Characterization of the Roles of Xrn1p in Small-RNA–Mediated Gene-Silencing
Pathways
by
Matthew Aaron Getz B.S. Biochemistry
M.S. Marine and Environmental Biology University of Southern California, 2008
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY SEPTEMBER 2019
© 2019 Massachusetts Institute of Technology All rights reserved
Signature of Author: _____________________________________________________________ Matthew A. Getz Department of Biology September 3, 2019 Certified by: ___________________________________________________________________ David P. Bartel Professor of Biology Member, Whitehead Institute Investigator, Howard Hughes Medical Institute
Thesis Supervisor Accepted by: ___________________________________________________________________
Stephen P. Bell Uncas and Helen Whitaker Professor of Biology
Investigator, Howard Hughes Medical Institute Co-Director, Biology Graduate Committee
Characterization of the Roles of Xrn1p in Small-RNA–Mediated Gene-Silencing
Pathways
by
Matthew Aaron Getz
Submitted to the Department of Biology on September 3, 2019
in partial fulfillment of the requirements for the degree of Doctor of Philosophy ABSTRACT
RNA interference (RNAi) is a small-RNA–mediated gene-silencing pathway that is involved in viral defense, transposon silencing, heterochromatin formation, and post-transcriptional gene silencing. Most RNAi pathways are initiated by long, dsRNA which is processed by Dicer into small interfering RNAs (siRNAs). These siRNAs are then loaded into an effector protein
Argonaute forming the RNA-induced silencing complex (RISC). RISC is then able to silence its target RNAs in a variety of fashions, including by slicing them. RNAi is ubiquitous in
eukaryotes with pathways found in plants, animals and fungi, suggesting its early origins and importance. Despite the usefulness of this pathway as a mechanism of genomic defense, the model budding yeast species, Saccharomyces cerevisiae, does not possess an RNAi pathway. Although S. cerevisiae does not possess an RNAi pathway, several related budding yeast species do, including Naumovozyma castellii and Vanderwaltozyma polyspora. Each of these species possesses orthologs of Dicer and Argonaute and has a population of 21–23-nt siRNAs that map to repetitive regions of the genome, including Ty transposable elements and Y¢ subtelomeric repeats. Disrupting either Dicer or Argonaute causes the loss of these small RNA populations. Additionally, RNAi in N. castellii can silence an exogenous GFP gene. Over-expressing Dicer and Argonaute in S. cerevisiae restores a functioning RNAi pathway that can silence endogenous transposable elements and an exogenous GFP gene.
To identify other factors that act in the budding-yeast silencing pathway, we performed an unbiased genetic selection in N. castellii. This selection identified Xrn1p, the cytoplasmic 5¢-to-3¢ exoribonuclease, as a cofactor of RNAi in budding yeast. Deletion of XRN1 impaired gene silencing in N. castellii and this impaired silencing was the result of multiple functions of Xrn1p. These functions include affecting the amount of different siRNA species in the cell, influencing the efficiency of loading these siRNAs into Argonaute, degradation of cleaved passenger strand, and degradation of cleaved target RNA. XRN1 has also been implicated in miRNA-mediated silencing in human cells. We found that disrupting XRN1 in a human cell line had no effect on the levels of mature miRNAs or their passenger strands but did de-repress miRNA targets, suggesting that in the miRNA pathway, XRN1 functions to degrade target mRNAs. Thesis Supervisor: David P. Bartel
Acknowledgements
There are a number of people who have enabled me to reach this juncture in graduate school whom I need to acknowledge. First, I would like to thank my advisor, Dave. I’m
tremendously grateful for the opportunity to work the lab with wonderful colleagues and to have had the experience of executing a scientific project from its nascent stages to its conclusion. I have appreciated his patience as I have run into obstacles with my project, and I have learned from him how to approach scientific questions with rigor and how to present data more persuasively.
I am deeply appreciative to Gerry Fink and Steve Bell, both of whom have been on my
thesis committee for the entirety of my graduate career. They have given excellent advice about directions to take in my project and ways to present my results in a more compelling fashion. I also enjoyed the experience of serving as teaching assistant for each of them.
I owe a huge debt of gratitude to a number of my scientific mentors from the start of my scientific training until I arrived at MIT. My time in Susan Forsburg’s lab at USC was
instrumental in starting my scientific career and her level of mentorship of undergraduate students was extraordinary, as was the mentorship provided by my immediate adviser, Angel Tabancay. I am also thankful for having the opportunity to participate in an immersive semester of marine microbiology on Catalina Island, where I was taught and mentored by John
Heidelberg, Jed Fuhrman, Eric Webb, Wiebke Ziebis, and Dave Caron. I am grateful for the opportunity given to me by Stephen Leppla to work in his lab at the NIH and for the mentorship I received from Haijing Hu, Andrei Pomerantsev, Clint Leysath, and Mahtab Moayeri.
I am also appreciative of the efforts that the faculty and staff of the MIT Biology department make to create and maintain such a wonderful scientific community. Personally, I am thankful for the outreach that Lenny Guarente, Alan Grossman, and Tony Sinskey made before, during and after graduate recruitment events; to Amy Keating and Angelika Amon for providing great rotation experiences in my first year; to Peter Reddien, Adam Martin, and Troy Littleton for good experiences as a TA; and to Frank Solomon for being a wonderful teacher and very helpful during my preliminary exam. I also want to thank Betsey Walsh and Janice Chang for all of their work to keep the graduate program running smoothly and to keep me adhering to deadlines. I am grateful for the Whitehead sixth floor staff, especially Dubi and Tsering, for making enormous amounts of yeast media that made my life substantially easier. I am also thankful for George Bell and Prat Thiru of BaRC for their help with computational questions related to my project.
I am very glad to have been a part of Biograd Class of 2010, as not only did I learn a tremendous amount from my extremely talented classmates, but I became good friends with them, as well. Thank you to members of the “Bio Mansion” and its associated members (Isaac, Ethan, Rob, Kevin, Brian, Greg, Vlad), as well as Mark, Peter, Kaitlin, Simina, Monica, Sara, and many others.
I spent a majority of my years in graduate school as a GRT for two vibrant communities, Bexley Hall and Senior House. My experiences as a GRT certainly enriched my time at MIT, and I want to thank all of the residents, the heads of house, and my wonderful fellow GRTs for making it a memorable experience. I also want to thank my roommates, including Brett,
Himanshu, Jacob, Jordan, Leerang, Tapovan, and Tim, for the good times and meals that I have shared with them.
I have overlapped with a large number of people during my time in the Bartel lab,
including (in alphabetical order) Alex, Anna, Asia, Ben, Charlie, Coffee, Dan, Danny, David G., David K., David W., Elena, Emir, Gina K., Gina L., Glenn, Grace, Huili, Igor, Jamie, Jarrett, Jeff, Jinkuk, Jin-Wu, Junjie, Justin, Kathy, Katrin, Laura, Lena, Lori, Michael N., Michael S., Namita, Noah, Olivia, Peter, Rosaria, Sean, Stephen, Sue-Jean, Thy, Tim, Vikram, Vincent, Wendy, Wenwen, and Xuebing. I especially would like to thank my “623” lab roommates, Alex, Jamie, Tim, and Glenn, and my baymates, David W., Sue-Jean, and Danny. I have spent more time around them than anyone else during my graduate career, and they have been a great group of people to spend time with and talk about things both science- and non-science-oriented. The lab was incredibly welcoming when I joined, and my initial mentors in the lab, including Anna, David W., and Vincent, really helped me to get my project off of the ground. I would also like to thank Sean, Katrin, Tim, Stephen and Jamie for lots of fun outings over the years, and to the rest of the lab for being an incredible group of people to work with. I have shared meaningful scientific discussions with almost everyone in the lab over the years, and I have been constantly awed by how deep everyone’s passion for science is. I also want to thank the members of the Fink Lab from over the years for treating me as one of their own, especially Doug, Valmik, Guy, Lindsey and Anna, and Felix. I would also like to thank David Pincus for a number of helpful discussions about my project.
I also have a large number of friends to thank for their support throughout graduate school and for good times we’ve shared. I’ve developed a network of good friendships with members of my graduate class, as well as some of their acquaintances, and I’ve had wonderful outings, “hygges,” and cabin trips with Ben, Kara, Stacie, Stephen, Megan, Tim, Grace, Jonars, Tomasz and Kristin. I am happy that I’ve been able to maintain close friendships with my college friends, including the SCuperfriends (Alice, Ashley, Brian K., Chris, Caterina, Cody, Jeremy, Kate, Katie, Melissa, Michelle, Miranda, Nic, Nick, Peter, Rob, and Sonya), as well as Brian C., Christina C., Arthi, and Chad. I cherish the friendships that I’ve been able to maintain since my time at the NIH with Zach, Allie, and Luhua, and they’ve been gracious hosts on my trips out to the Bay Area. There are also the friends, near and far, that I’ve made while living in Boston, including Shuyu, Priyanka, Shivan, Vineeta, Parag, David T., Jim, Kate, Dan D., Matt Bond, Vy, Allan, and Jason P. Finally, I would also like to thank some of my oldest friends from high school, including Chevy and Niti, Kavita, and Julie. These friendships have truly sustained me during trying times.
