HAL Id: hal-02795556
https://hal.inrae.fr/hal-02795556
Submitted on 5 Jun 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0
International License
Le programme MetEpiC : Etude d’une programmation
nutritionnelle chez le canard. Contribution à l’analyse
de l’expression de gènes cibles dans le foie de canetons
nouveau-nés
Mireille Morisson, Justine Grosjean
To cite this version:
Mireille Morisson, Justine Grosjean. Le programme MetEpiC : Etude d’une programmation
nutrition-nelle chez le canard. Contribution à l’analyse de l’expression de gènes cibles dans le foie de canetons
nouveau-nés. Autre [q-bio.OT]. 2016. �hal-02795556�
Remerciements
Je tiens tout d’abord à remercier Mireille MORISSON, ingénieure de recherche supervisant
le projet de recherche de m’avoir donné l’opportunité d’y participer. Ce stage de biologie
moléculaire a su attiser ma curiosité par une mise en pratique des cours vus cette année.
Merci pour le temps consacré à répondre à mes questions tout au long du stage, pour vos
relectures du rapport et vos précieux conseils.
Mille mercis à Aurélie SECULA, technicienne de recherche,
pour m’avoir transmis ses
connaissances et son savoir-faire qui m’ont aidée à effectuer les manipulations en
laboratoire. Merci également pour ta disponibilité et toutes tes explications enrichissantes.
Je tiens également à remercier Laurence LIAUBET, chargée de recherche, animatrice de
l’équipe GENOROBUST de m’avoir accueillie au sein de l’unité de recherche GenPhyse.
Je remercie aussi, l’ensemble des membres de l’unité mixte de recherche GenPhyse pour
leur sympathie et leur bonne humeur. Plus particulièrement, merci à Lisa BLUY et Laure
GRESS avec qui j’ai été amenée à collaborer, pour leurs renseignements instructifs.
1
Sommaire
ARTICLE SCIENTIFIQUE
I. Contexte et objectifs de l’étude
1.1 La programmation nutritionnelle
………...2
1.2 La programmation nutritionnelle chez le canard mulard
...2
1.3 Les objectifs du stage
……….3
II. Matériels et méthodes
2. 1 Matériels
……….……….4
2. 1. 1 Biologie moléculaire : extraction de l’ARN et contrôle qualité
………...4
2. 1. 2 Bioanalyse
………...……….……….5
2. 2 Méthodes
……….5
2. 2. 1 Biologie moléculaire : extraction de l’ARN et contrôle qualité
………...……5
2. 2. 1. 1 Extraction de l’ARN
……….5
2. 2. 1. 2 Estimation de la quantité d’ARN par dosage au Nanodrop
………….6
2. 2. 1. 3 Contrôle de la qualité des échantillons d’ARN
………...7
a) Electrophorèse sur gel d’Agarose………...7
b) Puces Agilent………..…8
c) Vérification de l’absence d’ADN génomique par PCR……….9
2. 2. 1. 4 Traitement à la DNase
………...9
a) Sur colonne……….9
b) Par précipitation………...10
2. 2. 2 Bioanalyse
….………..10
III. Résultats et discussion
3. 1 Biologie moléculaire
……….………..……12
3. 1. 1. Extraction de l’ARN et dosage au Nanodrop
………...12
3. 1. 2 Contrôle de la qualité des échantillons d’ARN
……….14
a) Electrophorèse sur gel d’Agarose……….14
b) Puces Agilent………14
c) Vérification de l’absence d’ADN génomique par PCR………...15
3. 1. 3. Comparaison des traitements à la Dnase
………16
a) Electrophorèse sur gel d’Agarose……….17
b) Vérification de l’absence d’ADN génomique par PCR………..17
c) Comparaison………18
3. 2 Bioanalyse
………...18
IV. Conclusion
………20
V. Bibliographie
……….20
PRESENTATION DU LABORATOIRE D’ACCUEIL
………....21
COMPTE-RENDU DES COURS DE L’UE STAGE
1. L’organisation de la recherche……….23
2. Expérimentation animale………..23
3. Initiation à la recherche clinique………..24
2
I. Contexte et objectifs de l’étude
1.1 La programmation nutritionnelle
Lors du développement embryonnaire, des marques épigénétiques sont apposées de façon différenciée en fonction du devenir cellulaire, conduisant à l’expression différentielle des gènes d’un même génome. Une fois posées, ces marques épigénétiques constituent les bases d’une « mémoire cellulaire » et sont transmises à travers les mitoses aux cellules filles. La modification de l’environnement de l’embryon, telle que la disponibilité de certains nutriments, peut modifier ces marques épigénétiques et cette « mémoire cellulaire » affectant durablement le métabolisme des tissus.
L’exemple le plus connu de programmation nutritionnelle, se trouve chez les abeilles. Chez ces insectes, les larves nourries exclusivement à la gelée royale deviennent des reines qui présentent des ovaires hypertrophiés et pondent jusqu’à 2000 œufs par jour. A l’inverse, les ouvrières qui, elles, n’ont reçu de la gelée royale que pendant les 3 premiers jours larvaires (leur régime alimentaire est ensuite composé de pollen et de nectar) ont des ovaires atrophiés non fonctionnels. C’est donc bien le régime alimentaire appliqué pendant le développement embryonnaire qui détermine le phénotype à l’âge adulte. [1]
De très nombreux exemples de programmation nutritionnelle sont également décrits chez les mammifères. Chez le rat, une carence maternelle en donneurs de méthyle (molécules impliquées dans le métabolisme du mono-carbone) au cours de la gestation et de la lactation, induit une stéatose hépatique chez les jeunes observée au sevrage, à 3 semaines d’âge [2]. La littérature est pauvre en exemples chez les oiseaux. Cependant, chez la poule pondeuse, Willems et ses collaborateurs ont montré que la carence précoce en protéines, obtenue en ponctionnant 3mL de l’albumen de l’œuf avant la mise en incubation, impacte le phénotype à l’âge adulte et modifie le transcriptome hépatique ; certains ARN (Acide RiboNucléique) messagers du foie sont présents en quantité différente selon que l’animal ait ou non subi une programmation nutritionnelle précoce. [3] [4]
Les mécanismes moléculaires impliqués dans la mise en place d’une programmation nutritionnelle relèvent de l’épigénétique et de la régulation de l’expression des gènes. Ils mettent en jeu les « marques épigénétiques » incluant des modifications de méthylation de l’ADN (Acide DesoxyriboNucléique), des modifications d’histones (acétylation, méthylation, phosphorylation), des petits ARN interférents (siRNA) et des très grands ARN non-codants (ARNnc). [5]
1.2 La programmation nutritionnelle chez le canard mulard
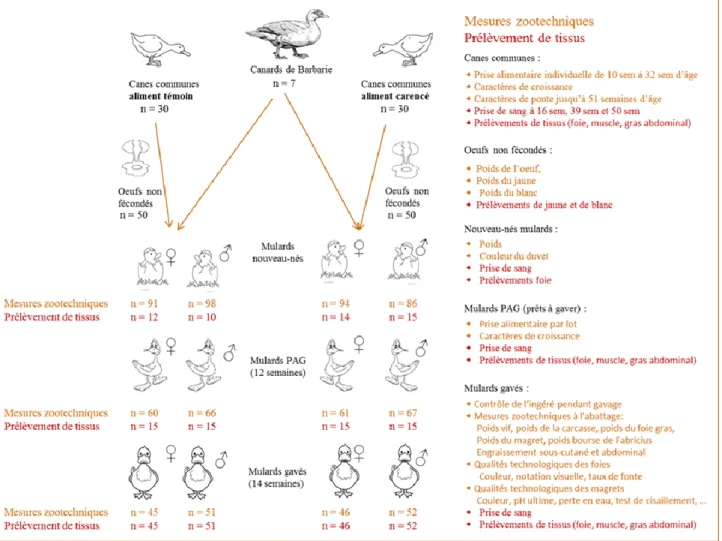
Les chercheurs de l’équipe GENOROBUST étudient dans le cadre du projet MetEpiC un mécanisme de programmation nutritionnelle chez le canard mulard. (Figure 1)
Le canard mulard est obtenu en croisant la cane commune (Anas platyrhynchos) et le canard de Barbarie (Cairina moschata). Pour cette étude, ils ont alimenté 30 canes communes avec un aliment carencé en méthionine et ont observé que ces canes carencées pondent des œufs de plus petit poids que les 30 canes témoins (65,6 grammes versus 71,2 grammes). De plus, les œufs des canes carencées ont moins de blanc (32,7g vs 37,1g) et moins de jaune (20,8g vs 21,7g). Les descendants mulards de ces canes carencées se sont donc développés dans un environnement carencé en nutriments, comparativement aux mulards témoins qui se sont développés dans des œufs de plus gros calibre. Les scientifiques ont également noté qu’après gavage, les mulards femelles ont un poids de foie gras diminué de près de 90g par rapport au poids de foie gras des femelles témoins (488g vs 575g). Chez les mulards mâles, la perte de poids de foie gras avoisine les 40g (514 vs 552g). La carence embryonnaire a donc impacté le métabolisme hépatique puisque les foies des mulards carencés ne répondent pas de la même façon à l’étape de gavage. Les scientifiques posent
3
l’hypothèse que la carence nutritionnelle appliquée lors du développement embryonnaire a induit une programmation nutritionnelle qui a réorienté le métabolisme hépatique. Cette programmation nutritionnelle doit pouvoir être détectée chez les foies des nouveau-nés mais aussi chez les foies des mulards prêts à gaver (PAG) à 12 semaines d’âge et chez les foies des mulards après gavage à 14 semaines. Pour mettre en évidence cette programmation nutritionnelle, les chercheurs espèrent observer dans les foies des mulards, des différences d’expression de gènes connus pour être impliqués dans le développement de la stéatose hépatique. [6]
Figure 1 : Schéma expérimental du programme MetEpiC
L’autorisation de projet par le ministre chargé de la recherche qui a saisi le comité d’éthique concerné est référencée sous le numéro APAFIS#1847-2015092213418825.
