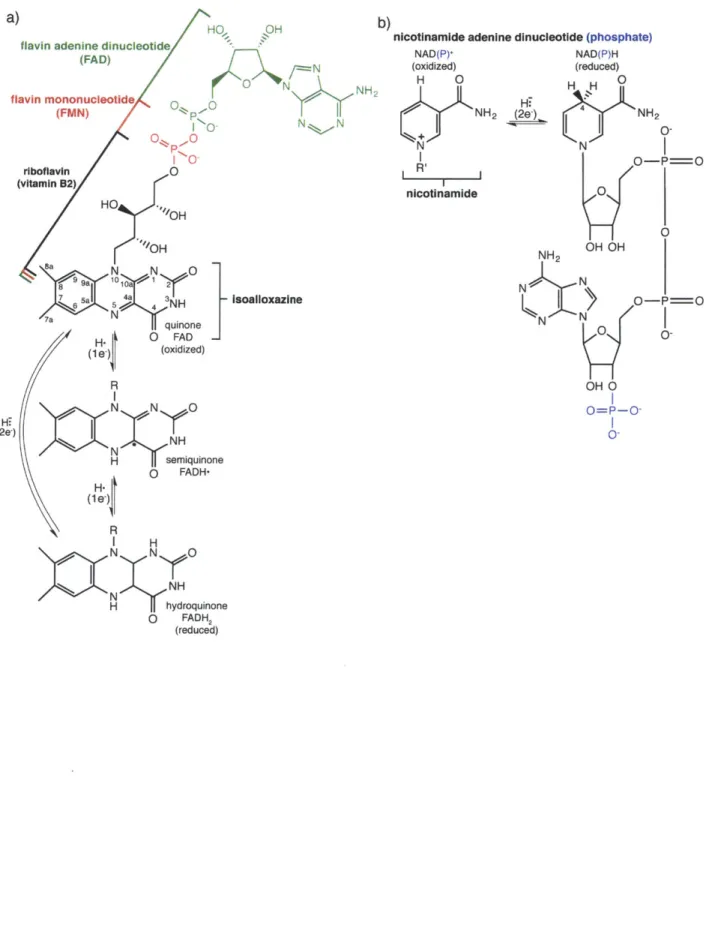
Figure 13 Introduction to the flavin and nicotinamide cofactors.
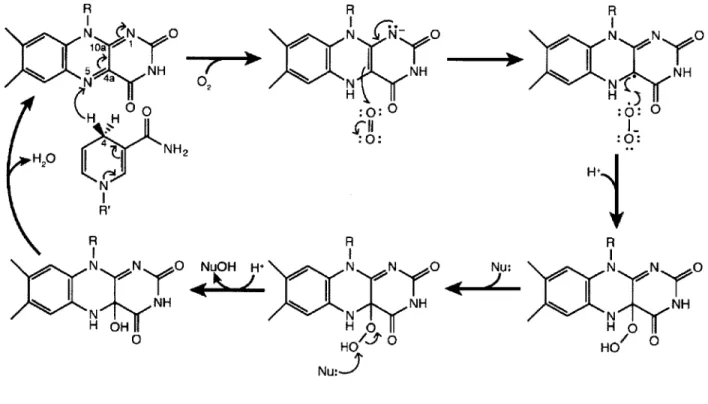
Figure 1.4 General mechanism of flavin-dependent monooxygenases.
I.V REFERENCES 23
Chapter 2. Structural Basis for the Inhibition of Human Alkyladenine DNA
Glycosylase (AAG) by 3,N4-Ethenocytosine-containing DNA
11.1 SUMMARY 27
11.11 INTRODUCTION 28
11.111 RESULTS 30
AAG binding studies
Catalytic ability of AAG for eC containing DNA Inhibition of AAG by cC containing DNA
Overall structure of the JI79AAG-EC DNA inhibitor complex Protein-DNA interactions
Metal ion Mn2
, in the A79AAG-EC:G structure
Active site architecture of JI79AAG-EC DNA complex
II.IV DISCUSSION 34
II.V MATERIALS AND METHODS 37
J 79AAG plasmid construction, creation of mutants, and protein preparation AAG protein expression and purification
Preparation of oligonucleotides and 32P-labeling Gel mobility shift assays
DNA glycosylase assays
Competition DNA glycosylase assays
Crystallization of the I79AAG-EC:G complex Data collection and structure determination
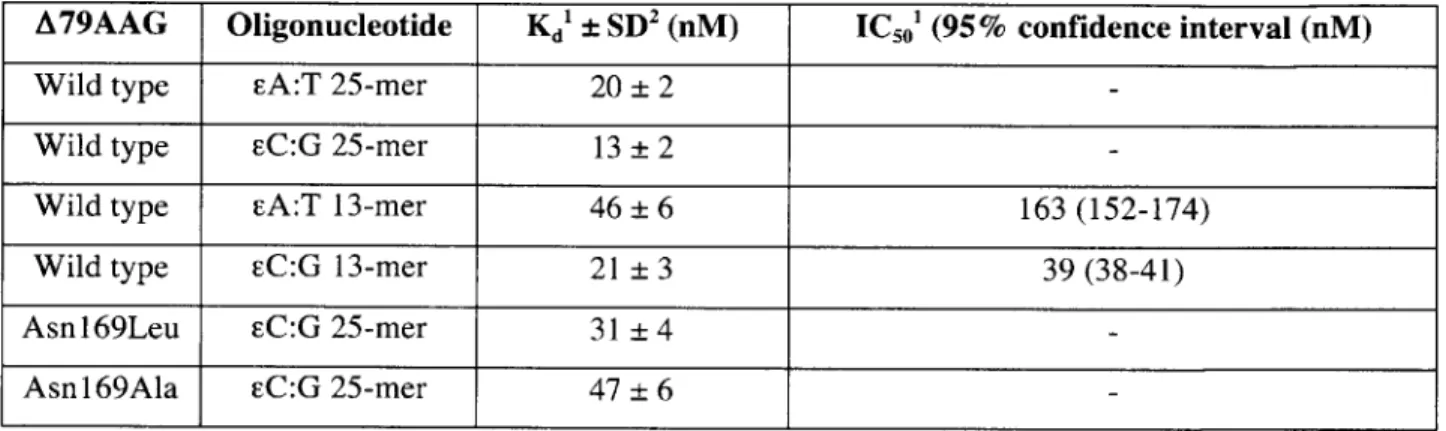
Table 11.1 Dissociation constant (Kd) values measured using gel shift assays; and 50% inhibitory concentration (IC50) for the inhibition of A79AAG activity on EA:T 25-mer, measured
using competition DNA glycosylase assay at 37'C, in the presence of increasing concentration of cold competitor 13-mer duplexes.
Table 11.2 Data collection and refinement statistics of the A79AAG-DNA complex.