I would like to thank my entire extended family for being incredibly supportive and loving throughout the years. My parents have always encouraged me to pursue my interests and have lovingly supported me as I have done so. They instilled in me the importance of education, but more importantly taught me to love learning for learning’s sake. I cannot thank them enough for the sacrifices they have made for my brother and me, as well as the unconditional love they have showed us.
Finally, thank you June for your love and support. I know being apart during graduate school was not ideal, but it was very helpful to go through this process together with someone who understood it. I look forward to more wonderful trips and new experiences together.
Table of Contents
Abstract ...3
Acknowledgements ...5
Table of Contents ...7
Chapter 1. Introduction ...9
Part 1: A short history of RNA interference ...9
The Discovery of RNAi ...9
Small RNAs as a Silencing Intermediate ...11
Dicer as the dsRNA Endonuclease...12
Guide RNAs as Effectors of Silencing ...14
RISC and Argonaute as ‘Slicer’...15
Part 2: The Components of RNAi Pathways...19
Dicer and RNase III Enzymes ...19
RISC Architecture and Function ...23
General mRNA Decay Factors and their Role in RNAi ...28
Other Protein Co-factors of RNAi ...30
Part 3: Other Classes of Small RNAs...34
miRNAs ...34
piRNAs ...38
Endogenous siRNAs ...41
Heterochromatic siRNAs in Fission Yeast ...42
Part 4: RNAi in Budding Yeast ...43
Discovery and Characterization of the Budding-yeast RNAi Pathway ...43
The Non-canonical Dicer of Budding Yeast ...50
Characterization of Budding-Yeast Argonaute ...51
References ...55
Chapter 2. Xrn1p Enhances Multiple Steps of the Budding Yeast RNAi Pathway ...71
Chapter 3. Future Directions and Conclusions ...131
Are There Additional Components of the Budding-Yeast RNAi Pathway? ...131
RNAi Pathways in Other Budding-Yeast Species ...134
Interactions Between Argonaute and Xrn1p and Their Cellular Localization ...136
The Role of Xrn1p in RISC Loading ...139
The Role of XRN1 in the Mammalian miRNA Pathway ...141
Conclusion ...144
Supplemental Material ... 145
References ...165
Appendix 1: Methods ...171
Appendix 2: New CRISPR Mutagenesis Strategies Reveal Variation in Repair Mechanisms among Fungi...217
Chapter 1 Introduction
Part 1: A Short History of RNA Interference
The Discovery of RNAi
The process now known as RNA interference (RNAi) was first observed separately in plants, fungi, and nematodes. In studying the pigmentation pathway of petunias, transgenes meant to overexpress chalcone synthase1 were introduced into the plants with the hope of seeing
whether this enzyme was important in anthrocyanin biosynthesis, which is responsible for the purple color of the petals (Napoli et al., 1990; van der Krol et al., 1990). Surprisingly, the introduction of this transgene resulted in >40% variegated or unpigmented petunias, and the levels of the endogenous and introduced chalcone synthase were 50-fold lower than wild-type petunias (Napoli et al., 1990). This phenomenon was termed “co-suppression” (Napoli et al., 1990). A similar observation was later made in studying pigmentation in the filamentous fungus Neurospora crassa, in which the introduction of a homologous RNA sequence resulted in a decrease in expression of the endogenous gene, a process they termed “quelling” (Romano and Macino, 1992). Both of these processes were reversible, and revertants that displayed either wild-type or intermediate phenotypes also exhibited increases in both the transgenic and endogenous transcripts (Napoli et al., 1990; Romano and Macino, 1992).
Antisense inhibition of messenger RNA (mRNA) had been shown to be an effective means of reducing or eliminating gene expression in a number of systems, including the nematode Caenorhabditis elegans (Izant and Weintraub, 1984; Harland and Weintraub, 1985;
Melton, 1985; Rosenberg et al., 1985; Fire et al., 1991). In the use of antisense inhibition in C. elegans, it was noted that the introduction of sense control constructs, which were designed to express the sense transcript, could also result in inhibition, although his result was initially posited to be due to “indiscriminate transcription” of these sense constructs that resulted in antisense RNA inhibition (Fire et al., 1991). Similar to the previously reported “co-suppression” and “quelling” phenomena in plants and fungi, respectively, it was also observed in C. elegans that the par-1 mRNA was degraded upon the direct injection of a sense (as well as an antisense) RNA (Guo and Kemphues, 1995), which directly implicated the sense transcript in inhibition. Inhibition using both sense and antisense RNA was seen in further studies in C. elegans (Lin et al., 1995; Mello et al., 1996; Powell-Coffman et al., 1996; Guedes and Priess, 1997), and this phenomenon was termed RNAi2 to distinguish it from antisense inhibition (Rocheleau et al.,
1997).
Mechanistic insight into the process underlying some of these previously surprising results came when it was discovered that double-stranded RNA (dsRNA) served as a more potent trigger than either single-stranded RNA (ssRNA) for silencing in C. elegans (Fire et al., 1998). Thus, it was surmised that the prior observations of inhibition with sense and antisense RNA were likely due to the unintentional formation of dsRNAs (Fire et al., 1998). Injection of gel-purified ssRNA was 10- to 100-fold less effective at triggering silencing compared to dsRNA that had been annealed in vitro (Fire et al., 1998). This silencing was also incredibly effective and specific, in many instances reducing the expression of a target gene to the point where it phenocopied the null-mutant. Furthermore, this silencing process seemed to be affecting transcripts post-transcriptionally, as dsRNAs targeting their promoters and introns had no effect
on expression (Fire et al., 1998). Further evidence supporting this post-transcriptional
mechanism of function was that dsRNA interference of the upstream gene of a polar operon had no effect on the downstream gene, arguing against the possibility of it affecting transcriptional initiation or elongation (Montgomery et al., 1998). RNAi also appeared to be a systemic response, as silencing spread beyond the cells that the RNA was first injected into in C. elegans (Rocheleau et al., 1997; Fire et al., 1998; Grishok et al., 2000), and its effects could “persist well into the next generation” (Fire et al., 1998). Finally, the dsRNA trigger seemed to work at sub-stoichiometric concentrations, as only a few molecules of dsRNA were required per cell for silencing to occur, which implied that there could be “a catalytic or amplification component in the interference process” (Fire et al., 1998).
Small RNAs as a Silencing Intermediate
Upon discovering that dsRNA was the triggering molecule of silencing, the focus of RNAi research became to understand how this dsRNA targeted a specific sequence and effected silencing systemically. As it was shown to be a post-transcriptional process, one proposed mechanism of how RNAi functioned was through the formation of a duplex between the target mRNA and the antisense strand of the dsRNA trigger (Palauqui and Balzergue, 1999). As this postulated long antisense strand was not found, researchers instead looked for the presence of small RNA species in RNAi. In plants, a population of ~25-nt RNAs was discovered that was associated with posttranscriptional gene silencing (PTGS3) of transgenes and viruses (Hamilton
and Baulcombe, 1999). These ~25-nt RNAs were antisense to the targeted genes and their
production required the sense transcription of the transgene or replication of the virus (Hamilton and Baulcombe, 1999). Importantly, they were also believed to be the cause for the specificity of RNAi (Hamilton and Baulcombe, 1999).
A number of studies set out to address whether these small RNAs were effector
molecules of RNAi derived from the dsRNA trigger or whether they happened to be byproducts of the pathway. A cell-free system using extracts from Drosophila cells was developed that enabled biochemical analysis of the RNAi pathway (Tuschl et al., 1999). In in vitro assays using this system, incubation of a target luciferase reporter gene with a corresponding dsRNA trigger in the lysate led to decreases in both the luciferase protein and RNA levels (Tuschl et al., 1999). Importantly, a long pre-incubation of the dsRNA with lysate prior to incubation with the target increased target-mRNA degradation compared to a non-pre-incubated control (Tuschl et al., 1999). This suggested that this pre-incubation might lead to some modification of the dsRNA that resulted in greater silencing. Further biochemical studies of RNAi using radiolabeled dsRNAs showed that each strand was processed into RNAs that were 21–23-nt in length and that this processing did not require the target mRNA to be present (Zamore et al., 2000).
Additionally, it was later shown that the dsRNA substrates had to be a certain length (~38 bp) to produce these small dsRNA fragments and allow for effective silencing (Elbashir et al., 2001a).