1.3 Les objectifs du stage
La recherche d’un différentiel d’expression nécessite plusieurs étapes. (Figure 2)
Il convient d’une part d’extraire les ARN totaux à partir d’échantillons de foies puis d’en vérifier la qualité et quantité avant de synthétiser les ADN complémentaires (ADNc) lors d’une transcription inverse réalisée à l’aide d’une réverse transcriptase. En parallèle, il faut réaliser une fouille des données dans différentes bases de données internationales afin de connaître la structure des gènes candidats (impliqués dans la stéatose hépatique) et, à partir de ces connaissances, définir pour chacun des gènes, un couple d’amorces nucléotidiques pouvant être utilisé lors d’une PCR quantitative pour comparer le niveau d’expression du gène d’intérêt entre les deux états physiologiques (foies carencés vs foies témoins).
4
Figure 2 : Schéma synoptique des étapes pré-PCR quantitative
Les étapes encadrées en rouge correspondent aux travaux que j’ai réalisés : des manipulations sur les échantillons de foie des nouveau-nés puis la définition d’un couple d’amorces pour 3 gènes candidats.
Dans le cadre de mon stage de 5 semaines, j’ai participé à ces deux démarches. J’ai extrait les ARN messagers des 51 mulards nouveau-nés du programme MetEpiC et j’en ai vérifié et assuré la qualité. Puis, j’ai utilisé les bases de données internationales pour connaître la structure de 3 gènes candidats (SIRT1, CEBPA et APOC3) et définir un couple d’amorces nucléotidiques pour chacun d’entre eux.
II. Matériels et méthodes
2. 1. Matériels
2. 1. 1. Biologie moléculaire : extraction de l’ARN et contrôle qualité
Des canetons mulards nouveau-nés ont été divisés en deux groupes : ceux dont les mères ont eu une nutrition normale faisaient partie du groupe T “témoin” et ceux dont les mères ont été carencées faisaient partie du groupe C “carencé”.
Des échantillons de foie de canetons nouveau-nés témoins (12 femelles et 10 mâles) et carencés (14 femelles et 15 mâles) ont été prélevés dès l’abattage, et plongés rapidement en azote liquide pour préserver les ARN. Puis, ils ont été stockés à -80°C avant d’être pulvérisés en poudre en présence d’azote liquide pour empêcher une décongélation qui pourrait provoquer la réactivation des RNases. Cette étape de pulvérisation ayant été réalisée auparavant, le matériel utilisé correspondait aux échantillons de foie réduits en poudre stockés dans un congélateur à -80°C. Des prises d’essai de 30mg ont été nécessaires pour les manipulations
.
5
Des recherches bibliographiques ont permis de trouver des gènes ayant potentiellement une variation d’expression au niveau du foie des canards en fonction du sexe, de l’âge et de la nutrition. Les critères de sélection des gènes cibles étaient : un rôle dans le métabolisme lipidique du foie [6] ou un rôle dans le métabolisme mono-carboné donneur de méthyle. [7] Aussi, d’autres gènes ont été sélectionnés pour l’intérêt suscité dans d’autres recherches où la mère avait été carencée (chez le rat [2] et la poule pondeuse [4] par exemple). En effet, une différence d’expression retrouvée chez d’autres espèces pourrait également exister chez le canard.
2. 2. Méthodes
2. 2. 1. Biologie moléculaire : extraction de l’ARN et contrôle qualité
2. 2. 1. 1. Extraction de l’ARN
L’extraction de l’ARN a été réalisée avec le kit « NucleoSpin® RNA » (du fournisseur Macherey-Nagel) décrit dans la figure 3.
Au vu de la toxicité des solvants utilisés, cette manipulation est effectuée sous une sorbonne afin de protéger le manipulateur. De plus, le port de gants et d’une blouse ainsi que l’utilisation de RNase Zap sont nécessaires pour travailler dans des conditions RNase-free. L’ARN étant fragile, les manipulations de l’ARN s’effectuent dans une pièce de laboratoire spécifique à l’ARN et les échantillons doivent rester dans de la glace jusqu’à l’ajout du tampon de lyse qui contient des inhibiteurs de la RNase.
Dans un premier temps, les cellules sont lysées dans un tampon de lyse contenant du thiocyanate de guanidine à 30-60% et du β-mercaptoéthanol (1%). Le mélange est ensuite vortexé, déposé sur un filtre et centrifugé à 11000g pendant 1 minute. Les
membranes cellulaires et
nucléaires ainsi que les protéines sont retenues sur le filtre tandis que l’ADN génomique (ADNg) et l’ARN sont récupérés dans le filtrat.
De l’éthanol à 70% est ajouté au filtrat pour ajuster les conditions de fixation des acides nucléiques (ADNg et ARN) à la membrane utilisée lors de la prochaine étape. Le mélange est ensuite placé sur le haut d’une colonne contenant une membrane de silice, cette colonne étant elle-même placée dans un tube de collecte. La centrifugation à 11000g pendant 30 secondes permet de faire tomber le mélange dans le tube de collecte tandis que les acides nucléiques restent fixés à la membrane de la colonne.
Un tampon dessalant les membranes est ajouté dans la colonne qui est ensuite centrifugée à 11000g pendant 1 minute, cette étape permet d’éliminer les inhibiteurs de DNase. Pour la digestion de l’ADNg, un dépôt de DNase est réalisé sur la membrane et incubé 30 minutes à température ambiante.
Par la suite, plusieurs lavages sont effectués. Le premier lavage consiste à ajouter sur la colonne un tampon contenant du guanidine hydrochloride à 24-36% et de l’éthanol à 20-35% pour inactiver la DNAse puis à centrifuger à 11000g pendant 30 secondes. L’ADNg digéré qui était resté accroché à la membrane se retrouve alors dans le filtrat et seul l’ARN reste fixé sur la membrane de silice. Pour le deuxième lavage, du tampon composé pour 80% d’éthanol est ajouté puis l’ensemble est centrifugé à 11000g pendant 30 secondes pour sécher la membrane. Le troisième lavage utilise le même tampon que le deuxième mais la centrifugation dure 2 minutes pour sécher complètement la membrane.
6
La dernière étape est l’élution de l’ARN. De l’eau RNase-free est ajoutée sur la membrane qui est centrifugée à 11000g pendant 1 minute. Le filtrat est déposé de nouveau sur la membrane pour récupérer tout l’ARN après une dernière centrifugation à 11000g pendant 1 minute.
Figure 3 : Protocole d’extraction d’ARN avec le kit NucleoSpin® RNA
L’extraction de l’ARN se déroule en neuf étapes. L’étape 5 de fixation de l’ARN à la membrane est l’étape clé. Auparavant des étapes de lyse cellulaire et d’ajustement des conditions de fixation sont nécessaires. Par la suite, l’ADNg restant est éliminé par la DNase et les lavages successifs. Enfin, l’ARN est élué lors de l’étape 9.
Macherey-Nagel ; RNA isolation : User manual NucleoSpin® RNA; June 2015; rev 17; p
2. 2. 1. 2. Estimation de la quantité d’ARN par dosage au Nanodrop
Le dosage des ARN est réalisé avec le modèle NanoDrop 8000 qui est un spectrophotomètre mesurant dans le domaine de l’ultraviolet (UV) (20-400nm) et du visible (380-780nm). Le Nanodrop
7
8000 est à échantillons multiples et à micro-volumes ; seulement 1 µL est nécessaire pour la mesure de l’échantillon. Avant la mesure de l’absorbance des échantillons, le blanc est réalisé avec de l’eau RNase-free (solution d’élution des ARN). De plus, le port de gants et d’une blouse, l’utilisation de RNase Zap ainsi qu’un maintien des échantillons dans la glace sont nécessaires pour travailler dans des conditions RNase-free.
La quantité d’ARN est déterminée par l’absorbance des acides nucléiques à 260nm. Grâce à la Loi de Beer-Lambert, des relations entre l’absorbance et la concentration en ADN et ARN ont été trouvées : 1 u.a (unité d’absorbance) correspond à 50ng/µL d’ADN et à 40ng/µL d’ARN. Vu que les protéines absorbent à 280 nm, le dosage des ARN au NanoDrop nous permet aussi d’évaluer la qualité de l’extraction préalablement réalisée grâce au ratio R (A260/A280) qui calcule le rapport de quantité d’acides nucléiques sur la quantité de protéines. Ce ratio doit se situer entre 1,8 et 2,2.