Table 11.3 List of oligonucleotide primers used for the creation of A79AAG mutants by PCR based site directed mutagenesis.
Figure 11.1 Biochemical characterization of AAG variants with oligomers containing etheno lesions.
Figure 11.2 Gel results of DNA glycosylase assays for truncated A79AAG on EA:T and EC:X
(X=G/A/T/C) 13-mer DNA duplexes used for crystallization. Figure II.3 Structure of A79AAG with EC inhibitor DNA.
Figure 11.4 Comparison of the overall structures of the A79AAG-DNA complexes.
Figure 11.5 Cation site in A79AAG structures.
Figure 11.6 Interaction of Tyr 162 and putative Mn2
' binding site in the structure of A79AAG-cC:G.
Figure 11.7 Active site architecture of AAG.
Figure 11.8 AAG binding pocket.
Figure 11.9 Activation of the leaving group by protonation.
Figure 11.10 A wall-eyed stereoview of the active site of AAG.
II.VII REFERENCES 58
Chapter 3. Searching for DNA Lesions: Structural Evidence for Lower- and
Higher-Affinity
DNA
Binding
Conformations
of
Human
Alkyladenine DNA Glycosylase
III. SUMMARY111.11 INTRODUCTION 111.111 RESULTS
Structural overview of asymmetric unit A79AAG pseudo-duplex structure A79AAG lower-affinity structure
III.IV DISCUSSION
III.V MATERIALS AND METHODS
AAG plasmid construction and protein preparation Crystallization of A79AAG with single-stranded EC DNA Data collection and structure determination
III.VI ACKNOWLEDGEMENTS TABLES & FIGURES
Table 111.1 Figure 111.1 Figure 111.2 Figure 111.3 Figure Figure Figure III.4 111.5 111.6 61 62 64 67 70 72 73 Data collection and refinement statistics for A79AAG-DNA complex.
DNA adducts to which AAG binds with high affinity: Lesions (A) EC and (B) EA and (C) one-base loop structures.
Structures of A79AAG bound to EC DNA.
Wall-eyed stereoview of the disordered loops of the lower-affinity A79AAG structure in the context of the crystal lattice.
A79AAG shows no affinity for ssFC 13-mer by gel shift. A79AAG structural comparisons.
Tyr162 contacts in lower-affinity A79AAG.
Figure 111.7 Comparison of the lower-affinity A79AAG structure with the high-affinity
A79AAG-EA:T structure.
Figure 111.8 Wall-eyed stereoviews of electron density in the lower-affinity A79AAG structure.
Figure 111.9 Proposal for how AAG can recognize DNA with two different affinities.
III.VIIREFERENCES
Crystallographic
in the Active Site
Evidence for Drastic
of a Flavin-Dependent
Conformational Changes
N-hydroxylase
IV.I SUMMARY IV.II INTRODUCTION IV.III RESULTS Quaternary structure Active site of reduced KtzI Active site of oxidized KtzIIn crystallo conformational changes
IV.IV DISCUSSION
IV.V MATERIALS AND METHODS
Protein expression and purification Reconstitution of KtzI for crystallization Crystallization of KtzI
Re-reduction of oxidized KtzI crystals Re-oxidation of reduced KtzI crystals Data collection, structure determination,
IV.VI ACKNOWLEDGEMENTS TABLES & FIGURES
Table IV.1 Table IV.2 Table IV.3 Table IV.4 Table IV.5 Table IV.6 Table IV.7 Table IV.8 Figure IV.1 Figure IV.2 Figure IV.3 Figure IV.4 Figure IV.5 Figure IV.6 Figure IV.7 Figure IV.8 Figure IV.9 Figure IV.10 87 88 91 97 106
and structural analysis
Structures of KtzI and their respective resolution.
Data collection and refinement statistics for KtzI-FADred-NADP'-L-orn. Data collection and refinement statistics for KtzI-FADox-NADP*-L-orn. Data collection and refinement statistics for KtzI-FADred-ox-NADP'-L-orn. Data collection and refinement statistics for KtzI-FADred-NADP'-Br. Data collection and refinement statistics for KtzI-FADox-Br.
Data collection and refinement statistics for KtzI-FADox-red-NADP*-Br. List of ordered residues and those modeled as alanine in KtzI structures. Kutzneride scaffold cryptically incorporates the product of KtzI.
Sequence alignment of L-orn N-hydroxylases.
Proposed kinetic mechanism for L-orn N-hydroxylases. Crystals of reconstituted KtzI.
Structure of KtzI in reduced and oxidized states. Tetrameric assembly of L-orn N-hydroxylases.
Wall-eyed stereoview of the electron density for bent isoalloxazine ring. Wall-eyed stereoviews of the active sites of reduced L-orn N-hydroxylases. Wall-eyed stereoviews of the active sites of oxidized KtzI.
Structurally-based proposal for L-orn N-hydroxylase mechanism. II.VII REFERENCES
Chapter 4.
84 112 113 138Chapter 5. Outstanding Questions for AAG and KtzI
V.1 SUMMARY 143
V.II Repairing DNA with AAG 143
V.111 Antibiotic biosynthesis with KtzI 145
FIGURES 149
Figure V.1 Sequence alignment of our N-terminal truncation mutant (A79) and full-length (FL) alkyladenine DNA glycosylase (AAG).
Figure V.2 Kinetic mechanism for hydroxylation in (a) bold and (b) cautious flavin-dependent monooxygenases.
Figure V.3 Diverse flavin conformations are found in (a) KtzI and (b) para-hydroxybenzoate hydroxylase (PHBH).
V.IV REFERENCES 152
Chapter 1. The Dynamism of DNA-binding and Flavin-Dependent Enzymes
I.I SUMMARY
Chemical reactions are at the heart of life. Molecules combine and change to create new entities, and life has emerged from, and mutated in response to, these chemicals. Many chemical reactions are not favored, with some likely to occur only on the timescale of millions of years. To accelerate these reactions in biological systems, enzymes are used. These biological catalysts function by binding a substrate and hastening its transformation into a product. Many enzymes depend on dynamic movements to catalyze their chemical reactions, and this dissertation will examine two such cases. In Chapters 2 and 3, an enzyme that repairs DNA in humans will be discussed. The subject of Chapter 4 will be a member of an assembly line of enzymes responsible for synthesizing an antibiotic in a soil bacterium. These dissimilar systems are linked by their reliance on conformational flexibility.
1.11 INTRODUCTION
Proteins comprise a significant portion of the cellular makeup, up to 55% in Escherichia coli (1). These entities perform functions as wide-ranging as acting as a cellular garbage disposal, in the case of proteolytic species like the 26S proteasome, to determining which cellular entities are produced by binding DNA sequences, in the case of transcription factors like NF-KB, to synthesizing vital components of the cell, like the over 30 enzymes involved in the biosynthesis of cholesterol. Dysfunctions in any of these pathways are linked to disease in humans, underlining their practical importance for our health and well-being. The proteins that function to take a hypothetical substrate 'S', to its product 'P', are known as enzymes, and are an extremely active area of research.