Dicer as the dsRNA Endonuclease
That short RNAs of discrete sizes were produced from a long dsRNA substrate suggested that a ribonuclease III (RNase III) enzyme was involved in their generation, as these enzymes were the only class of nuclease known to have this activity (Nicholson, 1999; Bass, 2000). The short dsRNA duplexes were found to possess a 5¢ monophosphate and a 3¢ hydroxyl on each
strand, and each strand contained a 2-nt 3¢ overhang, which indicated they were produced by an RNase III enzyme (Elbashir et al., 2001a). Concurrently, another study combined candidate gene and biochemical analysis to discover the RNase III enzyme responsible for the processing of dsRNAs (Bernstein et al., 2001). They first identified RNase III proteins in the C. elegans and Drosophila genomes and separated them into three classes: the first class, or canonical RNase III, which resembled the bacterial enzymes in having a single RNase III motif and a double-stranded RNA-binding domain (dsRBD); the second class was represented by Drosha, a Drosophila enzyme (with a C. elegans ortholog), and contained two RNase III repeats and a dsRBD; and the third class, which had orthologs in C. elegans and Drosophila, which contained two RNase III motifs, a dsRBD, and an amino-terminal (N-terminal) helicase domain (Bernstein et al., 2001). Next, they immunoprecipitated Drosha, this class III enzyme, and Homeless, a protein containing only a helicase domain, and incubated long dsRNA with lysate from either Drosophila S2 cells or embryos, or one of these immunoprecipitated proteins. Only the lysates and this unnamed RNase III enzyme had the ability to produce fragments ~22-nt in length from this dsRNA and did so in a sequence-independent manner, an activity for which this enzyme was named Dicer (Bernstein et al., 2001). Depleting Dicer in Drosophila S2 cells by transfection of a dsRNA targeting Dicer impaired RNAi function in vivo (Bernstein et al., 2001). Similar results were observed in C. elegans dcr-1 mutants, as these mutants lacked the ability to silence a GFP transgene, as well as some endogenous genes, and also had abnormal germ line development and were sterile (Knight and Bass, 2001). Dicer proteins were also found to be widely conserved in a number of organisms, including Arabidopsis thaliana, the fission yeast Schizosaccharomyces pombe, and mammals (Bernstein et al., 2001). Notably, Dicer homologs were not found in the model budding yeast species Saccharomyces cerevisiae.
Guide RNAs as Effectors of Silencing
Studying RNAi in the in vitro cell-free Drosophila system, it was recognized that
cleavage of the target was restricted to the region where the long dsRNA was complementary to it (Hammond et al., 2000; Zamore et al., 2000). Furthermore, the cleavage intervals of the target were 21–23-nt in length, which were the same length as the products of Dicer processing
(Zamore et al., 2000). Additionally, the target nuclease activity could be biochemically purified by fractionation, and the fractions possessing nuclease activity also contained ~25-nt RNAs that were specific to the target sequence (Hammond et al., 2000). Together, these data suggested that these small RNAs might act to guide cleavage specifically.
It was shown definitively that these short dsRNAs were the effectors of RNA silencing through the use of synthetic 21- and 22-nt dsRNAs that had sequence complementarity to a firefly luciferase target (Elbashir et al., 2001a). In the Drosophila in vitro system, these short dsRNAs were able to guide the cleavage of sense and antisense targets within this region of complementarity (Elbashir et al., 2001a). Short duplexes that contained 2-nt 3¢ overhangs, a characteristic of the small RNA species produced by Dicer, were more effective at silencing their targets than those with either blunt-ends or long overhangs (Elbashir et al., 2001a). These RNAs were named short interfering RNAs (siRNAs) (Elbashir et al., 2001a).
Using a biochemical system in which siRNA duplexes that were biotinylated on either the sense strand, the antisense strand, or both, were affinity selected after being incubated with HeLa extract, it was shown that the strand antisense to the target, the guide, remained associated with the active nuclease, while the other, the passenger, was discarded (Martinez et al., 2002). While less effective, single-stranded antisense strands could also guide target cleavage in HeLa extracts and mediated gene silencing in HeLa cells, which suggested only one strand was needed
for the nuclease activity (Martinez et al., 2002; Schwarz et al., 2002). It was also shown that the 5¢ monophosphate of the guide strand was required for target slicing, whereas the 3¢ hydroxyl was dispensable (Nykanen et al., 2001; Martinez et al., 2002; Schwarz et al., 2002).
RISC and Argonaute as ‘Slicer’
Although early research into RNAi had established that dsRNA served as the trigger for RNAi, that Dicer served as the enzyme that produced siRNAs, and that the guide strand of the siRNA imparted specificity of the nuclease activity, the identity of the effector nuclease remained unknown. Based on the data that target-RNA nuclease activity purified with small RNAs (Hammond et al., 2000) and that it cleaved at 21–23-nt intervals (Zamore et al., 2000), an early model of RNAi suggested that Dicer might also serve as the effector nuclease of RNAi (Bass, 2000). This model invoked a nuclease-associated RNA-dependent ATPase or helicase that would help to unwind the siRNA duplex to allow for exchange of the passenger strand for mRNA (Bass, 2000), which would be consistent with the ATP-dependence of RNAi in vitro (Zamore et al., 2000). An alternative model proposed that the dsRNA processing activity of Dicer was separate from the target-RNA cleavage activity. Consistent with this second model, it was discovered that the activity of Dicer was separable from the activity of the enzyme
responsible for target cleavage by fractionation (Bernstein et al., 2001). This target-RNA nuclease was named RNA-induced silencing complex (RISC) (Hammond et al., 2000).
Additionally, in vitro experiments in Drosophila S2 extract showed that target cleavage occurred at the phosphodiester bond positioned across from nucleotides 10–11 with respect to the 5¢ terminus of the guide RNA (Elbashir et al., 2001b). This further argued against Dicer, as an RNase III, acting as the target ‘Slicer’ enzyme (Hutvagner and Zamore, 2002b).
As RISC activity had already been shown to exist in the extract of Drosophila S2 cells transfected with dsRNA (Hammond et al., 2000), biochemical fractionation was used to purify RISC to near-homogeneity (Hammond et al., 2001). These RISC-purified fractions were subjected to mass spectrometry, and a member of the Argonaute4 protein family, Argonaute-2
(Ago-2), was identified (Hammond et al., 2001). Human Argonaute proteins were later found to purify with RISC-activity in HeLa cell extract, as well (Martinez et al., 2002). Argonaute proteins had previously been identified as being involved in RNAi by genetic studies in fungi, animal and plant species (Cogoni and Macino, 1997; Tabara et al., 1999; Catalanotto et al., 2000; Fagard et al., 2000). While it was recognized that Argonaute family members possessed two domains, PAZ5 and PIWI, their functions were largely unknown (Cerutti et al., 2000), and thus
the role of Argonaute in RNAi remained enigmatic (Hammond et al., 2001).
Using biochemical purification of RISC from Drosophila cells, attempts were made to identify the potential ‘Slicer’ as well as other proteins that associated with both the cleavage activity and Ago-2 protein of RISC. RISC-associated proteins discovered included the
Drosophila homolog of the fragile X mental retardation protein (dFXR) and Vasa intronic gene (Vig), which were both RNA binding proteins (Caudy et al., 2002). Additionally, Tudor-SN (TSN), a micrococcal nuclease homolog, was isolated, which, as a nuclease, was an intriguing ‘Slicer’ candidate (Caudy et al., 2003). However, its mechanism of action, substrates, and products were inconsistent with what was known about the activity of RISC, thus making it unlikely that it was the ‘Slicer’ enzyme (Caudy et al., 2003).
4 Argonaute was the description of a mutant of the Arabidopsis AGO1 gene whose flowers looked like the pelagic
octopus Argonauta argo (Bohmert et al., 1998).
Further biochemical analyses of the activity of the ‘Slicer’ enzyme showed that target cleavage by RISC was a magnesium-dependent hydrolysis reaction that generated fragments with 5¢-monophosphate and 3¢-hydroxyl termini (Martinez and Tuschl, 2004; Schwarz et al., 2004). Double-radiolabeled, ligated substrates6 showed two discrete product bands upon
incubation with human RISC, demonstrating endonucleolytic cleavage by ‘Slicer’ (Martinez and Tuschl, 2004). Additionally, treatment of Drosophila embryo lysates with an inhibitor
nonsequence-specific ribonuclease activity (N-methylmaleimide) allowed the 5¢ and 3¢ cleavage fragments of the target to be visualized, which was further evidence that the ‘Slicer’ enzyme was an endonuclease (Schwarz et al., 2004). RISC in a number of systems was shown to function as a multiple-turnover enzyme in vitro (Hutvagner and Zamore, 2002a; Tang et al., 2003; Haley and Zamore, 2004). Kinetic analysis of target cleavage by RISC in vitro revealed that product
release was the rate-limiting step of the reaction (Haley and Zamore, 2004). Finally, the RISC endonuclease was not inhibited by addition of a Tudor-SN inhibitor (pdTp) (Schwarz et al., 2004), which, combined with other biochemical data on target cleavage on RISC, demonstrated that the micrococcal nuclease TSN was not ‘Slicer.’