2. 2. 1. 3 Contrôle de la qualité des échantillons d’ARN
a)
Electrophorèse sur gel d’agarose
Pour vérifier que les ARN ne se sont pas dégradés, une électrophorèse sur gel d’agarose est réalisée. C’est une méthode séparant les ARN en fonction de leur taille (poids moléculaire). Les acides nucléiques sont chargés négativement, l’application d’un champ électrique entraîne donc leur migration vers le pôle positif. Plus l’ARN est de bas poids moléculaire, plus sa migration sera importante car en se polymérisant le gel d’agarose crée un réseau de mailles dense.
Plus de 80% de l’ARN total est de l’ARN ribosomique (ARNr) dont les fragments 18S et 28S, tandis que les ARN messagers (ARNm) ne représentent que 3 à 4% des ARN totaux. De plus, le fragment d’ARNr 28S est le premier à être dégradé en cas de dégradation de l’ARN. La présence sur le gel de deux bandes représentant les fragments 18S et 28S, avec une bande plus intense pour le fragment 28S, est le signe de la qualité et non dégradation des ARN totaux et donc également le signe de la qualité des ARNm. Si l’ARN est dégradé, un « smear » (trainée), représentant les fragments d’ARN dégradés à différentes tailles, apparait.
Au vu de la toxicité des solvants utilisés, cette manipulation est effectuée sous une sorbonne afin de protéger le manipulateur. De plus, le port de gants et d’une blouse ainsi que l’utilisation de RNase Zap sont nécessaires pour travailler dans des conditions RNase-free. Enfin, pour éviter la dégradation des ARN, les échantillons en attente de manipulation sont conservés dans de la glace.
Un gel à 0,8% d’agarose Seakem RNase free est préparé dans un tampon TBE (Tris, Borate, EDTA (Éthylène Diamine Tétra-Acétique)) avec une goutte de BET (bromure d’éthidium). Le BET est un agent intercalant facilitant la visualisation des bandes d’ARN après exposition du gel sous lumière UV. En effet, le BET est excité par les rayons UV et devient fluorescent lorsqu’il est intercalé entre les bases azotées de l’ARN. La classification du BET en tant qu’agent mutagène impose des procédures adaptées lors de sa manipulation et son élimination.
La concentration des échantillons étant connue grâce au dosage au NanoDrop, les échantillons sont dilués pour obtenir 2µg d’ARN dans QSP 8µL d’eau RNase free. Ils sont ajoutés à un mélange contenant du formaldéhyde, du formamide désioinisé qui dénaturent l’ARN, de l’eau ultra pure et un tampon MOPS (3-(N-morpholino) propanesulfonic acid) qui permet de neutraliser le pH. Comme les ARN ont tendance à former des épingles, ils sont dénaturés à 65°C pendant 10 minutes puis refroidis rapidement dans la glace pour empêcher leur renaturation et une nouvelle formation d’épingles. Après centrifugation et ajout du bleu-CHO (tampon de charge) contenant également du BET, les échantillons sont déposés dans les puits du gel placé dans une cuve d’électrophorèse. Le champ électrique à 60 Volts entraîne la migration des ARN pendant environ 1 heure.
8
b)
Puces Agilent
La puce Agilent est la méthode de référence pour vérifier la qualité des ARN. Le principe de la puce Agilent est le même que celui de la migration sur gel d’agarose : les produits sont séparés selon leur poids moléculaire mais ce processus est miniaturisé et utilise la microfluidique. Le kit employé est l’Agilent RNA 6000 Nano Kit (du fournisseur Agilent Technologies).
Pour cette manipulation, le port de gants et d’une blouse, l’utilisation de RNase Zap et le maintien des échantillons dans la glace sont nécessaires pour travailler dans des conditions RNase-free. Le matériel et l’analyseur des puces Agilent se trouvent au niveau de la plateforme de génomique GeT-PlaGE de la génopole toulousaine.
D’un côté, le gel est préparé avec une molécule intercalante et fluorescente qui permet la détection des ARN. D’un autre côté, les échantillons sont dénaturés à 70°C pendant 2 minutes et refroidis immédiatement dans la glace avant d’être centrifugés. Le gel est délivré dans les puits désignés tandis qu’un calibrateur est chargé dans tous les autres puits. Par la suite, pour chaque échantillon ainsi que pour le ladder (marqueur de taille), 1 µL est prélevé puis déposé dans un puits. La puce est ensuite installée sur un vortex pendant 1 minute avant de passer dans la machine d’analyse Agilent 2100. Douze échantillons peuvent être traités par puce.
L’électrophorégramme permet de visualiser trois pics principaux. Le premier correspond au calibrateur montrant le départ du signal. Le deuxième représente le fragment d’ARN ribosomique 18S et le troisième pic, plus élevé, désigne le fragment d’ARN ribosomique 28S. Comme pour l’électrophorèse sur gel d’agarose, c’est la qualité (non dégradation) des ARN 18S et 28S qui informe sur la qualité des ARNm. Dans le cas d’une dégradation d’ARN, le pic du fragment 28S s’effondre en premier et d’autres pics apparaissent.
Pour attester de la qualité des ARN, le RIN (RNA Integrity Number) compris entre 1 et 10 doit être supérieur à 7. Un RIN égal à 10 représente un ARN intact. La qualité des ARN est donc d’autant plus forte que le RIN est élevé. (Figure 4)
Figure 4 : Exemples d’electrophorégrammes avec des RIN différents
L’électrophorégramme ayant un RIN égal à 10 présente trois pics représentant : le calibrateur, le fragment 18S et le fragment 28S. Avec un RIN égal à 6, le pic du fragment 28S diminue, signe d’un début de dégradation. Avec un RIN égal à 3, de nombreux pics apparaissent formant un pic plus large. Avec un RIN égal à 2, deux pics sont présents : le calibrateur et un ensemble de petits fragments d’ARN fortement dégradés.
Agilent Technologies; RNA Integrity Number (RIN): Standardization of RNA Quality Control; 2016; p.3; Publication Number 5989-1165EN
9
c)
Vérification de l’absence d’ADN génomique par PCR
Une PCR (Réaction en Chaîne par Polymérase) est effectuée afin de mettre en évidence d’éventuels résidus d’ADN génomique (ADNg) pouvant être présents dans les échantillons. En effet, la présence d’ADNg dans nos échantillons serait gênante pour l’étape de PCR quantitative. Les amorces choisies pour s’hybrider avec l’ADN complémentaire obtenu par rétro-transcription de l’ARN, s’hybrideraient aussi avec l’ADNg. Toute mesure quantitative serait alors faussée.
On utilise un couple d’amorces permettant d’amplifier un fragment d’ADN situé dans une région non exprimée du génome. Il s’agit pour cette manipulation du couple d’amorces CAM 167 qui amplifie un fragment d’ADN intergénique. D’autres couples d’amorces amplifiant un intron de l’ADNg auraient également pu être choisis.
Après la PCR, une électrophorèse sur gel d’agarose à 2% permet de visualiser la présence ou l’absence du fragment amplifié et donc la présence ou l’absence d’ADNg.
Sur une plaque PCR, 3µL de chaque échantillon sont déposés ainsi que deux échantillons d’ADNg représentant les témoins positifs (ART20032823 et ART20039668) et deux témoins négatifs (eau ultra pure). Un mélange contenant du tampon 5X PCR, des dNTP (désoxyribonucléotides), un couple d’amorces CAM 167, de l’eau ultra pure et la Taq polymérase (5U/µL) est ajouté sur la plaque après centrifugation du mélange. Le tampon de PCR contient du magnésium dont la Taq polymérase a besoin pour polymériser et deux colorants qui facilitent la visualisation de la migration. Chaque composant du mélange ainsi que les échantillons et témoins sont au préalable vortexés et centrifugés. Les enzymes ne devant pas être vortexées, la Taq polymérase est seulement centrifugée. La plaque de PCR est centrifugée puis placée dans le thermocycleur. Dans un premier temps, la température monte à 94°C pendant 5 minutes pour dénaturer tout l’ADNg. Ensuite un enchaînement de 42 cycles est lancé. Un cycle se compose d’une première phase de 30 secondes à 94°C pour dénaturer l’ADN, une deuxième phase de 30 secondes à 55°C permettant l’hybridation des amorces et une troisième et dernière phase à 72°C pendant 30 secondes pour la polymérisation par la Taq polymérase. Enfin, la température chute à 4°C pour conserver les échantillons jusqu’à récupération de ces derniers.
Le gel d’agarose à 2% est réalisé en chauffant du TBE et de l’agarose. Deux gouttes de BET sont ensuite ajoutées au mélange avant polymérisation du gel. En plus du port des gants et d’une blouse, l’ajout du BET doit se faire sous une sorbonne car il possède un effet mutagène important. Après la PCR, les échantillons et les témoins sont centrifugés une dernière fois puis déposés dans les puits du gel d’agarose placé dans une cuve d’électrophorèse. Un marqueur de taille est également déposé. Le champ électrique à 180 Volts entraîne la migration des produits PCR pendant environ 45 minutes.
2. 2. 1. 4. Traitement à la DNase
a) Sur colonne
Le traitement à la DNase sur colonne est réalisé avec le kit « RNeasy minElute Cleanup » du fournisseur Qiagen.