Enzymes use their amino acids, and in some cases, the help of organic or inorganic species known as cofactors, to accelerate chemical reactions, thus acting as biological catalysts. These biocatalysts have traditionally been viewed as having a fixed structure that is tuned to precisely fit its substrate molecule, stemming from their initial likening to a "lock and key" by H. Emil Fischer in the late 19 th century. In more recent years, however, proteins have been understood to be dynamic, and an emphasis has been placed on studying both small-scale (short timescale) and large-scale (long timescale) movements, as this conformational flexibility is often vital to a protein's function (reviewed in (2)). Various techniques including nuclear magnetic resonance (NMR), small-angle X-ray scattering (SAXS), and fluorescence methods like those
based on Fbrster resonance energy transfer (FRET), are well-suited to study large-scale conformational changes, as these techniques occur while the protein is in solution, mimicking the freedom found in the cellular milieu (2). X-ray crystallography, which requires a protein to be trapped in a crystalline lattice (often frozen at 100K), is less useful in this context, as the protein's degrees of freedom are limited. What this method does provide, however, is a much higher-resolution picture than the other techniques mentioned above. Therefore, X-ray crystallography tends to trade the vital information on conformational flexibility for more detailed molecular-level understanding. Indeed, a crystal structure provides very little in the way of dynamic information. As we will show in this thesis, however, a combination of crystal structures can illustrate the flexibility of a protein. In the following chapters, we describe the ability to get the best of both worlds, obtaining information on protein dynamics by combining a series of atomic-level snapshots, using two enzymes with very dissimilar roles as case studies.
In Chapters 2 and 3, we analyze the human DNA repair protein alkyladenine DNA glycosylase (AAG). In Chapter 4, we will examine the L-ornithine N-hydroxylase from Kutzneria sp. 744, KtzI. These two enzymes function in very different worlds. AAG repairs damaged DNA in humans, and KtzI is a member of a host of enzymes that synthesize natural product defenses for a soil actinomycete. Through our structural work, however, we have found that these systems are linked through their reliance on conformational changes. For AAG, we have snapshots of the enzyme both bound to an inhibitor DNA base (Chapter 2) and interacting with DNA in a nonspecific manner (Chapter 3). These structures allow us to observe the differences between these higher- and lower-affinity complexes, with most alterations found in a region that is highly flexible in the absence of a bound DNA base. By compiling our data with that of the field in general, we propose this dynamic region to be vital for AAG's ability to search for its damaged-DNA substrates. For KtzI, we have determined several structures of this enzyme along its reaction path, which indicate that a highly mobile active site, including movements of the protein backbone and the flavin cofactor, is required for catalysis (Chapter 4). We have been able to recapitulate these proposed conformational interchanges in the protein crystal, showing that the required conformational flexibility is both chemically competent, and able to occur in the "rigidified" environment of the crystalline lattice. Together, these studies show large-scale dynamics to be a crucial aspect of enzyme catalysis, and further, that this
information can be obtained by a collection of X-ray crystallography snapshots put together in a 'flip-book'-type methodology.
1.111 DNA repair by human alkyladenine DNA glycosylase (AAG)
Damaged DNA must be repaired to allow faithful replication of genetic information for healthy cell division and, thus, for life to occur. Ultraviolet light, ionizing radiation, xenobiotic chemicals such as alkylating agents, and endogenous metabolites such as reactive oxygen and nitrogen species (RONS) or methylating agents (e.g. S-adenosylmethionine, AdoMet) can harm nucleic acids (3). The most common manifestations of DNA damage are known as DNA lesion bases (4), where a canonical DNA base (Figure 1.1, top) is chemically altered (Figure 1.1, bottom). These lesion bases occur at a rate of 10,000 per cell per day (4, 5), and affect base pairing interactions and DNA energetics, causing transition (A<-+G; C<-+T) and transversion
(A,G-*C,T) mutations and, as errors are compounded, cell death (5, 6). Most DNA lesions are
repaired via base excision repair (BER), an endogenous pathway that removes and replaces the damaged base in a series of steps (3). BER is highly conserved in all kingdoms of life (7), and in humans these steps are catalyzed by the sequential action of a DNA glycosylase, AP endonuclease I, DNA polymerase P, and DNA ligase I/III ((8, 9); Figure 1.2).
Alkyladenine DNA glycosylase (AAG) is a monofunctional DNA glycosylase involved in the human BER process. Monofunctional glycosylases, which lack inherent endonuclease activity, initiate BER by binding specific lesion bases and catalyzing the cleavage of the bond between the base and its ribose sugar (N-glycosidic bond), releasing the free base and leaving behind an apurinic/apyrimidinic (AP) site (Figure 1.2, 1). The repair of the AP site is completed
by the action of three subsequent enzymes as mentioned above (Figure 1.2, 2-4), which finally
restores the DNA to its undamaged state. AAG has been of particular interest due to its involvement in remediating lesions brought on by chronic inflammatory diseases like ulcerative colitis (10-12), which predispose individuals to cancer. Further, AAG represents an interesting biochemical problem as it recognizes a wide variety of lesion bases (13-15), including substrates that it can excise, such as 1,M-ethenoadenine (EA) and 7-methylguanine (7-meG) (Figure I.1), and those that it cannot remove, including the inhibitory 3,N 4-ethenocytosine lesion (EC) (Figure
Samson groups here at MIT, and together we were able to biochemically and structurally characterize AAG.
In one study (Chapter 2, (16)), we examined the ability of AAG to bind EC with high-affinity, while being unable to excise this lesion, and determined the molecular structure of this AAG-EC inhibitory complex. These data allowed us to propose a chemical rationale for the inability of AAG to excise the eC lesion. We were also able to obtain a fortuitous crystallographic snapshot of this enzyme with a self-assembled, "pseudo-duplex" piece of DNA. In this structure, one AAG molecule in the asymmetric unit recognizes a EC lesion in a manner similar to the high-affinity depiction described in Chapter 2, while the other AAG monomer is making only nonspecific contacts with DNA, and thus is in a lower-affinity state. Taking these and other studies together, we were able to provide evidence for a mechanism by which AAG can find its lesion base substrates in the million-fold excess of undamaged DNA bases using a flexible DNA binding region (Chapter 3, (17)).