As Argonaute was a conserved component of RISC and possessed domains of unknown function, a number of structural studies of prokaryotic Argonautes, which share similar domain architectures to their eukaryotic counterparts, helped to illuminate its role in RNAi. The crystal structure of the Argonaute of the archaeon Pyrococcus furiosus showed that it possessed four main domains: the Argonaute-defining PAZ and PIWI domains, as well as the N-terminal and middle (MID) domains (Figure 1A) (Song et al., 2004). The N-terminal, middle and terminal
domains formed “a crescent-shaped base” with the PAZ domain being held above this crescent by a “stalk-like” structure comprised by the region in between the N-terminal and PAZ domains (Figure 1A) (Song et al., 2004).
Figure 1. Crystal structures of bacterial Argonaute proteins.
A) Crystal structure of Argonaute from Pyrococcus furiosus along with its domain structure from Song et al., 2004. The structure shows the “crescent-shaped base” of the N, Mid, and PIWI domains with the PAZ domain being held above by a “stalk-like” structure.
B) Crystal structure of Argonaute bound to 5¢-monophosphorylated 21-nt guide RNA from
Thermus thermophilus from Wang et al., 2008b. The structure shows the characteristic bilobal architecture of Argonaute proteins.
The domain architectures of the Argonautes of the bacteria Aquifex aeolicus and Thermus thermophilus were later found to be very similar to that of P. furiosus, and these structures were characterized as being bilobal with one lobe consisting of the N and PAZ domains and the other lobe consisting of the MID and PIWI domains (Figure 1B) (Yuan et al., 2005; Wang et al., 2008b). Structural studies were particularly illuminating when it came to understanding the role of the PIWI domain. The Argonaute PIWI domain had the same tertiary structure as the RNase H family of enzymes (Song et al., 2004). RNase H domains were known to have an active site comprised of highly conserved carboxylate residues (Yang and Steitz, 1995). Reactions
involving RNase H enzymes require divalent cations, such as magnesium or manganese, and its cleavage products possess 5¢ monophosphate and 3¢ hydroxyl termini (Wintersberger, 1990). As
these were the same properties of the ‘Slicer’ enzyme, the structural similarities between the RNase H domain and the PIWI domain highly implicated Argonaute, when bound to a guide RNA, as being ‘Slicer’ (Parker et al., 2004; Song et al., 2004; Yuan et al., 2005).
Biochemical analyses of three of the four human Argonaute proteins (Argonaute-1, -2, and -3)7 showed that only Argonaute-2 had slicing activity (Liu et al., 2004). Changing two
residues in Argonaute-2 (Q633 and H634) to the amino acids present in the cleavage
incompetent Argonautes eliminated its ability to slice targets (Liu et al., 2004). Additionally, mutating either of two catalytic aspartate residues in Argonaute-2, which were in the same position in the RNase H domain, abolished slicing activity of Argonaute-2 (Liu et al., 2004). Cleavage-competent Drosophila RISC was purified to near homogeneity and was found to contain only Argonaute-2 (Rand et al., 2004). Recombinant human Argonaute-2 purified from Escherichia coli, which does not possess RNAi, was able to bind to a single-strand of an siRNA and cleave a target (Rivas et al., 2005). In total, these data definitively showed that Argonaute proteins bound to siRNA guides were responsible for the slicing activity of RISC.
Part 2: The Components of RNAi Pathways
Dicer and RNase III Enzymes
RNase III enzymes recognize, bind, and cleave dsRNA substrates at specific locations to transform them into functional RNA (Zamore, 2001; MacRae and Doudna, 2007). Members of this family have at least one RNase III nuclease domain that catalyzes the cleavage of dsRNA into products that possess 5¢ monophosphate and 3¢ hydroxyl termini and a characteristic 2-nt 3¢ overhangs (Robertson et al., 1968). The catalytic domain of all RNase III enzymes is dimeric in
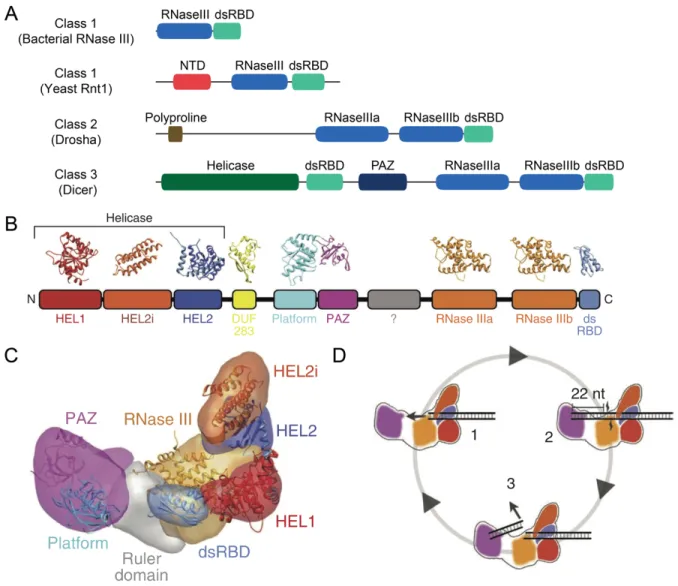
nature, allowing each RNase III domain to cleave one strand of a dsRNA substrate (MacRae and Doudna, 2007). Enzymes in this family are typically separated into three classes depending on their domain composition (Lamontagne et al., 2001). Class I enzymes contain one RNase III domain and one dsRBD (Figure 2A). Enzymes of this class are found in bacteria, bacteriophage, and fungi, and are involved in the processing and maturation of ribosomal RNA (rRNA), as well as other types of small non-coding RNAs in eukaryotes (Lamontagne et al., 2001). The S. cerevisiae protein Rnt1p belongs to this class and processes pre-rRNA, small nuclear RNA (snRNA) and small nucleolar RNA (snoRNA) (Lamontagne et al., 2001).
Class II enzymes possess two RNase III domains, a dsRBD, and an extended N-terminal region lacking any known domains (Figure 2A). Drosha, a nuclear protein first implicated in rRNA processing in humans and later shown to be essential to microRNA (miRNA) biogenesis in metazoans (Wu et al., 2000; Lee et al., 2003), belongs to this class. Drosha functions as a monomer but requires interaction with another dsRNA-binding protein, DGCR8, to bind and cleave its substrates properly (Han et al., 2004). These two proteins form the Microprocessor complex that produces pre-microRNA (pre-miRNA) hairpins from a longer primary-microRNA (pri-miRNA) transcript (Denli et al., 2004; Gregory et al., 2004; Kim et al., 2009).
RNase III Class III enzymes, like Class II proteins, typically have two RNase III domains and a dsRBD, and additionally possess a PAZ domain and frequently also possess an N-terminal DExD/H box helicase domain, as well (Figure 2A) (Lamontagne et al., 2001; MacRae and Doudna, 2007; Song and Rossi, 2017). Canonical Dicers, defined as those with PAZ domains (Weinberg et al., 2011), are class III enzymes that are present in most eukaryotic species (Figure 2A–2C) (MacRae et al., 2007). Each RNase III (RNase IIIa and RNase IIIb) domain of a Dicer
Figure 2. Domain structure of RNase III enzymes and the molecular architecture of Dicer. A) Classification of RNase III enzymes (adapted from MacRae and Doudna, 2007 and
Weinberg, 2013). Shown are the characteristic domain architectures for each class of RNase III enzymes, with the corresponding proteins indicated in parentheses. The N-terminal domain (NTD) is unique to yeast RNase III enzymes.
B) Schematic of the conserved domain structure of metazoan Dicer with the corresponding crystal structures for each domain from Lau et al., 2012.
C) Segmented map of human Dicer with crystal structures of each domain docked from Lau et al., 2012.
D) Schematic of processive dicing from Lau et al., 2012. The helicase domains move the dsRNA substrate into the nuclease core (1). The PAZ domain (purple) recognizes the end of the dsRNA (2). The siRNA product is released and the dsRNA substrate remains bound to the helicase (3).
molecule cleaves one strand of a dsRNA substrate containing a 2-nt 3¢ overhang at its terminus, which is recognized by the PAZ domain (Zhang et al., 2002; Zhang et al., 2004). In this way, the two RNase III domains act as an intramolecular homodimer to form one catalytic center, which resembles the catalytic center of bacterial RNase III enzymes that are comprised of
intermolecular homodimers (Zhang et al., 2004).
The discrete size of the Dicer products is determined by the distance between the PAZ domain and the RNase III active sites, which acts as a molecular ruler (Figure 2D) (Zhang et al., 2004; Macrae et al., 2006; MacRae et al., 2007). Dicer typically measures from the free 3¢ end of dsRNAs, which helps to determine the length of the product (MacRae et al., 2007). Although Dicer prefers to measure from a free, 2-nt 3¢ overhang, it can also bind to the 5¢ end of dsRNA possessing a 5¢ monophosphate and measure from this end (Park et al., 2011). This allows Dicer to process substrates that have their 3¢ ends occluded by modifications (Park et al., 2011).