Au vu de la toxicité des solvants utilisés, cette manipulation est effectuée sous une sorbonne afin de protéger le manipulateur. De plus, le port de gants et d’une blouse ainsi que l’utilisation de RNase Zap sont nécessaires pour travailler dans des conditions RNase-free. L’ARN étant fragile, les manipulations de l’ARN s’effectuent dans une pièce de laboratoire spécifique à l’ARN et les échantillons doivent rester dans de la glace.
Dans un premier temps, les échantillons sont préparés pour contenir 10 µg d’ARN puis un mélange contenant de la RNasine (inhibiteur des RNAses), de la DNase, du tampon adapté aux
10
besoins de l’enzyme, du MgCl2 et de l’eau ultra pure RNase free est ajouté. Une incubation de 15 minutes à 37°C permet de dégrader l’ADNg.
Ensuite, un premier tampon contenant du thiocyanate de guanidine (dénaturant les RNases) puis de l’alcool à 100% sont ajoutés en mélangeant par pipetage. Les conditions de fixation de l’ARN sont ainsi améliorées. L’ensemble est déposé sur une colonne positionnée sur un tube collecteur. Après une centrifugation de 15 secondes à 10000rpm, le liquide est jeté et un tampon contenant de l’alcool est ajouté dans la colonne. Une nouvelle centrifugation de 15 secondes à 10000rpm permet de laver la membrane de la colonne en éliminant les sels.
De l’éthanol à 80% est déposé dans la colonne pour sécher la membrane. Après une centrifugation de 2 minutes à 10000rpm suivie d’une seconde centrifugation de 5 minutes à 15000rpm dans un nouveau tube collecteur, la colonne est placée sur un tube propre de 1,5mL. Finalement, l’ARN de l’échantillon est élué dans le tube par 2 ajouts d’eau RNase free suivis chacun d’une centrifugation à 15000rpm pendant 1 minute.
b)
Par précipitation
Dans un premier temps, les échantillons sont préparés pour contenir 10 µg d’ARN puis un mélange contenant de la RNasine (inhibiteur des RNases), de la DNase, du tampon adapté aux besoins de l’enzyme, du MgCl2 et de l’eau ultra pure RNase free est ajouté. Une incubation de 15 minutes à 37°C permet de dégrader l’ADNg.
Ensuite, un mélange contenant de l’acétate de sodium, de l’éthanol à 100% froid et du glycoblue est ajouté et mélangé en pipetant plusieurs fois. L’acétate de sodium permet de tamponner la solution et neutralise les phosphates, ce qui favorise la précipitation de l’ARN dans l’éthanol à 100%. Le glycoblue permet de voir le culot et favorise la précipitation. L’ensemble est stocké 1 heure à -20°C pour favoriser la précipitation. Les échantillons sont ensuite centrifugés à 14000rpm pendant 15 minutes à 4°C. L’ARN se retrouve dans le culot tandis que le surnageant contenant les DNases et les bases libres est éliminé. Le culot est lavé avec de l’éthanol à 75% : les sels utilisés lors de la précipitation sont dissous puis éliminés par une centrifugation à 7500rpm pendant 10 minutes à 4°C. Par la suite, les échantillons sont laissés à l’air libre pour que l’éthanol s’évapore jusqu’à ce que le culot soit sec, il devient alors blanc. Enfin, le culot est repris dans de l’eau RNase free.
2. 2. 2. Bioanalyse : design des amorces
La recherche d’amorces nécessite l’utilisation de plusieurs logiciels et bases de données complémentaires.
Tout d’abord, la séquence d’ARNm du gène cible du canard mulard (Anas platyrhynchos) est recherchée. Pour cela, deux bases de données peuvent être utilisées. La première « Gene » est développée par le NCBI (National Center for Biotechnology Information) l’institut national américain pour l'information biologique moléculaire (source : http://www.ncbi.nlm.nih.gov/gene/). La seconde « Ensembl » est développée par l’institut européen de bio-informatique et le centre Sanger (source : http://www.ensembl.org). Ce sont des bases de données qui permettent entre autre, la lecture des séquences d’ARNm et d’ADN mais aussi la lecture des séquences protéiques pour de nombreuses espèces. Lors d’absence de résultats, la base de données du génome humain « Genecards » (source : http://www.genecards.org) permet de connaître les autres noms donnés au gène recherché et ainsi de relancer la recherche de la séquence avec ceux-ci. Toutefois, le génome du canard n'étant pas complètement annoté, la séquence d’ARNm du gène peut ne pas être connue chez le canard, la séquence est alors cherchée chez la poule (Gallus gallus).
Pour définir les exons et leur position, l’ARNm (canard ou poule) est comparé et aligné à l’ADN génomique du canard avec le programme « BLAST » (Basic Local Alignment Search Tool)
11
développé par le NCBI. (Source : http://blast.ncbi.nlm.nih.gov/Blast.cgi). Cette étape est primordiale car les amorces choisies doivent s’hybrider au niveau des exons et non au niveau des introns. En effet, avant l’étude d’expression des gènes cibles par PCR quantitative (qPCR), l’ARN est rétrotranscrit en ADNc (ADN complémentaire) qui a une séquence ne comportant que les exons. Contrairement à l’ADNc, l’éventuel ADNg contaminant les préparations d’ADNc a une séquence composée d’exons et d’introns. Or, pour quantifier l’expression de l’ARN, seul l’ADNc doit être amplifié lors de la qPCR. En optant pour des amorces présentes sur deux exons et en choisissant une taille d’amplicons (fragments amplifiés) entre 100 et 150 paires de base (pb), seul l’ADNc sera amplifié lors de l’étape de qPCR (Figure 5).
Figure 5 : Design d’amorces autour d’un intron
Les 2 amorces, figurées par les 2 flèches vertes, sont définies au sein de 2 exons, de part et d’autre d’un intron. Cela permet de différencier, lors de l’étape d’amplification sur ADNc (qPCR), l’amplification d’éventuelles traces d’ADNg contaminant car le fragment présent entre les 2 amorces est de plus grande taille.
Pour le design des amorces proprement dit, l’interface Primer 3 Plus est utilisée [8] (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/). Cette interface permet de visualiser quelles amorces peuvent s’aligner avec la séquence d’ARNm. Elle permet aussi de choisir sur quels exons les amorces doivent s’aligner, la température d’hybridation (entre 58°C et 60°C) et la taille du fragment amplifié (amplicon) (100 – 150 pb). Ces critères de sélection sont les mêmes pour tous les gènes testés car lors de l’étape de qPCR tous les gènes seront amplifiés en même temps, selon les mêmes conditions expérimentales. Des amorces s’hybridant avec les exons proches de l’extrémité 3’ seront préférentiellement choisies car la reverse transcription est faite à partir d’oligo dT en extrémité 3’ et elle risque de se décrocher avant d’arriver à l’extrémité 5’.
Enfin, pour vérifier la spécificité de l’amplification des amorces choisies, un test d’alignement est réalisé entre les amorces et l’ADNg du canard avec le programme « BLAST ».
12
III.
Résultats et discussion
3. 1 Biologie moléculaire
Dans cette étude, 51 foies de canards mulards nouveau-nés et sacrifiés dès l’éclosion ont été utilisés. L’échantillonnage compte 12 foies de canards TF (Témoin - femelle), 10 foies de canards TM (Témoin - mâle), 14 foies de canards CF (Carencé - femelle), 15 foies de canards CM (Carencé - mâle).
3. 1. 1. Extraction de l’ARN et dosage au Nanodrop
Les ARN de chaque échantillon ont été extraits en 4 séries avec le kit NucléoSpin® RNA puis dosés au Nanodrop. Grâce à la loi de Beer-Lambert, le Nanodrop a pu estimer les concentrations des ARN (entre 179,7 ng/µL et 1224 ng/µL) qui sont rapportées dans le tableau 1. Ce tableau montre également la quantité d’ARN extraits (entre 9,0 µg et 61,2 µg) calculée à partir de leur concentration, pour chaque échantillon. Les volumes de dilution nécessaires pour la préparation des échantillons au contrôle de la qualité ont aussi été calculés. Une quantité de 2 µg est demandée pour l’électrophorèse sur gel d’agarose et une concentration de 100 ng/µl est nécessaire pour les puces Agilent et pour la vérification de l’absence d’ADNg par PCR.
Une première idée de la qualité de l’extraction a pu être présumée grâce au ratio R (A260/A280) dont les valeurs de références sont comprises entre 1,8 et 2,2.
Trois échantillons (n° 551 TF, 555 TF et 563 TM) ont posé problème lors de l’étape d’extraction. Après la centrifugation suivant l’étape de fixation de l’ARN, leurs colonnes étaient bouchées probablement suite à une quantité trop importante d’ADNg, elle-même due à une prise d’essai excessive. Ces échantillons ont reçu une dose supérieure de DNase lors de l’étape 7 afin d’éliminer tout l’ADNg. Après le premier lavage, plus aucune colonne n’était bouchée. Ces échantillons ont été extraits de nouveau en refaisant une prise d’essai afin de confirmer que l’erreur provenait bien de celle-ci. Aucune colonne n’a été bouchée lors de la seconde extraction. Pour être identifiés, ces échantillons sont marqués de leur numéro suivi de « bis ».