I.IV Natural product biosynthesis with FAD-dependent N-hydroxylases
Flavin-containing enzymes, or flavoenzymes, have been an intense area of study for nearly a century (18). From the initial discovery of a yellow pigment in cow's milk in 1879 (19)
(which we now know to be riboflavin or vitamin B2 (Figure I.3a)), to the elucidation of flavin's chemical structure (20, 21) and the first description of a flavoenzyme (22, 23) in the 1930s, this
field has grown and matured to the point that today over 18,000 articles found on PubMed use the term "flavin". Flavoenzymes utilize either the flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD) forms of flavin (Figure I.3a), and these cofactors can be used to perform a wide variety of reactions. This chemical diversity is due to the reactive isoalloxazine ring of the flavin, which can adopt three oxidation states differing by 1-electron, making it one of the few species in biology that can mediate between both 1- and 2-electron transfers (Figure I.3a). Herein, we will concentrate on a class of FAD-dependent enzymes known as hydroxylases or monooxygenases, and more specifically a recently discovered sub-class that act upon the amino acid L-ornithine (L-orn).
The FAD-dependent hydroxylases use molecular oxygen and reducing equivalents from nicotinamide adeninine dinucleotide (phosphate) (NAD(P)H; Figure I.3b) to install a hydroxyl (-OH) group on a nucleophilic substrate (as outlined in Figure 1.4). The catalytic cycle of these
enzymes is initiated by hydride transfer from the Pro-R C4 position of the nicotinamide of
NAD(P)H to the N5 position of the oxidized flavin isoalloxazine ring. Molecular oxygen can
then add to the reduced isoalloxazine via a radical-mediated reaction, generating a C4a-hydroperoxy species. The hydroxylated product is created after nucleophilic attack by the bound substrate on the distal oxygen of the C4a-hydroperoxy, leaving a C4a-hydroxyflavin intermediate. Finally, dehydration of the hydroxyflavin readies this cofactor for another round of catalysis. Although this general mechanism is conserved in the majority of monooxygenase enzymes (reviewed in (24-27)), there are intriguing differences observed in the N-hydroxylase class. Herein, we explore the L-orn N-hydroxylase from Kutzneria sp. 744, called KtzI. This enzyme is involved in the biosynthesis of cyclic peptides that have antimicrobial and antifungal activity, which made it of great interest to both the Walsh and Drennan Labs. We have been able to structurally characterize KtzI in several states along its reaction path, and by pairing these snapshots with the biochemical and structural data already available for this enzyme-class, we propose a structurally-based reaction mechanism that includes never-before-seen conformational changes of both the protein backbone and the flavin cofactor. Further, we were able recapitulate these conformational changes in the protein crystal, displaying their chemical competence. These results are presented in Chapter 4.
FIGURES
Figure 1.1. The damage of DNA bases. Canonical DNA bases (top) are acted upon by a variety of
endogenous and exogenous sources, some of which cause damage. These damaged, 'lesion bases' (bottom) are chemically altered (in red), commonly by alkylation (7-meG, 3-meT) or through interaction with peroxide and aldehyde species (EA, EC). Endogenous pathways must repair damaged bases such that mutagenesis and cytotoxicity are minimized.
NH2 N N IN DNA Adenine (A) N N N N N DNA 1 ,M-ethenoadenIne (sA) 0 N N NH2 DNA Guanine (G) OH3 +N NH N N NH2 DNA 7-methylguanine (7-meG) 0 CH3 NH N A DNA Thymine (T) 0 CH3 N CH 3 N O DNA 3-methylthymine (3-meT) NH2 N N K DNA Cytosine (C) N NA N 0 DNA 3, A-ethenocytosine (CC)
Figure 1.2. Base excision repair (BER) in humans. The major BER pathway in humans begins with the
action of a monofunctional DNA glycosylase like AAG (1). After lesion (red circle) removal, the apurinic/apyrimidinic (AP) site left behind is recognized by AP endonuclease I, which clips the phosphodiester DNA backbone leaving a 3'-hydroxyl group and a 5'-deoxyribosephosphate (dRP) moiety
(2;(28)). The dual-function DNA polymerase $ attaches a new template base (in blue) to the 3'-OH and
removes the 5'-dRP, leaving a free 5'-phosphate (3;(29)). To complete the repair, the nick left behind is sealed using DNA ligase I or III (4;(9, 30)).
4
9
4
9
3'
4
9
6-1 - I I I I 511. DNA
glycosylase
5'2. AP Endonuclease I
dRP OH 3'3. DNA Polymerase
p tPO 32 H o4. DNA Ligase
1/11t
4
9
9
4
4
9
4
19
4
9t
5
3'
5'
3'
5'
3'
5 13'
5'
3'
4
9
4
9
4
4
9
3'5'
4
4
4
3'
5'
TFigure 1.3. Introduction to the flavin and nicotinamide cofactors.
a)
flavin adenine dinucleotide
(FAD) 0 N flavin mononucleotide NNH 2 (FMN) p -0 1 0 N N O 0P.-. 0-riboflavin 0 (vitamin B2) HO "OH "OH Sa N 0 81959a101y~ 21 4 3 Isoalloxazine 7a quinone H. jj O FAD (1 e. (oxidized) R N N 0 Hi (2e-) N NH NH H semiquinone (1 e) 0 FADH-R
nicotinamide adenine dinucleotide (phosphate)
NAD(P)' (oxidized) H 0 NH2 N nicotinamide H.) (2L) NAD(P)H (reduced) H H 0 NH2
K)
0-N O-P=O 0 1 0 NH2 OH OH N N-O-P==OKN
N0 0-OH 0 O=P-0-I H NNyO NH H hydroquinone 0 FADH2 (reduced) b)Figure 1.4. General mechanism of flavin-dependent monooxygenases. R R 'N N 0 H N N N NH NANHN N 02N CH H :0: N H H N N 0H
I
I~
)aN N 0 ~~HO .., Nu:J R N N o H D :0: 0 I-:0: A I0 Nu: N N 0 *z X N NH HOt 0I.V REFERENCES
1. Moran, U., Phillips, R., and Milo, R. (2010) SnapShot: key numbers in biology., Cell 141,
1262-1262.
2. Henzler-Wildman, K., and Kern, D. (2007) Dynamic personalities of proteins, Nature 450,
964-972.
3. Zharkov, D. 0. (2008) Base excision DNA repair, Cell Molec Life Sci 65, 1544-1565.
4. Lindahl, T. (1993) Instability and decay of the primary structure of DNA, Nature 362, 709-715.
5. Lindahl, T., and Barnes, D. (2000) Repair of endogenous DNA damage, Cold Spring Harb Symp
Quant Biol 65, 127-134.
6. Shrivastav, N., Li, D., and Essigmann, J. M. (2010) Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation, Carcinogenesis 31, 59-70.
7. Robertson, A. B., Klungland, A., Rognes, T., and Leiros, I. (2009) DNA repair in mammalian cells: Base excision repair: the long and short of it, Cell Mol Life Sci 66, 981-993.
8. David, S. S., and Williams, S. D. (1998) Chemistry of Glycosylases and Endonucleases Involved in Base-Excision Repair, Chem Rev 98, 1221-1262.