The helicase domain of Dicer, which has been shown to be auto-inhibitory (Ma et al., 2008), is proposed to allow for the ATP-dependent translocation of Dicer along a long dsRNA substrate, which would allow for processive production of siRNAs by Dicer without dissociation (Cenik et al., 2011; Welker et al., 2011; Lau et al., 2012). A recent study of the structure and function of Drosophila Dicer-2 found that its helicase domain was required for binding double-stranded RNA substrates with blunt ends, but not those with 2-nt 3¢ overhangs (Sinha et al., 2018). These blunt dsRNAs are locally unwound and threaded through the Dicer helicase domain in a processive, ATP-dependent manner (Sinha et al., 2018). In constrast, dsRNA substrates with 3¢ overhangs are processed by Drosophila Dicer-2 in a distributive, ATP-independent manner (Sinha et al., 2018).
In addition to the canonical C-terminal dsRBD, which is important in binding dsRNA substrates, and is involved in the nuclear localization of Dicer in S. pombe and in humans (Barraud et al., 2011; Doyle et al., 2013), Dicer possesses DUF283 (Domain of Unknown Function) (Figure 2B). DUF283 has a double-stranded RNA-binding fold and that has weak dsRNA-binding affinity but is involved in the protein–protein interactions of Dicer and other proteins in A. thaliana and humans (Dlakic, 2006; Qin et al., 2010; Ota et al., 2013). The DUF283 domain of human Dicer has also been implicated in the annealing of complementary ssRNAs (Kurzynska-Kokorniak et al., 2016).
RISC Architecture and Function
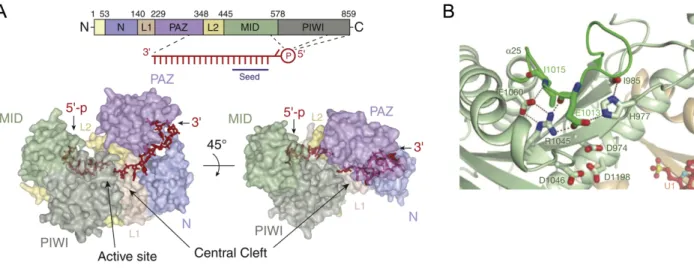
Members of the Argonaute protein family have a highly conserved bi-lobal architecture composed of four globular and two linker domains which fashion a “central RNA-binding cleft” (Figure 3A) (Sheu-Gruttadauria and MacRae, 2017). The four conserved domains of Argonaute are N, PAZ, MID, and PIWI (Song et al., 2004), with the N and PAZ domains forming the N-terminal lobe and the MID and PIWI domains forming the C-N-terminal lobe (Figure 3A) (Yuan et al., 2005). In the N-terminal lobe, the N domain serves an important role in RISC assembly by wedging into one end of a small-RNA duplex to pry it open and facilitate unwinding of the duplex (Kwak and Tomari, 2012). The PAZ domain is an RNA-binding domain that has a variant of the OB (oligonucleotide/oligosaccharide-binding) fold (Lingel et al., 2003; Song et al., 2003; Yan et al., 2003). Structural studies of the PAZ domain in isolation showed that it
possesses a hydrophobic nucleotide-binding pocket that binds to the two 3¢-terminal nucleotides of the guide strand (Lingel et al., 2004; Ma et al., 2004). The C-terminal tail and several other
positively-charged !-strands of the PAZ domain interact with the backbone of the guide strand, and the !2-!3 loop of the domain caps the 5¢-end of the other strand (Ma et al., 2004).
Figure 3. Molecular architecture of RISC
A) Linear schematic of the domains and surface representation of the structure of human Argonaute2 bound to guide RNA from Sheu-Gruttadauria and MacRae, 2017. Regions that contact the 5¢ and 3¢ nucleotides of the guide RNA are indicated. Structure also displays the central cleft and active site.
B) Crystal structure of the active site of V. polyspora Argonaute from Nakanishi et al., 2012. The catalytic tetrad of budding-yeast Argonaute proteins is displayed (D974, E1013, D1046, D1198).
In the C-terminal lobe, the MID domain recognizes the 5¢-terminal nucleotide of the guide strand, which is unpaired in RISC, via a Rossman-like fold, which is a nucleotide-binding motif (Ma et al., 2005; Parker et al., 2005; Boland et al., 2010). This conformation of the 5¢ nucleotide is in accordance with data that indicate that the first nucleotide of the guide is not important for target recognition (Lewis et al., 2005). The MID domain also has a nucleotide specificity loop, which imparts a bias for certain nucleotides at the 5¢ terminus of the guide by interacting with the Watson-Crick face of this first nucleotide, and has been proposed to be involved in small RNA sorting into different Argonaute proteins in Arabidopsis (Boland et al., 2010; Frank et al., 2010; Frank et al., 2012; Zha et al., 2012; Sheu-Gruttadauria and MacRae, 2017). The nucleotide specificity loop makes the only known sequence-specific contact between
Argonaute and the guide (Sheu-Gruttadauria and MacRae, 2017). The 5¢ phosphate of the guide strand is buried in a conserved hydrophilic (basic) pocket present at the interface of the MID and PIWI domains (Parker and Song, 2004; Ma et al., 2005; Parker et al., 2005), which is in line with the necessity of this phosphate for RISC loading and function in vitro (Nykanen et al., 2001).
The two lobes of Argonaute are connected by the two linker domains (L1 and L2), which together comprise the central cleft, also known as the nucleic-acid-binding channel (Figure 3A) (Wang et al., 2008b). The guide RNA extends through this channel and makes contacts with all domains of Argonaute with a majority of these interactions with the protein occurring via the RNA sugar-phosphate backbone, which allows RISC to contain small RNAs without any sequence specificity (Schirle et al., 2014). Nucleotides 2–7 of the guide strand (g2–g7), which are known as the “seed sequence,” are organized in a helical conformation via protein contacts with the phosphate backbone with several of these nucleotides solvent exposed (Nakanishi et al., 2012; Schirle and MacRae, 2012; Nakanishi et al., 2013; Schirle et al., 2014). This
pre-organization of the guide within Argonaute facilitates the binding of target and gives a structural explanation as to why the seed contributes significantly to this guide-target binding (Lewis et al., 2003; Bartel, 2004; Doench and Sharp, 2004).
Upon target binding, Argonaute undergoes a conformational change. The central cleft widens and accommodates an A-form helix that forms between the seed of the guide and the complementary nucleotides of the target, forming a ternary complex of Argonaute-guide-target (Wang et al., 2008a). In the binary complex (Argonaute-guide) and ternary complex with short RNA targets, the 5¢ terminal nucleotide of the guide is buried in the MID domain and its 3¢ end is bound to the PAZ domain (Wang et al., 2008a; Wang et al., 2008b; Wang et al., 2009). In contrast, when the guide binds to a longer target that involves more than one helical turn of
pairing, the 3¢ end of the guide is released from the PAZ domain (Wang et al., 2009). In total, these structural insights are in accordance with a ‘two-state model’ for target recognition, which involves a nucleation step and a propagation step (Bartel, 2004; Tomari and Zamore, 2005). During the nucleation step, only the seed of the guide and the target interact, as the guide is bound on each of its ends to the MID and PAZ domains. During the propagation step, the 3¢ end of the guide releases from the PAZ domain and is able to extend its pairing with the target RNA, which is accompanied with “pivot-like domain movements” within Argonaute and positions a phosphate in the target RNA across from guide nucleotides 10–11 at the active site (Wang et al., 2009).
The PIWI domain of Argonaute harbors the active site, which resembles that of RNase H (Song et al., 2004). RNase H enzymes catalyze phosphodiester bond hydrolysis using a two metal-ion-mechanism (Steitz and Steitz, 1993). In the catalysis of these reactions, the divalent metal ions are stabilized by carboxylate residues at the active site (Nowotny and Yang, 2006). Argonaute was shown to possess two conserved aspartate residues in the same position as RNase H (Parker et al., 2004; Song et al., 2004), and a glutamate residue was proposed to complete a triad of conserved carboxylates in the active site (Liu et al., 2004; Song et al., 2004). It was later found that mutating this glutamate residue had no effect on slicing and that the triad was
apparently completed by a proximal histidine residue, making a ‘DDH’ catalytic triad (Rivas et al., 2005).
As a ‘DEDD’ tetrad motif is involved in the metal-ion coordination of RNase H
(Nowotny et al., 2005), it seemed unlikely that the Argonaute ‘DDH’ triad could accomplish this function (Hall, 2005). A number of studies proposed other conserved residues that might take the place of the glutamate in the tetrad, but functional analysis of these residues showed they
were not involved in catalysis (Rivas et al., 2005). It was later discovered with the crystal structure of budding-yeast Argonaute, that the fourth residue of the ‘DEDD’ catalytic tetrad was, in fact, an invariant glutamate (E1013) that exists on a loop (L2) in the MID-PIWI lobe that can insert into the catalytic pocket near three aspartate residues (D974, D1046, D1198) upon the movement of another loop (L1) in this lobe (Figure 3B) (Nakanishi et al., 2012). Human Argonaute-2 protein (HsAGO2) also contains this “plugged-in glutamate finger” in its ‘DEDH’ catalytic tetrad, demonstrating the conserved nature of this catalytic glutamate in
slicing-competent Argonautes (Nakanishi et al., 2012). In zebrafish Ago2 (dsAgo2), which is severely impaired in its ability to slice targets, this glutamate is substituted for an aspartate8 (Chen et al.,
2017).