Quelques modifications ont été apportées au protocole pour le rendre plus efficace. Lors de l’étape 9, après l’élution de l’ARN une première fois, le contenu du tube a été déposé de nouveau sur la colonne puis centrifugé pour éluer le maximum d’ARN. De plus, le protocole préconise d’utiliser 60µL d’eau RNase free, mais seul 50µL ont été ajoutés pour ne pas avoir à concentrer nos échantillons en ARN lors de la préparation au contrôle qualité. Ainsi les échantillons étaient plus concentrés en ARN et ont pu être préparés par une simple dilution.
13
Tableau 1 : Résultats obtenus par dosage au Nanodrop
Pour les 51 échantillons le ratio R est compris entre 2,0 et 2,1, signe d’une bonne extraction. Les volumes de dilution nécessaires pour la préparation des échantillons au contrôle de la qualité ont été calculés grâce à la concentration des ARN extraits estimée par Nanodrop.
Volume en µl nécessaire pour 2µg (Dépôt gel) QSP H2O à 8µl Volume en µl nécessaire pour 10µl à 100ng/µl QSP H2O à 10µl 518 642,4 16,1 7,5 2,2 32,1 3,1 4,9 1,6 8,4 519 654,1 16,4 7,6 2,1 32,7 3,1 4,9 1,5 8,5 520 611,1 15,3 7,0 2,2 30,6 3,3 4,7 1,6 8,4 521 326 8,2 3,9 2,1 16,3 6,1 1,9 3,1 6,9 521b 239 6,0 2,9 2,1 12,0 8,0 0,0 4,2 5,8 522 976 24,4 11,6 2,1 48,8 2,0 6,0 1,0 9,0 523 454,7 11,4 5,5 2,1 22,7 4,4 3,6 2,2 7,8 524 757,8 18,9 8,8 2,2 37,9 2,6 5,4 1,3 8,7 525 179,7 4,5 2,1 2,1 9,0 8,0 0,0 5,6 4,4 526 213,1 5,3 2,5 2,1 10,7 8,0 0,0 4,7 5,3 527 670 16,7 7,7 2,2 33,5 3,0 5,0 1,5 8,5 528 499,1 12,5 6,1 2,0 25,0 4,0 4,0 2,0 8,0 529 435,2 10,9 5,4 2,0 21,8 4,6 3,4 2,3 7,7 530 526,3 13,2 6,5 2,0 26,3 3,8 4,2 1,9 8,1 531 494,7 12,4 6,1 2,0 24,7 4,0 4,0 2,0 8,0 532 265,7 6,6 3,1 2,1 13,3 7,5 0,5 3,8 6,2 533 438,6 11,0 5,3 2,1 21,9 4,6 3,4 2,3 7,7 534 811,6 20,3 9,6 2,1 40,6 2,5 5,5 1,2 8,8 535 1137 28,4 13,3 2,1 56,9 1,8 6,2 0,9 9,1 536 324,8 8,1 3,9 2,1 16,2 6,2 1,8 3,1 6,9 537 491,3 12,3 6,0 2,1 24,6 4,1 3,9 2,0 8,0 538 1118 27,9 13,1 2,1 55,9 1,8 6,2 0,9 9,1 539 198,7 5,0 2,4 2,1 9,9 8,0 0,0 5,0 5,0 540 219,2 5,5 2,6 2,1 11,0 8,0 0,0 4,6 5,4 541 520,7 13,0 6,4 2,0 26,0 3,8 4,2 1,9 8,1 542 794,2 19,9 9,3 2,1 39,7 2,5 5,5 1,3 8,7 543 486,1 12,2 6,0 2,0 24,3 4,1 3,9 2,1 7,9 544 399,6 10,0 4,8 2,1 20,0 5,0 3,0 2,5 7,5 545 862,2 21,6 10,0 2,2 43,1 2,3 5,7 1,2 8,8 546 752,3 18,8 8,9 2,1 37,6 2,7 5,3 1,3 8,7 547 335,4 8,4 4,0 2,1 16,8 6,0 2,0 3,0 7,0 548 364,3 9,1 4,4 2,1 18,2 5,5 2,5 2,7 7,3 550 500,9 12,5 6,1 2,0 25,0 4,0 4,0 2,0 8,0 551b 235,7 5,9 2,8 2,1 11,8 8,0 0,0 4,2 5,8 551 775,2 19,4 9,2 2,1 38,8 2,6 5,4 1,3 8,7 552 409,2 10,2 4,9 2,1 20,5 4,9 3,1 2,4 7,6 553 488,7 12,2 5,9 2,1 24,4 4,1 3,9 2,0 8,0 554 380,6 9,5 4,5 2,1 19,0 5,3 2,7 2,6 7,4 555 636,5 15,9 7,3 2,2 31,8 3,1 4,9 1,6 8,4 555b 262,6 6,6 3,1 2,1 13,1 7,6 0,4 3,8 6,2 556 696,4 17,4 8,1 2,2 34,8 2,9 5,1 1,4 8,6 557 587,8 14,7 6,9 2,1 29,4 3,4 4,6 1,7 8,3 559 691,2 17,3 8,0 2,2 34,6 2,9 5,1 1,4 8,6 560 475,9 11,9 5,8 2,1 23,8 4,2 3,8 2,1 7,9 561 822,5 20,6 9,6 2,1 41,1 2,4 5,6 1,2 8,8 562 515 12,9 6,3 2,0 25,8 3,9 4,1 1,9 8,1 563 1224 30,6 14,5 2,1 61,2 1,6 6,4 0,8 9,2 564 528,5 13,2 6,5 2,0 26,4 3,8 4,2 1,9 8,1 565 276,3 6,9 3,3 2,1 13,8 7,2 0,8 3,6 6,4 567 437,1 10,9 5,3 2,1 21,9 4,6 3,4 2,3 7,7 569 682,6 17,1 7,9 2,2 34,1 2,9 5,1 1,5 8,5 570 669,8 16,7 7,9 2,1 33,5 3,0 5,0 1,5 8,5 571 670,5 16,8 7,9 2,1 33,5 3,0 5,0 1,5 8,5 573 611,3 15,3 7,2 2,1 30,6 3,3 4,7 1,6 8,4 563b 321,1 8,0 3,8 2,1 16,1 6,2 1,8 3,1 6,9 565b 507,5 12,7 6,2 2,0 25,4 3,9 4,1 2,0 8,0 N° Echantillon Quantité totale d'ARN extraits en µg
Gel Agarose qualité
Dosage AGILENT + Contrôle contamination Concentration (ng/µl) DO A260 (Acides nucléiques) DO A280 (Protéines) Rapport R 260/280 M â l e s
T
é
m
o
i
n
s
F e m e l l e s M â l e s F e m e l l e sC
a
r
e
n
c
é
s
14
3. 1. 2. Contrôle de la qualité des échantillons d’ARN
a)
Electrophorèse sur gel d’agarose
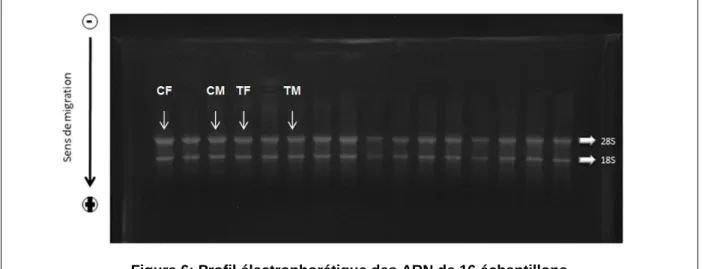
Pour chacun des 51 échantillons, 2µg d’ARN ont été déposés dans les puits du gel. La migration de l’ARN sur le gel d’agarose a permis de visualiser deux bandes : une première bande, plus intense, pour le fragment d’ARNr 28S et une deuxième bande pour le fragment d’ARNr 18S. Ce dernier est plus éloigné du puits car le fragment d’ARN étant plus petit, il est moins retenu par les mailles du gel et peut donc migrer davantage. Ainsi, le fait que les ARNr 28S et 18S ne soient pas dégradés permet d’en déduire une bonne qualité des ARN totaux car les ARNr représentent plus de 80% des ARN totaux et le fragment 28S est le premier à être dégradé. (Figure 6)
Figure 6: Profil électrophorétique des ARN de 16 échantillons
Les ARN, chargés négativement, migrent vers le pôle positif en fonction de leur taille. Le fragment d’ARN 18S migre davantage que le fragment d’ARN 28S dont la bande est plus intense. Seulement deux bandes sont présentes sur le gel, c’est un signe de qualité et de non dégradation des ARN.