9. Kubota, Y., Nash, R. A., Klungland, A., Schar, P., Barnes, D. E., and Lindahl, T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between
DNA polymerase beta and the XRCC1 protein, EMBO J 15, 6662-6670.
10. Meira, L. B., Bugni, J. M., Green, S. L., Lee, C.-W., Pang, B., Borenshtein, D., Rickman, B. H., Rogers, A. B., Moroski-Erkul, C. A., McFaline, J. L., Schauer, D. B., Dedon, P. C., Fox, J. G., and Samson, L. D. (2008) DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice, J Clin Invest 118, 2516-2525.
11. Nair, J., Gansauge, F., Beger, H., Dolara, P., Winde, G., and Bartsch, H. (2006) Increased
etheno-DNA adducts in affected tissues of patients suffering from Crohn's disease, ulcerative colitis, and
chronic pancreatitis, Antioxid Redox Signal 8, 1003-1010.
12. Hofseth, L., Khan, M., and Ambrose, M. (2003) The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation, J Clin Invest 112,
1887-1894.
13. Lee, C.-Y. I., Delaney, J. C., Kartalou, M., Lingaraju, G. M., Maor-Shoshani, A., Essigmann, J. M., and Samson, L. D. (2009) Recognition and Processing of a New Repertoire of DNA Substrates by Human 3-Methyladenine DNA Glycosylase (AAG), Biochemistry 48, 1850-1861. 14. O'Brien, P. J. (2006) Catalytic promiscuity and the divergent evolution of DNA repair enzymes,
Chem Rev 106,720-752.
15. O'Brien, P. J., and Ellenberger, T. (2004) Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase, J Biol Chem 279, 9750-9757.
16. Lingaraju, G. M., Davis, C. A., Setser, J. W., Samson, L. D., and Drennan, C. L. (2011) Structural basis for the inhibition of human alkyladenine DNA glycosylase (AAG) by 3,N4-ethenocytosine containing DNA, J Biol Chem 286, 13205-13213.
17. Setser, J. W., Lingaraju, G. M., Davis, C. A., Samson, L. D., and Drennan, C. L. (2012)
Searching for DNA lesions: structural evidence for lower- and higher-affinity DNA binding conformations of human alkyladenine DNA glycosylase., Biochemistry 51, 382-390.
18. Massey, V. (2000) The chemical and biological versatility of riboflavin., Biochem Soc Trans 28, 283-296.
19. Blyth, A. (1879) The composition of cow's milk in health and disease., J Chem Soc 35, 530-539.
20. Kuhn, R., Reinemund, K., and Weygand, F. (1934) Syntheses des lumi-lactoflavins (Synthesis of lumi-lactoflavins), Ber Deut Chem Gesell 67B, 1460-1462.
21. Karrer, P., Sch6pp, K., and Benz, F. (1935) Synthese des flavins IV (Synthesis of flavins IV),
22. Theorell, H. (1935) Reindarstellung der Wirkungsgruppe des gelben Ferments. (Purification of the active group of the yellow enzyme), Biochem Z 275, 344-346.
23. Warburg, 0., and Christian, W. (1933) Uber das gelbe Ferment und seine Wirkungen (About the yellow enzyme and its effects), Biochem Z 266, 377-411.
24. Palfey, B. A., and McDonald, C. A. (2010) Control of catalysis in flavin-dependent monooxygenases., Arch Biochem Biophys 493, 26-36.
25. van Berkel, W. J. H., Kamerbeek, N. M., and Fraaije, M. W. (2006) Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts., J Biotech 124, 670-689.
26. Entsch, B., Cole, L. J., and Ballou, D. P. (2005) Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase., Arch Biochem Biophys 433, 297-311.
27. Dym, 0., and Eisenberg, D. (2001) Sequence-structure analysis of FAD-containing proteins.,
Protein Sci 10, 1712-1728.
28. Robson, C. N., and Hickson, I. D. (1991) Isolation of cDNA clones encoding a human apurinic/apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E. coli xth (exonuclease III) mutants, Nucleic Acids Res 19, 5519-5523.
29. Matsumoto, Y., and Kim, K. (1995) Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair, Science 269, 699-702.
30. Fan, J., and Wilson, D. M. (2005) Protein-protein interactions and posttranslational modifications in mammalian base excision repair, Free Radical Biol Med 38, 1121-1138.
Chapter 2. Structural Basis for the Inhibition of Human Alkyladenine DNA
Glycosylase (AAG) by 3,N-Ethenocytosine-containing DNA
This research was originally published in the Journal of Biological Chemistry. "Gondichatnahalli M. Lingaraju, C. Ainsley Davis, Jeremy W. Setser, Leona D. Samson, Catherine L. Drennan. Structural Basis for the Inhibition of Human Alkyladenine DNA Glycosylase (AAG) by 3,N 4-Ethenocytosine-containing DNA. Journal of Biological Chemistry. 2011; 286(15):13205-13213" C the American Society for Biochemistry and Molecular Biology.
11.1 SUMMARY
Reactive oxygen and nitrogen species, generated by neutrophils and macrophages in chronically inflamed tissues, readily damage DNA, producing a variety of potentially genotoxic etheno base lesions; such inflammation-related DNA damage is now known to contribute to carcinogenesis. While the human alkyladenine DNA glycosylase (AAG) can specifically bind DNA containing either 1,N 6-ethenoadenine (FA) lesions or 3,N4-ethenocytosine (EC) lesions, it can only excise eA lesions.
AAG binds very tightly to DNA containing 8.C lesions, forming an abortive protein-DNA complex;
such binding not only shields EC from repair by other enzymes, but also inhibits AAG from acting on other DNA lesions. To understand the structural basis for inhibition, we have characterized the binding of AAG to DNA containing EC lesions and have solved a crystal structure of AAG bound to a DNA duplex containing the EC lesion. This study provides the first structure of a DNA glycosylase in complex with an inhibitory base lesion that is induced endogenously, that is by exposure to environmental agents such as vinyl chloride. We identify the primary cause of inhibition as a failure to activate the nucleotide base as an efficient leaving group, and demonstrate that the higher binding affinity of AAG for sC versus i A is achieved through formation of an additional hydrogen bond between Asn169 in the active site pocket and the 02 of 8C. This structure provides the basis for the design of AAG inhibitors currently being sought as an adjuvant for cancer chemotherapy.