Human Argonaute-1 (HsAGO1) and Argonaute-4 (HsAGO4) do not have a complete ‘DEDH’ catalytic tetrad and are catalytically inactive, (Liu et al., 2004; Meister et al., 2004). HsAGO1 and HsAGO4 were made to be slicing competent through several structural
substitutions, including completing the catalytic tetrad in the active site as well as swapping in either part of or the whole N domain of HsAGO2 (Faehnle et al., 2013; Hauptmann et al., 2014). Although human Argonaute-3 (HsAGO3) has a complete ‘DEDH’ catalytic tetrad, it was
initially believed to be catalytically inactive (Liu et al., 2004; Meister et al., 2004), but could be made slicing competent by swapping in the N domain of HsAGO2 (Faehnle et al., 2013). Recently, HsAGO3 was shown to have slicer activity, and this activity was shown to depend on the identity of the guide RNA and the presence of 5′- and 3′- flanking regions in the target RNA (Park et al., 2017). The differences in substrate requirements of slicing-competent HsAGO3
8 Zebrafish Ago2 also contains a second nearby phenylalanine to tyrosine substitution that also impairs its ability to
compared to HsAGO2 were suggested to be due, in part, to HsAGO3 lacking a well-defined nucleic acid-binding channel (Park et al., 2017). In total, these results showed the importance of the catalytic tetrad in Argonaute target slicing but demonstrated that other elements of the structure were important for this function, as well.
General mRNA Decay Factors and their Role in RNAi
RNA interference results in the destruction of target RNAs, which are frequently mRNAs. Independent of being targets of RNAi, eukaryotic mRNAs undergo a characteristic degradation process. Along with their production by transcription, this process of general mRNA decay is critical in regulating mRNA levels in the cell. After an mRNA has been transcribed, processed, and polyadenylated in the nucleus, it is exported to the cytoplasm where it can be translated by the ribosome into protein.
As general mRNA decay is an exonucleolytic process, mRNAs are protected by their 5′ N7-methyl guanosine cap (m7G) and 3′-end poly(A) tail (Figure 4) (Shatkin and Manley, 2000;
Balagopal et al., 2012). The first and frequently rate-limiting step of mRNA decay is shortening of the poly(A) tail by deadenylases (Figure 4) (Decker and Parker, 1993; Hsu and Stevens, 1993; Muhlrad et al., 1994, 1995). There are two cytoplasmic deadenylase complexes that shorten the poly(A) tails. The Pan2/Pan3 complex may cause the initial deadenylation of the poly(A) tail, while most of the deadenylation is caused by Ccr4–Not complex (Decker and Parker, 1993; Brown and Sachs, 1998; Tucker et al., 2001).
After deadenylation, the 3′ end of the mRNA is susceptible to degradation in a 3′-to-5′ direction catalyzed by the cytoplasmic exosome (Figure 4) (Chlebowski et al., 2011; Balagopal et al., 2012). The cytoplasmic exosome in yeast consists of a protein complex of 3′-to-5′ exo-
and endo-ribonucleases with Rrp44p/Dis3p being the catalytic subunit (Chlebowski et al., 2011). In yeast, the Ski complex is a heterotetrameric protein complex (comprised of one copy of the helicase Ski2p, one copy of Ski3p and two copies of Ski8p) that guides RNAs to the cytoplasmic exosome via a fourth protein called Ski7p and regulates their destruction (Araki et al., 2001; Wang et al., 2005). A majority of general mRNA decay in yeast occurs in a 5′-to-3′ direction after removal of the m7G cap.
Figure 4. Schematic of the general cytoplasmic mRNA decay pathway.
The general degradation pathways are initiated when deadenylation of the poly(A) tail occurs by the Ccr4–Not deadenylase complex. Once the poly(A) tail reaches a certain length (~10
adenosines), the mRNA can be exonucleolytically degraded from either the 5′ end by Xrn1 after decapping or the 3′ end by the cytoplasmic exosome.
Decapping occurs by the Dcp1/Dcp2 complex (Figure 4). Dcp2 is the catalytic subunit, and it requires its binding partner, Dcp1, to hydrolyze the cap in vivo (Beelman et al., 1996; Steiger et al., 2003; Mildvan et al., 2005; She et al., 2006). Decapping is irreversible, except under certain conditions in mammalian cells (Schoenberg and Maquat, 2009; Balagopal et al.,
rapidly and processively degraded by the 5¢-to-3¢ cytoplasmic exoribonuclease, Xrn1 (Figure 4) (Chang et al., 2011; Jinek et al., 2011).
Some of these general mRNA decay factors are involved in RNAi and other small-RNA– mediated gene-silencing pathways. Specifically, after activated RISC binds and cleaves its RNA target, exoribonucleases are involved in the removal of cleaved target fragments. Xrn1 degrades the 3¢-target-cleavage products of RISC, while the cytoplasmic exosome degrades the 5¢-target-cleavage fragments (Souret et al., 2004; Orban and Izaurralde, 2005; Lima et al., 2016).
Other Protein Co-factors of RNAi
The RNAi pathways of many species require proteins other than Dicer and Argonaute to function in vivo. These other protein factors play a role in RISC loading and maturation, modification and amplification of siRNAs, as well as removal of target-RNA cleavage
fragments. Early in vitro biochemical studies of RISC demonstrated that Argonaute is able to load a single-stranded RNA but not an siRNA duplex in vitro (Liu et al., 2004; Rivas et al., 2005), suggesting that other proteins may be involved in RISC loading. Despite evidence that Dicer and Argonaute might directly physically interact via their RNase III and PIWI domains, respectively (Tahbaz et al., 2004), Dicer does not directly deliver the small-RNA duplex to Argonaute during loading in metazoan RNAi pathways. The loading process relies on dsRBD-containing proteins that interact with Dicer and Argonaute and facilitate RISC assembly,
including R2D2 in Drosophila, HYL1/DRB19 in Arabidopsis, and TRBP in Homo sapiens (Liu
et al., 2003; Vazquez et al., 2004a; Chendrimada et al., 2005). These ternary complexes of
Figure 5. Schematic of the RNAi Pathway (adapted from Weinberg, 2013).
RNAi is initiated by either endogenous or exogenous sources of double-stranded RNA (dsRNA). After cleavage of the dsRNA by Dicer, an approximately 21–23-nt siRNA duplex with a 2-nt 3′ overhang is produced. The siRNA duplex is loaded into Argonaute to form the precursor RNA-induced silencing complex (pre-RISC). This loading requires a cofactor, usually a protein containing a double-stranded RNA binding domain (dsRBD), in some systems and when
combined with Dicer is known as the RISC loading complex (RLC). The passenger-strand of the siRNA duplex (blue strand) is then nicked by the endonucleolytic activity of Argonaute and the cleavage fragments are discarded. In some systems, the elimination of the passenger strand cleavage fragments is carried out by an endo- or exonuclease. This mature RISC can then bind and cleave target RNAs (long, curved cyan strand) that base pair extensively with the guide strand (red strand). Degradation of the target cleavage fragments by 5′-to-3′ and/or 3′-to-5′ exonucleases facilitates multiple turnover of RISC. Identified cofactors in each of the steps of the pathway are indicated in gray italics. The dashed lines represent 2′-O-methylation, which
Dicer, Argonaute, and a dsRBD-containing protein are known as the RISC-loading complex (RLC) (Maniataki and Mourelatos, 2005).
It was originally thought that the RLC not only facilitated the transfer of small-RNA duplexes from Dicer to Argonaute but also supported the asymmetric incorporation of these small-RNA duplexes into Argonaute (Tomari et al., 2004). It was thought that the dsRBD-containing proteins of this complex could function as asymmetry sensors that helped to select which small-RNA duplex strand becomes the guide strand that would ultimately influence what target RNAs got silenced (Tomari and Zamore, 2005). More recent work in Drosophila and mammalian systems has suggested these dsRBD-containing proteins are dispensable for
asymmetry sensing, and instead that Argonaute is the asymmetry sensor (Kawamata et al., 2009; Betancur and Tomari, 2012; Nishida et al., 2013; Noland and Doudna, 2013; Suzuki et al., 2015). The original models of asymmetry sensing in Drosophila proposed that R2D2 binds near the 5¢ end of the more thermodynamically stable strand, which is the passenger strand that will be ejected from RISC, and Dicer binds near the 5¢ end of the less thermodynamically strand, which is the guide strand (Tomari et al., 2004). Additionally, asymmetry sensing was proposed to be separated from processing, as an siRNA that is produced by Dicer in the opposite orientation with respect to its thermodynamic stability can release from Dicer and rebind the Dicer-dsRBD-containing protein heterodimer in the correct orientation (Vazquez et al., 2004b; Tomari and Zamore, 2005). The more recent model suggests that this asymmetry is caused by Argonaute selecting the less thermodynamically stable 5¢ end, whose 5¢ nucleotide would be more likely to be unpaired, thus facilitating the sequestration of its 5¢ phosphate in the MID-domain binding pocket (Suzuki et al., 2015).