Pour deux échantillons (n°521 CF et 565 TM), le profil électrophorétique présentait trois bandes au lieu des deux recherchées. Cette troisième bande, proche de la bande représentant le fragment 28S est apparue à cause d’un problème lors de la dénaturation des ARN à 65°C. En effet, les puits des plaques chauffantes étaient plus grands que les tubes contenant les ARN. Ces derniers ne touchaient pas les parois et n’étaient donc pas à 65°C mais plutôt aux alentours de 60°C. A cette température, certains ARN n’ont pas été dénaturés, sont restés « en épingle » et ont migré différemment que les ARN 28S et 18S dénaturés ce qui a entraîné l’apparition d’une troisième bande. Ces échantillons ont été extraits de nouveau puis dénaturés à 65°C dans des plaques adaptées. Dans ces conditions, leur profil électrophorétique ne présentait que les deux bandes recherchées, cela confirme que l’erreur provenait bien de l’étape de dénaturation. Pour être identifiés, ces échantillons sont marqués de leur numéro suivi de « bis ». Au final, 56 échantillons (51 + 5 bis) ont donc été traités.
b) Puces Agilent
Les cinq puces Agilent réalisées avec le kit « Agilent RNA 6000 Nano Kit » ont permis d’attester de la qualité des ARN extraits. En effet, les trois pics principaux recherchés (calibrateur, fragments 18S et 28S) sont visibles sur l’électrophorégramme et le pic du fragment 28S est supérieur au pic du fragment 18S. De plus, le RIN qui doit être supérieur à 7 pour affirmer une bonne qualité des ARN, est compris entre 7,40 et 9,20 pour l’ensemble de nos 56 échantillons. (Figure 7)
15
Figure 7: Electrophorégrammes par technique Agilent de douze échantillons
Pour chaque échantillon, le premier pic correspond au calibrateur, le second au fragment 18S et le troisième, plus grand, représente le fragment 28S. Pour ces douze échantillons, le RIN est compris entre 7,90 et 9,20, signe de qualité et de non dégradation des ARN.
c)
Vérification de l’absence d’ADN génomique par PCR
Nous avons utilisé un couple d’amorces (CAM 167) qui permet d’amplifier un fragment d’ADN intergénique, situé dans une région non exprimée du génome. Ce fragment a une taille de 298pb. Après une PCR de 42 cycles, les produits réactionnels des échantillons ainsi que des témoins positifs et négatifs ont migré sur un gel d’agarose par électrophorèse en présence d’un marqueur de taille qui présente des fragments d’ADN dont les tailles sont des multiples de 100pb. (Figure 8)
Le témoin négatif, l’eau ultra pure, n’a qu’une seule bande ayant fortement migré et qui correspond à des dimères d’amorces. Le témoin positif, quant à lui, montre une bande de forte intensité qui a migré vers les 300pb si l’on observe la migration du marqueur de taille. Les échantillons présentent la même bande que les témoins positifs mais avec une intensité moindre, ce qui est le signe de la présence d’ADNg dans nos ARN.
16
Figure 8 : Profils électrophorétiques des produits de PCR présents dans 56 échantillons des ARN extraits et des témoins positifs et négatifs, après une PCR de 42 cycles
La PCR a permis d’amplifier, dans chacun des 56 échantillons testés, le fragment de 300pb spécifique du marqueur CAM 167. Nos extraits d’ARN sont donc contaminés par de l’ADNg.
Pour autant, la quantité d’amplicon issu de l’ADNg augmentant exponentiellement avec la PCR et donc principalement lors des derniers cycles, une deuxième PCR avec 35 cycles a été programmée. En effet, lors de l’étape de qPCR seuls les gènes qui apparaissent avant 30 cycles sont sélectionnés. La présence d’amplicon issu de l’ADNg après une PCR de 42 cycles pourrait ainsi être négligeable lorsque les échantillons subissent une PCR de 35 cycles. Néanmoins, les résultats obtenus après la PCR de 35 cycles n’ont fait que confirmer la présence d’ADNg dans nos échantillons d’ARN. Un traitement supplémentaire à la DNase pour supprimer l’ADNg est donc nécessaire avant la poursuite des manipulations.
3. 1. 3. Comparaison des traitements à la DNase
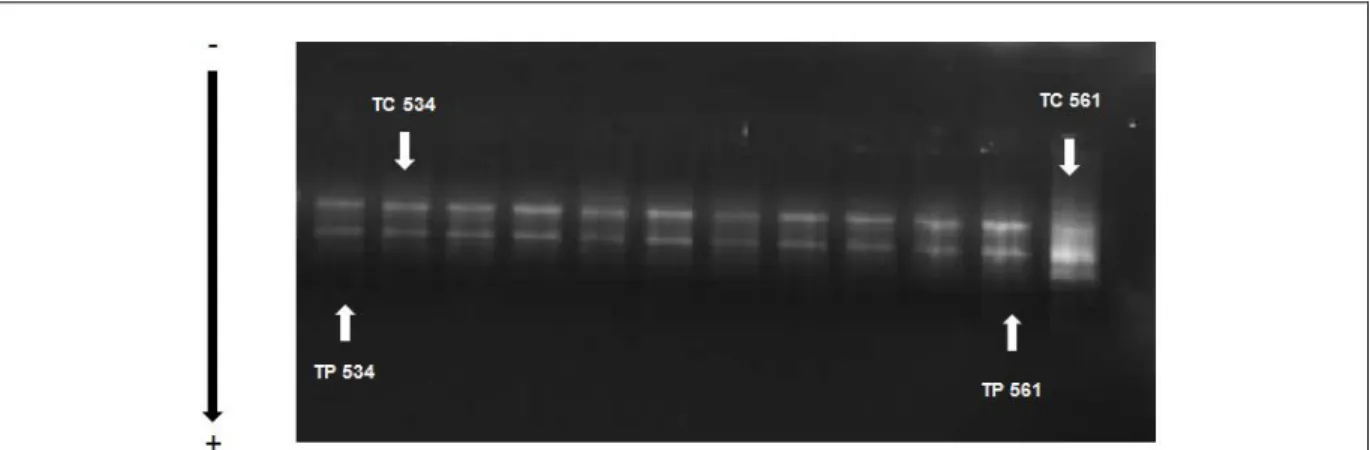
Cette étape, non prévue initialement a nécessité la recherche de protocoles de traitement à la DNase. Avant de traiter tous les échantillons, deux protocoles semblant prometteurs ont été testés sur six échantillons pour comparer leur efficacité. Ces protocoles sont semblables pour le traitement à la DNase proprement dit. C’est l’étape de purification de l’ARN avec élimination de la DNase et de l'ADN qui varie ; l’ARN est purifié soit sur colonne, soit par précipitation (voir méthode 2.2.1.4 : Traitement à la DNase). Ces deux traitements sont comparés sur le rendement, la qualité des ARN avec une électrophorèse sur gel d’agarose et sur l’absence d’ADNg avec une PCR de vérification.
Les échantillons choisis pour tester les deux méthodes sont les six échantillons avec le plus d’acides nucléiques avant traitement à la DNase. Il s’agit des échantillons 534, 538, 542, 545, 546, 561.
Pour déposer la même quantité entre tous les échantillons pour l’électrophorèse sur gel et la PCR de vérification, un dosage au Nanodrop a été effectué. Ce dernier a révélé que les échantillons ayant été traités par précipitation étaient plus concentrés en ARN.
17
a)
Electrophorèse sur gel d’Agarose
Les échantillons ont été dilués pour obtenir une quantité de 1µg d’ARN dans QSP 8µL qui a ensuite été déposée dans les puits du gel.
Tous les échantillons présentent bien les deux bandes attendues représentant les fragments d’ARNr 18S et 28S sauf l’échantillon 561 ayant subi le traitement par colonne (TC). Ce dernier présente un smear, signe d’une dégradation de l’ARN. (Figure9)
Figure 9: Profils électrophorétiques des échantillons traités à la DNase par deux méthodes
(TP : traitement par précipitation ; TC : traitement sur colonne)
Les échantillons classés par ordre croissant sont de bonne qualité, sans dégradation hormis l’échantillon TC 561 présentant une trainée, signe d’une dégradation de l’ARN.
b)
Vérification de l’absence d’ADN génomique par PCR
Figure 10: Profils électrophorétiques des produits de PCR présents dans les 6 échantillons d’ARN avant traitement à la DNase et après les deux méthodes,
Et témoins positifs et négatifs après une PCR de 35 cyles
Dans les échantillons après traitement à la DNase, il n’a pas été possible d’amplifier de fragments d’ADNg contrairement aux échantillons avant traitement.
18
Les échantillons traités par la DNase ont subi par la suite une PCR de 35 cycles. En plus des témoins positifs et négatifs, les échantillons ont été comparés aux échantillons avant traitement. Les profils électrophorétiques obtenus montrent une absence de bande d’ADNg amplifiée pour les échantillons après traitement à la DNase tandis que les témoins positifs et les échantillons avant traitement présentent la même bande à 300pb. (Figure 10)
c) Comparaison
Les deux traitements ont permis d’éliminer l’ADNg mais le traitement par colonne a engendré la dégradation d’un échantillon et les concentrations en ARN sont inférieures à celles des échantillons ayant été traités par précipitation. De plus, le traitement par précipitation engendre un coût moins élevé. Le traitement par précipitation est donc retenu pour l’ensemble des échantillons car il est plus efficient.