II.I INTRODUCTION
Genotoxic etheno (e)-lesions such as 3,N 4-ethenocytosine (EC) and 1,N-ethenoadenine
(eA) are endogenously generated when DNA is attacked by reactive aldehydes. These reactive
compounds are generated as by-products of lipid peroxidation that is induced by reactive oxygen and nitrogen species (RONS). Neutrophils and macrophages generate large quantities of RONS in tissues undergoing chronic inflammation (1, 2), and it is widely accepted that such inflammation increases the risk of colon cancer in ulcerative colitis (UC) and Crohn's disease patients, and increases the risk of liver cancer in Wilson's disease and Hemochromatosis patients
(1, 3). In fact, increased levels of F-lesions in the DNA of tissues undergoing chronic
inflammation have been reported for each of these diseases (4). Depending on the type of DNA polymerase, EC mispairs with A, T or C during DNA replication, resulting in both transition and transversion mutations (5). In contrast, EA primarily gives rise to A:T to T:A transversion mutations (6). These mutagenic E-lesions are generally removed via the base excision repair (BER) pathway, initiated by lesion-specific DNA glycosylases that cleave the N-glycosidic bond between the damaged base and the deoxyribose sugar (5, 7). In humans several DNA
glycosylases can excise eC, namely thymine DNA glycosylase, methyl-CpG binding domain protein and single strand monofunctional uracil DNA glycosylase (5). In contrast, there is only one DNA glycosylase known to excise EA lesions in humans, alkyladenine DNA glycosylase
(AAG) (5, 8).
AAG (also known as MPG and ANPG) has been previously characterized by
crystallography. The crystal structures of an N-terminally truncated, but catalytically active, construct of AAG (A79AAG), both bound to a pyrrolidine abasic site mimic (pyr) and bound to an EA containing piece of DNA, suggest a mode by which AAG recognizes a wide range of lesions, while still discriminating against undamaged bases (9, 10). When substrate is bound,
AAG can excise the damaged base through acid-base catalysis (11). A putative catalytic water
molecule, revealed by crystallographic studies, is proposed to act as a nucleophile, as it is ideally positioned to attack the N-glycosidic bond present between the EA base and the deoxyribose sugar (10). This water molecule is also in contact with the side chain of Glul25, which is proposed to be the catalytic base responsible for activating the water for nucleophilic attack. Consistent with this proposal, introduction of a E125Q mutation completely abolishes AAG activity (9-11). The identity of the general acid is unknown. Interestingly, in the structure of
A79AAG bound to FA:T containing DNA, the FA lesion remained intact (10). Later experiments
showed that the catalytic activity of A79AAG is significantly inhibited in the presence of a variety of divalent metal ions including Mn2*, Zn2*, Ca2+, Cd2*, Ni2+ and most importantly,
Mg2+,
which was contained in the crystallization buffer (9, 10, 12, 13). However, the structures did not show any Mg2+ ions bound (9, 10).
In addition to repairing cA, AAG can repair other RONS, and alkylation induced DNA damage, including lesions hypoxanthine (Hx), 1,N 2-ethenoguanine, 8-oxoguanine, 3-methyladenine (3-meA), 7-methylguanine (7-meG) and 3-methylguanine (2, 8). However, in spite of this broad substrate specificity, there is a growing list of lesions to which AAG can bind while failing to excise the lesion. In addition to FC containing DNA (14), this list now includes 3-methyluracil, 3-ethyluracil, 3-methylthymine and 3-methylcytosine (15). This binding without cleavage can result in the formation of stable abortive complexes between AAG and damaged
DNA. The EC-AAG abortive complex has been shown to inhibit AAG glycosylase activity in
vivo in human cells, and to result in replication blockage (14), increasing the genotoxicity of cC lesions in vivo. In tissues undergoing chronic inflammation, which have higher cC content (4), the formation of AAG-EC abortive complexes may significantly diminish the repair of other
AAG substrates, ultimately resulting in the accumulation of various DNA lesions in addition to cC. Interestingly, in ulcerative colitis patients, the colon epithelium undergoing chronic
inflammation was found to have increased AAG expression, perhaps indicating an adaptive response triggered by increased levels of DNA damage (16). This adaptation might compensate for the AAG hijacked by cC lesions.
In order to understand the structural basis for the inhibition of AAG by EC containing
DNA, we have carried out biochemical and crystallographic studies on AAG, using a truncated
form of the protein (A79) that was previously shown to have full catalytic activity (11, 15, 17). Our crystal structure of A79AAG bound to a DNA duplex containing cC:G (G paired opposite
EC), in combination with previous (11) and current (this work) biochemical analysis, suggests
that the failure of AAG to activate the leaving group (cC) by protonation is likely the primary reason for its inability to remove cC from the DNA. This structure also shows that a divalent metal ion, Mn2
, can bind to the base opposite the cC lesion, changing its sugar pucker, and
providing the first structural framework for considering the molecular basis for metal ion inhibition of AAG.
11.111 RESULTS
AAG binding studies
Th, binding affinity of A79AAG to the EA:T (EA paired opposite T) lesion containing 25-mer and 13-mer duplexes was compared with the binding affinity to cC:G (EC paired opposite
G) duplexes, using gel mobility shift assays (Figure 11.1, A and B; and Table 11.1). As shown in
Table II.1, when either FA or EC lesions are present in a given sequence context, A79AAG consistently binds the FC:G duplex with ~2-fold higher affinity compared to that of the EA:T duplex. In addition, A79AAG binds the EA:T 25-mer duplex (Kd = 20 ± 2 nM) with ~2-fold higher affinity, compared to the EA:T 13-mer duplex used for crystallization (Kd = 46 ± 6 nM).
Correspondingly, A79AAG also binds the eC:G 25-mer duplex (Kd = 13 ± 2 nM) with -2-fold
higher affinity, compared to the eC:G 13-mer duplex (Kd = 21 ± 3 nM). These results indicate
that the binding affinity of A79AAG to DNA containing the same lesion varies depending on the length of the DNA duplex. The binding studies also show that in a given sequence context,
A79AAG binds EC:G duplex with higher affinity compared to that of the eA:T duplex.
Catalytic ability of AAG for cC containing DNA
Following our binding studies, we tested the DNA glycosylase activity of both full-length and A79AAG on EA and PC residues present in the 25-mer oligonucleotide duplexes (Figure II.lE). As shown in the representative gel, both full-length and A79AAG robustly removed EA from the EA:T 25-mer duplex. However, the activity of both full-length and A79AAG on PC
residues from the same duplex was completely absent. In contrast, the positive control, E. coli Mismatch Uracil DNA Glycosylase (MUG) (Trevigen) shows robust catalytic activity on PC contained in an cC:G 25-mer duplex (Figure II.lE). Further, we tested the activity of A79AAG on PC containing 13-mer oligonucleotide duplexes used for crystallization, with cC paired opposite different bases. The results show A79AAG does not have activity on cC:G, cC:A,
EC:C, or cC:T 13-mer duplexes (Figure 11.2).