RNAi in vitro requires ATP hydrolysis (Zamore et al., 2000). While target RNA cleavage is an independent process (Nykanen et al., 2001), RISC assembly is
ATP-dependent with the duplex unwinding step being a passive process (Kawamata et al., 2009; Yoda et al., 2010). This suggested that the process of loading Argonaute with a small-RNA duplex requires ATP and led to the “rubber band” model of RISC maturation (Kawamata and Tomari, 2010). As the central cleft of Argonaute is too small to readily fit “rigid” and “bulky” A-form RNA duplexes, the protein would need to be actively opened to accommodate these small-RNA duplexes (Kawamata and Tomari, 2010). Any tension introduced into Argonaute by
incorporation of these small-RNA duplexes could be relieved with the release of the passenger strand (Kawamata and Tomari, 2010). The Hsp70/Hsp90 machinery associates with Argonaute and facilitates this ATP-dependent opening (Figure 5) (Tahbaz et al., 2001; Hock et al., 2007; Landthaler et al., 2008; Iki et al., 2010; Iwasaki et al., 2010; Miyoshi et al., 2010; Iwasaki et al., 2015; Tsuboyama et al., 2018).
Once the small-RNA duplex is loaded into Argonaute, RISC activation occurs by
Argonaute slicing its passenger strand and discarding the resulting cleavage fragments (Matranga et al., 2005; Miyoshi et al., 2005; Rand et al., 2005). In addition to the exonucleolytic activities of the cytoplasmic exosome and Xrn1, there are other exoribonucleases and endoribonucleases that are involved in RNAi by removing passenger strand or target cleavage fragments. In
Neurospora, the QIP exonuclease removes the passenger-strand cleavage fragments during RISC activation (Figure 5) (Maiti et al., 2007). In Drosophila and human, the C3PO endonuclease, which is a complex of the proteins Trax and Translin, is reported to have a similar activity that degrades the cleaved passenger strand (Figure 5) (Liu et al., 2009; Ye et al., 2011). Additionally,
the La autoantigen has been implicated in enhancing multiple turnover of RISC in Drosophila by removing the cleaved target fragments (Figure 5) (Liu et al., 2011).
A number of cofactors modify or amplify siRNAs in a way that can affect the RNAi response. Orthologs of the S-adenosylmethionine (SAM)–dependent methyltransferase Hen1 methylate the 2¢ oxygen of the 3¢-terminal nucleotide of Arabidopsis and Drosophila siRNAs, thereby protecting these small RNAs from 3¢-end modifications (e.g., tailing by uridylation and trimming) and degradation (Li et al., 2005; Yang et al., 2006; Horwich et al., 2007; Ameres et al., 2010). Additionally, other types of small RNAs, including plant microRNAs (miRNAs) and metazoan Piwi-interacting RNAs (piRNAs), are also methylated on their 3¢-terminal nucleotide (Li et al., 2005; Yu et al., 2005; Vagin et al., 2006; Houwing et al., 2007; Kirino and Mourelatos, 2007a, b; Saito et al., 2007; Kamminga et al., 2010; Ji and Chen, 2012). RNA-dependent RNA polymerases (RdRPs) can either initiate or amplify the RNAi response by either making or increasing the amount of dsRNA, which is important in processes such as systemic RNAi and the biogenesis of trans-acting siRNAs (ta-siRNAs) (Baulcombe, 2004; Cerutti and Casas-Mollano, 2006). These enzymes are conserved in all five eukaryotic supergroups and required for RNAi and related pathways in nematodes, some fungi, and plants (Cogoni and Macino, 1999; Dalmay et al., 2000; Smardon et al., 2000; Sijen et al., 2001; Volpe et al., 2002; Baulcombe, 2004; Xie et al., 2004; Cerutti and Casas-Mollano, 2006; Shabalina and Koonin, 2008).
Part 3: Other Classes of Small RNAs
miRNAs
Like siRNAs, miRNAs are a class of ~22 nt RNAs that are loaded into RISC (Bartel, 2004). However, these two classes of small RNAs differ primarily in their precursor transcripts.
MicroRNAs are produced from long primary miRNA transcripts (pri-miRNA), which are able to fold into stem-loop (or hairpin) structures (Bartel, 2004). The first miRNA discovered, lin-4, was the product of a C. elegans gene that influenced developmental timing that was shown to produce two short RNAs rather than a protein (Lee et al., 1993). These small RNAs were found to have antisense complementarity to the 3¢ UTR of the lin-14 gene (Lee et al., 1993; Wightman et al., 1993), a region which had previously been shown to be important in the regulation of lin-14 by lin-4 (Wightman et al., 1991). The discovery of the second miRNA, let-7, also came in C. elegans (Reinhart et al., 2000), but, unlike the C. elegans-specific lin-4, let-7 was found to be conserved in bilaterians and these RNAs together were referred to as small temporal RNAs (stRNAs) (Pasquinelli et al., 2000). Shortly thereafter, small-RNA probing or cloning and sequencing experiments discovered that lin-4 and let-7 were members of a large class of small RNAs present in nematodes, flies, and mammals (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001), and hundreds of miRNAs have been discovered in many species since (Bartel, 2004; Kim and Nam, 2006).
Canonical miRNA biogenesis in animals begins with transcription of the pri-miRNA in the nucleus, which is followed by the recognition of its stem-loop structure by a protein complex called the Microprocessor (Figure 6). This complex, which consists of the Class II RNase III
enzyme Drosha and its dsRNA-binding protein partner DGCR810, processes the pri-miRNA
stem-loop structure to form a precursor-miRNA (pre-miRNA) hairpin (Figure 6) (Lee et al., 2003; Denli et al., 2004; Gregory et al., 2004; Han et al., 2004; Landthaler et al., 2004). The pre-miRNA is exported from the nucleus to the cytoplasm by Exportin-5 (Figure 6) (Lund et al., 2004). In the cytoplasm, Dicer, which is in a complex with a dsRBD-containing protein such as
TRBP, cleaves the pre-miRNA producing a ~22-nt mature-miRNA duplex comprised of the guide and passenger (or miRNA star/miRNA*) strands (Lee et al., 2002). The miRNA duplex is then incorporated into Argonaute asymmetrically11 (Khvorova et al., 2003; Schwarz et al., 2003).
During loading, the duplex unwinds passively by a still unclear mechanism and the passenger strand is removed (Kawamata et al., 2009; Kawamata and Tomari, 2010). Plant miRNA biogenesis differs from animal miRNA biogenesis in having a single RNase III enzyme, Dicer-like protein (DCL1), that makes both cleavages that are necessary to make a mature miRNA in the nucleus (Park et al., 2002; Reinhart et al., 2002; Papp et al., 2003; Kurihara and Watanabe, 2004).
Figure 6. Canonical metazoan miRNA biogenesis pathway (adapted from Weinberg, 2013). The capped and polyadenylated pri-miRNA undergoes cleavage by the microprocessor in the nucleus resulting in the pre-miRNA. This pre-miRNA is then exported to the cytoplasm by Exportin-5. In the cytoplasm, the pre-miRNA is cleaved by Dicer to form the miRNA/miRNA* duplex. This duplex is then loaded into Argonaute, and after removal of the miRNA*, mature miRISC is formed.
Although there are instances12 of animal miRNAs repressing their targets by slicing, such
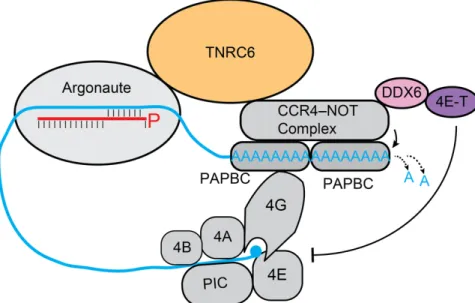
as when they contain sufficient complementarity with their targets and are loaded into a slicing-competent Argonaute protein (Hutvagner and Zamore, 2002a; Liu et al., 2004; Meister et al., 2004; Yekta et al., 2004), this is not their dominant mode of repression (Bartel, 2018). Instead, animal miRNAs typically repress their mRNA targets by destabilizing them or translationally repressing them (Guo et al., 2010; Bazzini et al., 2012; Djuranovic et al., 2012; Eichhorn et al., 2014). To cause repression of their targets, Argonaute-bound metazoan miRNAs typically bind via their seed to sites within the 3¢ UTR of the target mRNA (Bartel, 2009). This repression requires an adaptor protein, such as one of the paralogs of TNRC6 in mammals (Figure 7), its fly ortholog GW182, or one of its two orthologs in nematodes AIN-1/2 (Ding et al., 2005;
Rehwinkel et al., 2005; Jonas and Izaurralde, 2015; Bartel, 2018). TNRC6 interacts with Argonaute and poly(A) binding protein (PABPC) and recruits deadenylases, such as the CCR4– NOT and PAN2–PAN3 complexes, to the poly(A) tail, which shorten it and lead to mRNA destabilization (Figure 7) (Chen and Shyu, 2011; Jonas and Izaurralde, 2015; Bartel, 2018). Plant miRNAs bind and repress their targets in a manner similar to that of siRNAs, as they have extensive pairing with their targets and typically slice them (Llave et al., 2002; Rhoades et al., 2002; Tang et al., 2003).