3. 2 Bioanalyse
Chez le rat, l’équipe de Jean-Louis Guéant a montré qu’une carence en donneurs de méthyl (acteurs du métabolisme du mono-carbone) pendant la gestation et la lactation conduit au développement d’une stéatose hépatique chez les descendants. [2], [9]. Ils ont décrit une diminution de l’expression du gène SIRT1 (Sirtuin 1) et de l’activité desacétylase de l’enzyme. PGC1α (Peroxisome Proliferator-Activated Receptor Gamma, Coactivator 1 Alpha ) n’est plus désacétylé et ne peut pas être méthylé par PRMT1 (Protein Arginine Methyltransferase 1). Il ne peut plus jouer son rôle de co-activateur de transcription pour des gènes impliqués dans la β-oxydation des acides gras et la chaine respiratoire. Les acides gras s’accumulent alors dans le foie favorisant la mise en place d’une stéatose. Compte-tenu de son rôle dans la mise en place de la stéatose hépatique chez le rat, un différentiel d’expression du gène SIRT1 est recherché dans le foie des canards mulards ayant subi une programmation nutritionnelle comparativement aux animaux témoins. (Figure 11)
(NR : nuclear receptors ; MDD : Methyl donor deficiency)
Figure 11 : Rôlede SIRT1 dans l’expression de gènes de la -oxydation des acides gras et de la chaîne respiratoire [9]
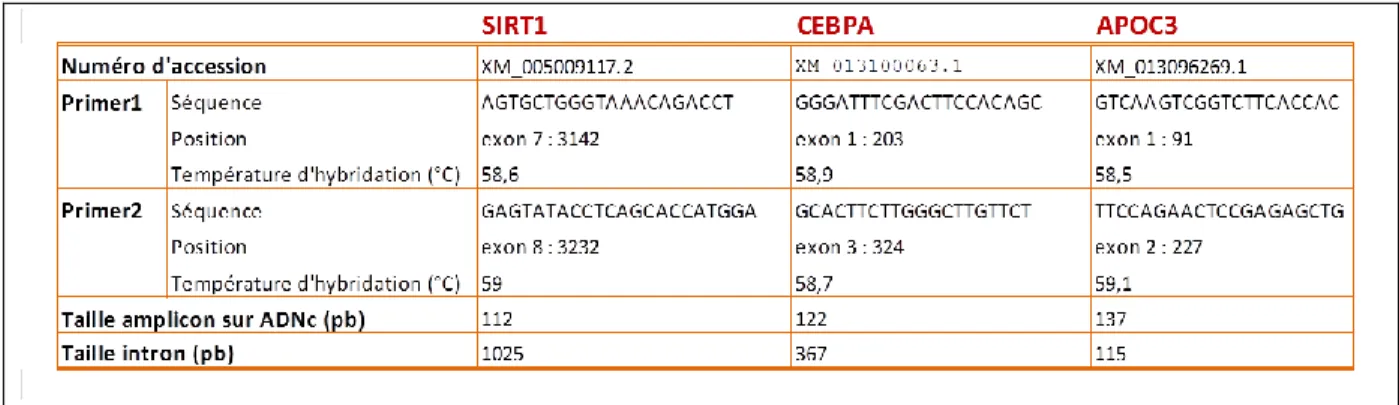
La séquence d’ARNm de SIRT1 chez le canard, existe sur la base de données de NCBI. Pour trouver les exons et leur position, elle a été alignée au génome du canard avec le programme BLAST de NCBI. Ce gène présente 8 exons. L’intron entre le 7ème et le 8ème exon étant assez grand avec 1025pb, les amorces ont été développées sur ces derniers avec Primer 3 Plus. Les critères de sélection étaient une hybridation à une température comprise entre 58°C et 60°C et une taille d’amplicon comprise entre 100 et 150pb. (tableau 2)
Le gène CEBPA (CCAAT/Enhancer Binding Protein Alpha) a été choisi car il code le facteur de transcription bZIP qui peut se fixer à certains promoteurs et réguler leur expression. Chez l’homme, CEBPA est exprimé dans le foie où il régule la gluconéogenèse et la lipogenèse par différents mécanismes. Au niveau de la gluconéogenèse, il est impliqué dans la régulation des gènes PCK1 (Phosphoenolpyruvate Carboxykinase 1) ou G6PC (Glucose-6-Phosphatase Catalytic Subunit). Au niveau de la lipogenèse, il intervient dans l’activation de gènes cibles comme ACAS2 (Acyl-CoA
19
Synthetase Short-Chain Family Member 2) par exemple. (Source : base de données sur les gènes humains nommée « GeneCards » : http://www.genecards.org/ consultée le 05/08/16).
La séquence d’ARNm de CEBPA chez le canard, existe sur la base de données de NCBI. Pour trouver les exons et leur position, elle a été alignée au génome du canard avec le programme BLAST de NCBI. Ce gène présente 4 exons. En règle générale, les amorces sont conçues du côté 3’ de l’ARNm. Pour ce gène il s’agirait donc d’amorces comprises entre les exons 3 et 4, mais l’intron entre ces exons n’est composé que de 48pb, ce qui est inférieur à la taille d’amplicon choisie (100-150pb). Si dans l’échantillon, de l’ADNg est présent à très faible quantité, la distinction entre celui-ci et l’ADNc sera difficilement faite. Aucun couple d’amorces n’a été trouvé entre les exons 2 et 3 avec les conditions souhaitées. Un couple d’amorces entre les exons 1 et 3 a donc été défini avec les mêmes critères de sélection que pour le gène précédant. Les introns entre ces deux exons mesurent 367pb (tableau 2). Pour ce cas, un couple d’amorces proche de la région 5’ n’est pas dérangeant car l’ARNm reste de petite taille, il est donc probable que la reverse transcriptase puisse rétro-transcrire l’ARNm en entier sans se détacher.
Le gène APOC3 a été sélectionné car il code l’apolipoprotéine C3 impliquée dans le métabolisme des triglycérides. En effet, cette lipoprotéine est l’un des constituants des VLDL (Very Low Density Lipoprotein : lipoprotéine de très basse densité) et des HDL (high density lipoprotein : lipoprotéine de haute densité) responsables du transfert et du transport des lipides. [10]
La séquence d’ARNm d’APOC3 chez le canard n’existe pas sur les bases de données de NCBI et Ensembl. Aucun résultat n’est trouvé avec les autres noms possibles du gène. La séquence d’ARNm chez la poule a donc été recherchée puis alignée aux séquences nucléotidiques chez le canard. La séquence ayant le plus d’homologie avec la séquence du gène APOC3 de la poule a été sélectionnée comme séquence d’ARNm prédictive chez le canard. Cette séquence a été comparée et alignée à la séquence génomique du canard pour trouver les exons et leur position. Ce gène présente 2 exons séparés par un intron de 115pb (tableau 2). Le couple d’amorces est développé entre ces deux exons avec les mêmes critères de sélection que précédemment. On note que l’intron est de petite taille (115pb seulement) mais dans le cas d’une éventuelle amplification de l’ADNg, la différence de taille des amplicons nous permettrait de repérer celui issu de l’ADNc.
20
IV. Conclusion
En vue de l’étude de l’expression des gènes influant sur le métabolisme hépatique dans le cadre d’une programmation nutritionnelle, des extractions d’ARN, un contrôle qualitatif et quantitatif des ARN et du design d’amorces ont été réalisés.
Le contrôle de l’ARN extrait a mis en évidence la nécessité d’une étape supplémentaire de traitement à la DNase afin d’éliminer l’ADNg restant dans les échantillons. Deux méthodes de traitement ont été testées et le traitement à la DNase suivi d’une précipitation pour purifier les ARN a été retenu pour son efficience. Par la suite, tous les échantillons seront traités avec ce protocole avant de poursuivre par une rétro-transcription et le contrôle quantitatifet qualitatif de l’ADNc.
Les amorces des 200 gènes cibles inscrits dans la liste réalisée par un travail bibliographique devront être conçues puis testées par PCR pour vérifier l’obtention d’un amplicon.
Enfin l’analyse de l’expression des gènes cibles sera réalisée par PCR quantitative.
Les mêmes manipulations seront effectuées sur des échantillons de foies de canards de 12 semaines (avant gavage) et 14 semaines (après gavage). Au final, l’expression des gènes cibles aura été étudiée en fonction de 3 paramètres : la nutrition des mères mais aussi en fonction de l’âge et du sexe du canard mulard. Ces travaux devraient permettre d’identifier des gènes différemment exprimés en fonction de ces trois paramètres.
V. Bibliographie
1. Chitka et Chitka. Epigenetics of Royalty. PLoS Biology. Nov 2010. Vol 8. e1000532
2. Pooya et al. Methyl donor deficiency impairs fatty acid oxidation through PGC-1α hypomethylation and decreased ER-α, ERR-α, and HNF-4α in the rat liver. Journal of Hepatology. 2012. Vol 57 344-351
3. Willems et al. Effects of nutritional programing on growth and metabolism caused by albumen removal in an avian model. Journal of Endocrinology. 2015. Vol 225. 89-100.
4. Willems et al. Differential Expression of Genes and DNA Methylation associated with Prenatal Protein Undernutrition by Albumen Removal in an avian model. Scientific Reports | 6:20837 | DOI: 10.1038/srep20837
5. Bomboy et Heard. Epigénétique : Comment se joue la partition du génome ? Science et santé, 11-12/2012, 11, p. 22-33
6. Baéza et al. La stéatose hépatique chez les palmipèdes. INRA Productions Animales. 2013. Vol 26 numéro 5. 403-414
7. Kalhan. One Carbon Metabolism, Fetal Growth and Long Term Consequences. Nestle Nutr Inst Workshop Ser. 2013 ; 74: 127–138. doi:10.1159/000348459
8. Untergasser et al. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012 Aug 1;40(15):e115.
9. Guéant et al. Nutritional models of foetal programming and nutrigenomic and epigenomic dysregulations of fatty acid metabolism in the liver and heart. Pflugers Arch - Eur J Physiol (2014) 466:833–850 DOI 10.1007/s00424-013-1339-4
21
PRESENTATION DU LABORATOIRE D’ACCUEIL
J’ai réalisé mon stage à l’INRA (Institut National de la Recherche Agronomique) au sein de l’équipe GenoRobust (Génétique des systèmes en lien avec l'adaptation et la robustesse) de l’UMR 1388 GenPhyse (Unité mixte de Recherche en Génétique, Physiologie et Système d’Elevage) sur le site d’Auzeville Tolosane.