Inhibition of AAG by eC containing DNA
We employed competition assays to measure the inhibition of A79AAG activity on cA:T 25-mers using cA:T and EC:G 13-mer competitor DNA oligonucleotides (Figure II.1, C and D; and Table II.1). The catalytic activity of A79AAG on labeled EA:T 25-mer duplex was measured
at 37*C in the presence of an increasing concentration of the aforementioned cold competitors (Figure II.1C). The cleavage data were fitted to equation (2) (Materials and Methods) in order to calculate the 50% inhibitory concentration (IC50) for each cold competitor used in the experiment
(Figure II.1D). The results obtained correlate with the binding measurements described above. As shown in Table 11.1, the EA:T 13-mer DNA duplex binds approximately 4-fold weaker (IC50
= 163 nM) than the cC:G 13-mer (IC5 0 = 39 nM).
Overall structure of the A 79AAG-EC DNA inhibitor complex
The crystal structure of A79AAG-E-C:G at 2.2
A
resolution was determined by molecular replacement using the structure of A79AAG-pyr:T complex as a search model (9). Difference electron density maps calculated in the absence of DNA show interpretable electron density forDNA backbone and for the EC base in the active site pocket of AAG. In comparison to the
previously published A79AAG-DNA complexes (9, 10), the A79AAG-eC inhibitor complex crystallized in a different space group (P1). The final model of A79AAG-EC:G has been refined to the R factor of 23.9 (Ree = 28.4) (Table 11.2).
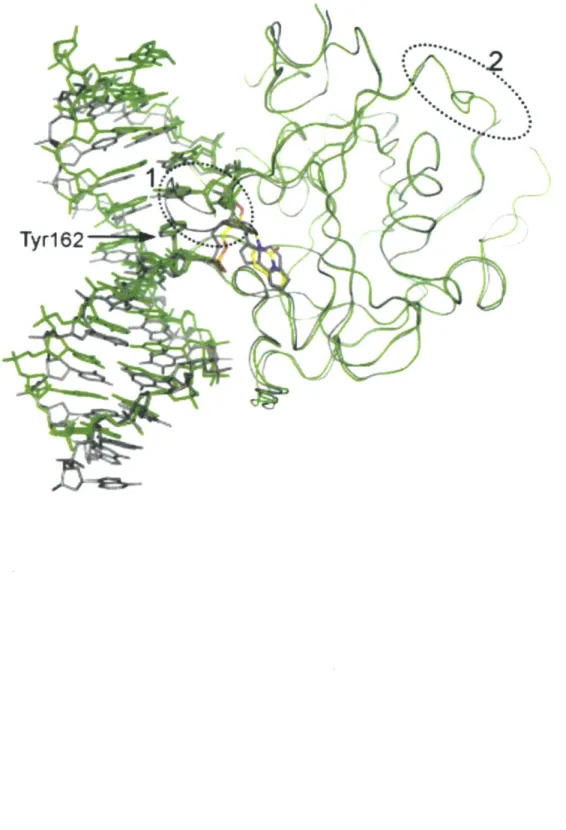
The overall structure of A79AAG bound to an EC DNA lesion includes the insertion of Tyrl62 into the DNA and the flip of the FC nucleotide into the enzyme active site (Figure 11.3). The two copies of this inhibitor-protein complex in the asymmetric unit are quite similar to each other, with a root mean square deviation (RMSD) for all alpha carbon atoms of approximately
0.6
A,
and to that of the A79AAG-cA:T substrate complex (10), also having a RMSD ofapproximately 0.6
A.
With the exception of some loops and two disordered regions (Figure II.4), the only notable difference in our inhibitor structure is that the octahedrally coordinated Na' metal ion modeled in the structure of the A79AAG-eA:T complex was found to be absent. Instead, this site is occupied by the N-terminal amino group of residue 80 of a symmetrically equivalent A79AAG molecule (Figure 11.5). Similar to the Na' ion, the N-terminal amine in this position also interacts with the main chain carbonyls of Metl49, Gly174, Ala177, Serl7l and theside chain of Ser172.
Protein-DNA interactions
In both molecules in the asymmetric unit of the A79AAG-C:G structure, the DNA duplex is largely B-form and is bent away from the protein by about 220, with the bend primarily
centered on the flipped EC nucleotide (Figure II.3A). For the most part, all protein-DNA interactions are similar between the A79AAG-EC:G inhibitor complex and the A79AAG-FA:T substrate complex (Figures 11.3 and 11.4). Tyr162 makes the most important protein contact, given that it is inserted into the DNA duplex, replacing the EC lesion, and forming Van der Waals contacts with the opposite base, G19 (Figure 11.3). A potential steric clash of Tyr162 with
G19 is prevented by shifting this opposite base out of the minor groove, leaving it without a base
pairing partner (Figure 11.3, B and C). This sliding rearrangement, in which G19 is orphaned in terms of hydrogen bonding, is disruptive and presumably has an energetic penalty associated with it. The deoxyribose of the orphaned base does make favorable Van der Waals interactions with Metl64 (Figures II.3B and II.6C), counteracting some of these energetic costs. As was the case in the A79AAG-EA:T substrate complex, no direct hydrogen bonding interactions are present between the protein and the base opposite the DNA lesion, indicating that specific recognition of this base is not required for the binding of AAG to DNA.
Metal ion Mn2" in the z79AAG-EC:G structure
The A79AAG-FC:G complex was crystallized in the presence of 200 mM MnCl2, a
condition under which AAG's ability to excise EA from a DNA duplex is impaired (12, 13). We find electron density consistent with a metal ion near the base opposite the EC lesion (Figures
II.3C and 11.6), at a distance of approximately 16
A
to the AAG active site (Cl' of EC). Mn2 ,refines well in this electron density with no positive or negative difference electron density. In contrast, refinement of a water molecule or a sodium ion (also present in the crystallization buffer) leads to positive difference electron density, suggesting that the correct atom in this site is heavier than water and sodium, consistent with Mn2
,. Anomalous difference density is also
present at both sites in the asymmetric unit at approximate sigma levels of 8 and 5 for chain A and chain B respectively, consistent with the presence of Mn2
1 ions (Figure II.6A). At the
wavelength of data collection (k = 1.116
A),
sodium would not give rise to an anomalous signal, ruling out the other metal ion contained in the crystallization buffer as being present in this site. The refined Mn2, is coordinated to the 06 of G19 (the base opposite SC), to the N7 of A 18, and
to three water molecules (Figure II.3C). The coordination of Mn2
' appears to induce a significant
change in the phosphodiester backbone in the region around G19, resulting in a change in the sugar pucker to a C2'-exo conformation (Figure II.6B).