Figure 7. Dominant modes of miRNA-mediated repression.
miRISC recruits the adapter protein TNRC6 that, in turn, recruits either the PAN2–PAN3 deadenylase complex or the CCR4–NOT deadenylase complex. These deadenylases shorten the poly(A) tail, which can ultimately result in either translational repression or degradation of the miRNA target depending on the cellular context. TNRC6 can repress translation initiation in post-embryonic cells through the recruitment of DDX6 and 4E-T by the CCR4–NOT
deadenylase complex (Bartel, 2018). This particular translation initiation typically involves the the 43S preinitiation complex (PIC) through the action of the translation initiation factors eIF4A, eIF4B, eIF4E, and eIF4G (Bartel, 2018).
piRNAs
Piwi-interacting RNAs (piRNAs) are a class of small RNAs in animals that are 21–35 nucleotides in length that primarily act as an adaptive immune response that silences
transposable elements, but can also act as an antiviral defense mechanism and regulate gene expression (Malone and Hannon, 2009; Siomi et al., 2011; Czech et al., 2018; Ozata et al., 2019). The precursors of piRNAs are single-stranded RNAs, unlike siRNAs and miRNAs, and do not require Dicer or RdRPs for their generation (Vagin et al., 2006). Piwi-interacting RNAs have 5ʹ monophosphate termini and 2ʹ-O-methyl-modified 3ʹ termini, and they associate with
PIWI-clade Argonautes (PIWI13 proteins), which are a class of Argonaute proteins whose expression is
primarily restricted to germ cells (Aravin et al., 2007; Ozata et al., 2019). While piRNAs were first identified in Drosophila melanogaster testis as “long siRNAs” which silenced the multi-copy Stellate gene on the X chromosome and were thus called repeat-associated siRNAs (rasiRNAs) (Aravin et al., 2001), they were later identified as a separate class of RNAs that associated with PIWI proteins in the mammalian testes (Aravin et al., 2006; Girard et al., 2006; Grivna et al., 2006; Lau et al., 2006; Watanabe et al., 2006). Piwi-interacting RNAs were also found in C. elegans, Drosophila, and zebrafish (Ruby et al., 2006; Vagin et al., 2006; Brennecke et al., 2007; Houwing et al., 2007; Yin and Lin, 2007; Batista et al., 2008; Das et al., 2008).
The characteristics of the genomic loci producing piRNAs varies from species to species, which results in the sequences of piRNAs being diverse and seldom conserved. A majority of the piRNA loci correspond to repetitive elements and are referred to as piRNA clusters in most species14 (Malone and Hannon, 2009; Ozata et al., 2019). In most animals, current evidence
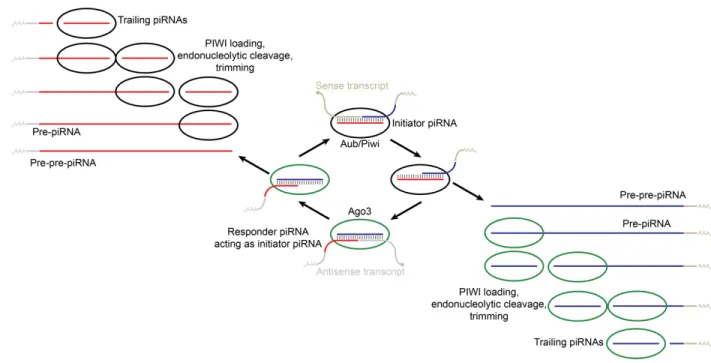
suggests that piRNAs are generated via two main mechanisms: the ping-pong amplification cycle and the phased-piRNA biogenesis pathway (Figure 8) (Ozata et al., 2019). In the ping-pong cycle (Brennecke et al., 2007; Gunawardane et al., 2007), existing piRNAs are amplified when a PIWI-bound initiator piRNA, which are maternally deposited in many animals, guide the
cleavage of complementary targets (Ozata et al., 2019). The subsequent cleaved pre-pre-piRNAs can bind to a PIWI-protein (e.g., Ago3 or Aubergine) after which endonucleolytic cleavage at its 3ʹ end produces a pre-piRNA (Figure 8). This pre-piRNA is trimmed 3ʹ-to-5ʹ by an exonuclease to produce
13 PIWI stands for “P-element induced wimpy testis,” which is a descriptor of the phenotype of the Drosophila
mutants whose germline lost the ability to proliferate (Lin and Spradling, 1997).
Figure 8. Schematic of piRNA biogenesis in most animals (adapted from Weinberg, 2013). The ping-pong amplification cycle and the phased piRNA biogenesis pathways work together to generate piRNAs. In the ping-pong cycle, an antisense initiator piRNA (red) is bound to the PIWI protein Aubergine (Aub) or Piwi. It guides cleavage of a sense transcript, which becomes the 5¢ end of a sense responder piRNA (blue). The formation of the 3¢ end of the sense responder piRNA occurs and then this piRNA binds to another PIWI protein, Ago3. It is able to cleave an antisense transcript, thus forming the 5¢ end of an antisense initiator piRNA, and the
amplification cycle restarts. In the phased piRNA biogenesis pathway, after cleavage by initiator piRNAs, pre-pre-piRNAs can be bound by PIWI proteins. The PIWI proteins direct
endonucleolytic cleavage that is then followed by 3′-to-5′ trimming to form phased trailing piRNAs.
a responder piRNA (Ozata et al., 2019). This responder piRNA can then act as an initiator piRNA for another cleavage event that propagates the ping-pong cycle (Figure 8) (Ozata et al., 2019). These initiator and responder piRNAs have historically been known as secondary piRNAs (Ozata et al., 2019). In the phased-biogenesis pathway (Han et al., 2015; Mohn et al., 2015), the endonuclease, thought to be Zucchini in flies or PLD6 in mammals (Haase et al., 2010; Ipsaro et al., 2012; Nishimasu et al., 2012), establishes the 3ʹ end of the responder piRNA, as mentioned above, and it also fragments the rest of the piRNA into phased trailing
pre-piRNAs which can be exonucleolytically trimmed to form trailing pre-piRNAs (Figure 8) (Ozata et al., 2019). These trailing piRNAs are also known as primary piRNAs (Ozata et al., 2019). In sum, the two piRNA biogenesis pathways work in concert: the ping-pong pathway produces piRNAs that cleave target RNAs and generate pre-pre-piRNAs with 5ʹ monophosphates that are then processed into either responder piRNAs or trailing piRNAs by the phased-piRNA pathway, and some of these responder piRNAs then become initiator piRNAs for the ping-pong pathway (Ozata et al., 2019).
Endogenous siRNAs
Endogenous sources of double-stranded RNA can produce siRNAs known as endogenous siRNAs (endo-siRNAs). Endo-siRNAs were initially discovered in plants, fungi and nematodes (Hamilton et al., 2002; Reinhart and Bartel, 2002; Ambros et al., 2003; Zilberman et al., 2003), and this class of small RNAs was initially thought to be present only in organisms that possessed RdRPs that could produce dsRNA from a ssRNA template (Nilsen, 2008). Examples of these RdRP-dependent endo-siRNAs included heterochromatic siRNAs in the fission yeast
Schizosaccharomyces pombe, cis-acting siRNAs (casiRNAs) and trans-acting siRNAs
(tasiRNAs) in Arabidopsis, and secondary siRNAs in C. elegans (Hamilton et al., 2002; Reinhart and Bartel, 2002; Zilberman et al., 2003; Peragine et al., 2004; Vazquez et al., 2004b; Xie et al., 2004; Sijen et al., 2007).
RdRP-independent endo-siRNAs were later discovered in human cultured cells,
Drosophila species and mouse oocytes (Yang and Kazazian, 2006; Czech et al., 2008; Ghildiyal et al., 2008; Kawamura et al., 2008; Okamura et al., 2008a; Okamura et al., 2008b; Tam et al., 2008; Watanabe et al., 2008). Sources of endogenous dsRNA for these RdRP-independent