L’INRA
L’INRA est un organisme français de recherche en agronomie ayant le statut d’établissement public à caractère scientifique et technique (EPST). C’est le premier institut de recherche agronomique en Europe et le deuxième au niveau mondial pour ses publications en sciences agricoles. Les recherches menées à l’INRA sont au service d’enjeux de société majeurs comme une agriculture compétitive et durable, une alimentation saine et de qualité et un environnement préservé et valorisé.
GenPhySE
GenPhySE (Génétique, Physiologie et Systèmes d’Elevage) est une Unité Mixte de Recherches INRA - INP ENVT (Institut National Polytechnique de Toulouse - Ecole Nationale Vétérinaire de Toulouse) - INP ENSAT (Institut National Polytechnique de Toulouse - Ecole nationale supérieure agronomique de Toulouse). Elle est rattachée à deux départements scientifiques de l’INRA : Génétique Animale (GA) et Physiologie Animale et Système d’Elevage (PHASE). L’Unité constitue un pôle de compétences dans le domaine de la génétique et de la biologie intégrative permettant d’étudier les mécanismes fins d’élaboration des caractères d’intérêt agronomique chez les animaux d’élevage. Dans ce contexte, les agents de l’unité étudient le fonctionnement du génome des espèces d’intérêt agronomique (principalement porcs, petits ruminants, lapins, volailles), identifient des zones du génome influençant les principaux caractères d’intérêt agronomique et développent des méthodes pour l’amélioration génétique et la gestion des populations animales. L’unité GenPhyse travaille également à l’élaboration de systèmes d’élevage innovants et développe des méthodes permettant d’évaluer l’impact de ces innovations sur la durabilité des systèmes d’élevage.
Dirigée par Xavier Fernandez, cette UMR est composée de dix équipes de recherche dont GenoRobust, une équipe service administratif et financier et une équipe informatique et automatisme. Une plateforme de contrôle chromosomique et une plateforme de génomique (GeT-PlaGe) sont également associées à l’unité.
L’unité GenPhySE a été évaluée par le HCERES (Haut Conseil de l’Evaluation de la Recherche et de l’Enseignement Supérieur) en décembre 2014. Elle est impliquée dans de nombreux projets en partenariat à l'échelle internationale en raison de son insertion dans des consortiums internationaux de séquençage de génomes complets et de développement et de la diffusion d’outils. L’unité est impliquée dans des projets européens, nationaux (financés par l’Agence Nationale de la Recherche) dont douze projets en tant que coordinateur, et dans des projets financés par la région Midi-Pyrénées. Cet ensemble participe fortement à son rayonnement scientifique. Au-delà des publications, dont 30 % sont co-signées avec des collègues étrangers, le rayonnement de l’unité se traduit par son implication dans les comités éditoriaux de 11 revues.
22
GenoRobust
Laurence Liaubet, chargée de recherche, dirige l’équipe GenoRobust dont les recherches visent à identifier ce qui permet d’augmenter la robustesse des animaux en rétablissant un meilleur équilibre entre la production et la capacité d’adaptation.
Le projet de recherche sur lequel j’ai travaillé est le programme MetEpiC (METabolisme et EPIgénétique chez le Canard mulard). L’objectif de MetEpiC est d’étudier la programmation nutritionnelle chez les canards mulards en vue d’améliorer la production de foie gras. Le but est de mesurer si la nutrition de la mère a un impact sur le phénotype du descendant (métabolisme hépatique, croissance du canard jusqu’à la mise en gavage à 12 semaines, poids du foie gras après gavage...), d’en expliquer les mécanismes et de comparer l’expression des gènes au niveau du foie en fonction de l’âge du canard, de son sexe et de la nutrition de sa mère.
Mireille Morisson, ingénieure de recherche dirigeant ce programme et Aurélie Secula, technicienne de recherche, m’ont encadrée lors de mon stage.
Prévention et qualité
Lors de mon arrivée, j’ai effectué une visite de prévention avec Laure Gress faisant partie du groupe prévention, hygiène et sécurité, ce qui m’a permis de connaître les consignes de sécurité mises en place et de faire le point sur les produits à risque que j’allais manipuler.
J’ai également eu l’opportunité d’avoir un entretien avec Kamila Canale-Tabet, responsable du groupe qualité. En effet, la qualité étant primordiale pour la validation des résultats, l’INRA a mis en place un référentiel qualité impliquant de nouvelles mesures. Entre autres, les pipettes doivent être vérifiées tous les ans afin d’améliorer la fidélité et la justesse. Les agents INRA doivent tenir un cahier de laboratoire expliquant les manipulations effectuées et un code barre est mis sur les échantillons pour améliorer la traçabilité. Les modes opératoires des manipulations sont standardisés et disponibles sur l’intranet de l’unité.
23
COMPTE-RENDU DES COURS DE L’UE STAGE
1. Organisation de la recherche
La recherche a une organisation impliquant différents acteurs : des étudiants (stagiaires, doctorants, post-doctorants), des techniciens, des ingénieurs, du personnel administratif et des chercheurs. Ces derniers exercent soit à temps plein, soit à temps partiel, le reste du temps étant occupé par l’enseignement et éventuellement une fonction clinique dans un CHU.
Les établissements publics scientifiques et techniques (EPST) font partie des structures possibles de la recherche. C’est le cas de l’INSERM (Institut National de la Santé et de la Recherche Médicale), le CNRS (Centre National de la Recherche Scientifique) et l’INRA (Institut National de la Recherche Agronomique). Mais l’université, l’industrie et les CHU sont également des structures majeures de la recherche. Ces différentes structures peuvent être complémentaires et apporter une richesse à la recherche en collaboration. C’est donc dans cette optique que les unités mixtes de
recherche ont été mises en place.
Les établissements de recherche et d’enseignement supérieur signent un contrat
quinquennal avec l’Etat. Ainsi tous les cinq ans, l’HCERES (Haut Conseil de l’Evaluation de la
Recherche et de l’Enseignement Supérieur) évalue les formations, les unités de recherche et les établissements. Ce contrat permet notamment le financement des objectifs de l’établissement fixés avec l’Etat. Le financement des unités de recherche est alors redéfini à chaque évaluation. La recherche peut également être financée par des organismes tels que l’ANR (Agence Nationale de la Recherche) et l’INCA (Institut national du Cancer) en répondant à des appels d’offres ou encore par des associations caritatives. Enfin, des programmes plus ambitieux peuvent être soutenus par l’Europe.
2. Expérimentation animale
Pour apporter des résultats de qualité, les expérimentations animales doivent être fiables, comparables et reproductibles. Pour cela, une harmonisation internationale a été mise en place : la génétique et le statut sanitaire de l’animal doivent être connus et le protocole expérimental doit être composé de procédures standardisées répondant aux bonnes pratiques de laboratoires de l’ANSM et à la démarche qualité (ISO 9001) afin d’améliorer la traçabilité. Une unité animale (zootechnie)
adaptée en fonction du type d’expérimentation, de l’espèce animale étudiée et de son état sanitaire, est également nécessaire.
Dans l’opinion publique, l’expérimentation animale est controversée. La reconnaissance de l’expérimentation animale passe alors par des règles d’éthique rigoureuses respectant le principe des 3 R : Remplacer par un modèle non animal dès que possible, Réduire le nombre d’animaux utilisés,
Raffiner la méthode utilisée en revoyant les critères d’interruption. Le bien-être animal est également
primordial que ce soit en ce qui concerne le stress, une souffrance physique ou psychologique. De plus, le respect du bien-être animal permet une réduction du risque de blessures du personnel et/ou de l’animal pendant les manipulations ce qui améliore la sécurité au travail et il diminue aussi la variabilité des résultats expérimentaux, ce qui améliore la qualité scientifique.
En France, le HCB (Haut Conseil des Biotechnologies) évalue le risque biologique et le niveau de confinement approprié en classant les unités animales de A1 à A4 et les laboratoires de L1 à L4.
Au niveau européen, les directives concernant l’expérimentation animale évoluent régulièrement. La dernière date de 2010 et va dans le sens d’une harmonisation européenne et du développement de l’Ethique. Tout protocole expérimental utilisant des animaux doit être soumis au préalable à un « comité d’éthique en expérimentation animale » qui donne un avis sur le projet en se



![Figure 11 : Rôle de SIRT1 dans l’expression de gènes de la -oxydation des acides gras et de la chaîne respiratoire [9]](https://thumb-eu.123doks.com/thumbv2/123doknet/14412734.704775/20.893.104.798.643.827/figure-rôle-expression-gènes-oxydation-acides-chaîne-respiratoire.webp)