Active site architecture of z179AAG-EC DNA complex
The PC base lesion is recognized and stabilized by hydrogen bonds, along with Van der Waals interactions (Figures 11.7 and 11.8). Similar to the recognition of FA (Figure II.7B), eC is stacked between Tyr127 on one side, and His136 and Tyr159 on the other side (Figure II.7A). Tyr159 makes edge-to-face contact with the lesion base. The specificity to discriminate between the EC lesion and undamaged cytosine appears to be achieved through a hydrogen bond donated from the main chain amide of His136 to the N' of the PC base. This interaction is similar to the damage-specific recognition of -A (versus undamaged adenine) by AAG, which is made through a hydrogen bond donated by the main chain amide of His136 to the N6 of EA. An additional
hydrogen bond is also observed between the carboxamide nitrogen of the side chain of Asn169 and the 02 of EC. Mutants of Asn169, namely Asnl69Leu and Asnl69Ala, show respective ~2-fold and ~4-fold reduced affinity for the EC:G 25-mer oligonucleotides, compared to that of wild type A79AAG (Table 11.1), indicating that the additional hydrogen bond donated from Asn169 to FC contributes to the increased affinity of A79AAG for EC inhibitor DNA.
Interestingly, in the A79AAG-FC:G inhibitor complex, the putative catalytic water molecule (proposed to act as a nucleophile) occupies the same position as in the A79AAG-eA:T substrate complex (Figure 11.7). Also in agreement with the structure of the A79AAG-EA:T substrate complex, this water molecule interacts with Glul25, Arg 182 and main chain carbonyl oxygen of Val262. Glul25, which is proposed to activate the water molecule for nucleophilic attack (10,
11), is held in its position through a hydrogen bond donated from Tyr127 to its carboxyl group.
The side chain of Arg 182 further contacts the 3'-phosphate of the EC nucleotide.
In comparison to the recognition of the EA substrate, the EC inhibitor recognition induces slight changes in the active site, mostly with respect to DNA backbone positions (Figure II.7C). Since FC is smaller than EA, the DNA backbone must be pulled farther into the active site such that EC is recognized with optimal molecular interactions. In a modeling exercise in which the
DNA backbone is held rigid, AAG fails to form optimal hydrogen bonding interactions between
both the main chain amide of His 136 and the N4 of FC, and the side chain of Asn 169 and the 02
of EC. Perhaps as a result of this slight repositioning of the DNA backbone, the side chain of Arg 182 adopts a different conformation (Figure II.7C). The side chain of Arg 182 still interacts with the 3' phosphate of the EC nucleotide and still hydrogen bonds to the putative catalytic water molecule. Overall, however, comparison of the active site of the A79AAG-eC inhibitor
complex with that of the A79AAG-EA:T substrate complex shows that, with the exception of Arg182, all the residues involved in lesion recognition and catalysis maintain similar orientations.
II.IV DISCUSSION
AAG plays an important role in the maintenance of genomic integrity, presumably
through its ability to recognize, bind and excise a wide-range of DNA base lesions. It was therefore surprising that AAG also has the ability to recognize and bind a number of DNA base lesions that it is incapable of excising, in particular the -C lesion. Moreover, the tight binding of
AAG to EC leads to the inhibition of its catalytic activity and, in addition, is known to shield EC
from ABH2-mediated direct reversal repair (18). In order to understand the structural basis for the inhibition of AAG by eC containing DNA, we solved the crystal structure of A79AAG bound to a 13-mer EC:G (EC paired opposite G) duplex.
Given that AAG can bind EC containing DNA, we anticipated that the lack of activity may be a result of one or more of the following factors: (i) AAG might fail to flip EC into its active site, or it might flip EC into an alternative binding pocket that lacks the appropriate catalytic residues; (ii) the binding mode of EC in the active site might not favor accommodation of the water molecule thought to act as a nucleophile in the reaction; (iii) the side chain of the putative catalytic base (Glul25) might adopt a nonproductive conformation that fails to activate the putative catalytic water molecule; (iv) AAG might be unable to protonate EC, failing to
activate it for departure.
The crystal structure shows that AAG successfully flips the EC inhibitor into the same active site pocket that binds the EA substrate, ruling out the first possibility. We also find that the putative catalytic water molecule is present in the inhibitor complex, ruling out the second possibility. Furthermore, as was observed in the structure of the A79AAG-EA:T substrate complex, this water molecule is in contact with Glul25, as would be required for its activation. The water molecules are ideally positioned to attack the N-glycosidic bond. Thus, we infer that AAG's inability to remove EC is unlikely to be due to a problem with nucleophilic activation or
attack, ruling out the third possibility from our list.
We next examined the fourth possibility, that the failure to excise EC is due to a problem with leaving group activation. In a previous biochemical study, O'Brien and Ellenberger
measured the pH-rate profiles for AAG's excision of neutral Hx and CA lesions, and its excision of the positively charged 7-meG lesion under single turnover conditions (11). They found that the pH-rate profiles for FA and Hx excision follow a bell-shaped curve, indicating that for the excision of neutral lesions, AAG uses the action of both a general acid and a general base (Glul25). The general base can activate a catalytic water molecule, while the general acid is expected to facilitate the protonation of neutral lesions, making the lesion base a better leaving group (11). In contrast, the pH-rate profile for the excision of 7-meG shows only a single ionization corresponding to a general base, suggesting that leaving group activation of 7-meG is not necessary because the base is already positively charged. To help pinpoint the site of protonation, the activity of AAG on Hx was compared to its activity on 7-deaza-Hx, and although AAG greatly enhances the rate of Hx excision (~10'), the same lesion with N7 changed to C7 is not cleaved by AAG, directly implicating the involvement of the N7 position in catalysis (See Figure II.9B for numbering) (11). While this study was unable to identify a specific residue as the general acid, the crystal structure of a A79AAG(E125Q)-EA:T substrate complex shows a water molecule in contact with the equivalent position to N7 of Hx, that is N7 of EA (Figure
II.9A) (10), raising the possibility that a protein-bound water molecule could be responsible for
protonation. Once protonated, the AAG active site is designed to stabilize the protonated form of the base through a hydrogen bond between N7H of FA and the backbone carbonyl oxygen of Ala134 (Figure II.9A). Given these findings on the catalytic significance of protonation of the
N7 of Hx and EA, it is important to consider the equivalent position in the EC base. A
superposition shows that unlike FA, cC has a carbon (C5) in the position equivalent to N7, and thus cannot be protonated at that site (Figure II.9A). Therefore, as opposed to our findings with respect to possibilities one through three, it appears that AAG's failure to cleave eC could be due to an inability to activate the EC leaving group by protonation. Since AAG is reported to bind and not cleave a number of different pyrimidine lesions, including 3-methyluracil, 3-ethyluracil, and 3-methylthymine (15), this mechanism of inhibition may be broadly applicable.
Given that AAG cannot repair EC lesions, it is interesting that AAG binds this lesion so tightly. The molecular basis for the approximate 2-fold higher affinity of AAG for the EC:G duplex (compared to the substrate cA:T duplex) can be attributed to an additional hydrogen bond formed between the carboxamide side chain of Asn 169 and the 02 of EC. Mutation of Asn169 to