Analytical and Bioanalytical Chemistry
Electronic Supplementary Material
CRM rapid response approach for the certification of arsenic species
and toxic trace elements in baby cereal coarse rice flour certified reference
material BARI-1
Zuzana Gajdosechova, Patricia Grinberg,
Kenny Nadeau Lu Yang, Juris Meija, Hakan Gürleyük,
Ben J. Wozniak, Joerg Feldmann, Laurie Savage, Suladda Deawtong, Paramee Kumkrong,
Kevin Kubachka, Zoltan Mester
Instrumentation used at Lab 1
All samples and standards preparations and dilutions was conducted in a class 100 clean room or in fume hoods of class 10 air quality. All plastic and glass labware, was cleaned by immersion in 5% (v v-1) HNO3 for at least 24 hours and thoroughly rinsed with DIW before use. Nitric acid was purified in-house by sub-boiling distillation of reagent grade feedstock in a quartz still. High purity deionized water (DIW) was produced by reverse osmosis of tap water followed by deionization (Barnstead/Thermolyne, Dubuque, IA, USA) to yield an 18 M.cm resistance. Analytical grade hydrogen peroxide (Sigma Aldrich, Canada), malonic acid (SigmaAldrich) and trifluoroacetic acid (SigmaAldrich) were also used. Natural isotopic abundance As, Cd, Hg and Pb stock solutions at 1000-3000 mg kg-1 were prepared by dissolving NRC traceable high purity metals, previously characterized by glow discharge mass spectrometry (GDMS) for purity (>99.9 %), in high purity HNO3 or mixture of HNO3 and HCl and diluted with DIW. For As speciation salts of arsenic pentoxide (AsV, AlfaAeser, purity 99.9%), dimethylarsenic acid (DMA, SigmaAldrich, purity 99.0+%) and arsenic trioxide (AsIII, Aldrich Chemical Company) were dissolved in DIW. Enriched 111Cd, 201Hg and 207Pb isotopes, purchased from Oak Ridge National Laboratory (Oak Ridge, TN, USA) or Trace Sciences International (Richmond Hill, ON, Canada), were dissolved in high purity HNO3 or mixture of HNO3 and HCl and diluted with DIW to prepare spike stock solutions. NIST SRM 1568b rice flour, NRC CRM DORM-4 fish protein, NRC CRM SQID-1 cuttlefish and NMIJ CRM 7532a Arsenic Compounds and Trace Elements in Brown Rice Flour were used for method validation.
Total metals analysis
A Multiwave 3000® microwave sample preparation system (Anton Paar, Graz, Austria), equipped with Conventional PTFE-TFM liner vessels was used for microwave-assisted acid digestion prior to total elemental analysis. Samples were digested in closed vessels for 15 min ramping to 1400 W and holding for 30 min, then 0 W for the cool down cycle (35 min). After digestion, samples were evaporated in class 10 fume hood and re-constituted in 2% HNO3. A high-resolution ICP-MS Element XR (Thermo Fisher Scientific, Bremen, Germany) equipped with a combination of cyclonic and scott-type spray chamber and 50 L min-1 MCN50 PFA nebulizer (Elemental Scientific, Omaha, USA) was used. A plug-in quartz torch with a quartz injector and a platinum guard electrode were also used. The Element XR was equipped with a Faraday cup detector in addition to the secondary electron multiplier (SEM) detector. A triple quadrupole ICPMS (Agilent 8800, Agilent Technologies Canada Inc., Mississauga, ON, Canada) was also used for the analysis of total trace elements and As speciation. The instrument was used in standard
set-up equipped with Ni cones. Prior to analysis, both ICP-MS were optimization for as recommended by the manufacturer. Individual analytes were detected in different modes as it can be seen in Table S1.
Table S1 Acquisition parameters of monitored analytes using Element XR and 8800 ICP-MS
Element XR resolution 8800 ICP-MS Collision/ Reaction mode
Low Medium High No gas O2
As x x
Cd x x
Hg x x
Pb x x
Determination of toxic metals by double Isotope dilution ICP-MS method
For the quantitation of Cd, Hg and Pb double ID [1] was applied using both high resolution (HR) ICP-MS and ICP-QQQ-MS. About 0.25 g of samples were gravimetrically spiked with known masses of enriched isotopes to achieve an approximately 1:1 ratio in intensities of selected isotope pairs. The following reference/spike isotope pairs were used: 114Cd/111Cd, 208Pb/207Pb and 202Hg/201Hg. Similarly, three procedural blanks were prepared by addition of only 10 % of the mass of enriched isotope spike used for the samples. After addition of 7 mL HNO3 and 0.5 mL H2O2, vessels were sealed and microwave digested as previously described. The final concentration of analysed samples was calculated using Equation 1 [1]. b r r y y z z x x y y y z x y z x w A A r K B A A r K B A r K B r K B A m m m D m w w (Z) (X) ' ' ' (1) where:
wx is the blank mass fraction of the analyte in the sample (mg/kg);
wz is the mass fraction of the analyte in primary standard solution (mg/kg);
my is the mass of spike solution used to prepare the mixture of sample and spike (g); mx is the mass of sample used (g);
D is the dry weight correction factor;
mz is the mass of primary assay standard used (g);
my is the mass of spike used to prepare the mixture of spike and primary assay standard (g); Ay is the abundance of the reference isotope in the spike;
By is the abundance of the spike isotope in the spike; Ax is the abundance of the reference isotope in the sample; Bx is the abundance of the spike isotope in the sample;
Az is the abundance of the reference isotope in the primary assay standard; Bz is the abundance of the spike isotope in the primary assay standard; K is the mass bias correction factor;
r is the measured reference/spike isotope ratio in the mixture solution of sample and spike; r is the measured reference/spike isotope ratio in the mixture solution of spike and primary
assay standard;
Ar(X) is the atomic weight of the analyte element in the sample;
Ar(Z) is the atomic weight of the analyte element in primary assay standard;
wb is the mass fraction of the analyte in the procedural blank (mg/kg) calculated using an expression identical to equation 1 wherein only the first term on the right hand side is used and the subscripted variables refer to the relevant blank parameters.
The measurement equation can be simplified to equation 2 for all elements whose isotopic abundances are invariant in nature (and therefore Ax = Az = Axz, Bx = Bz = Bxz, Ar(X) = Ar(Z)):
b y y xz xz xz xz y y y z x y z x w r K B A A r K B A r K B r K B A m m m D m w w ' ' ' (2)
Determination of total As by standard addition ICP-MS at NRC
For determination of As we used a three-point standard addition calibration method and Equation 3 was used for the calculation of mass fraction of As [2, 3]
a
m
m
m
m
I
b
m
m
m
w
m
s i x xf
i d0 i -df i s i f i s std i std and wxa (3) where:wx is the mass fraction of the analyte in the sample (µg kg-1);
wstd is the mass fraction of the analyte in the primary standard solution (µg kg-1);
mstd-i is the mass of natural abundance standard added to the spiked sample (g), i=1, 2;
ms-i is the mass of aliquot of diluted sample used to prepared spiked sample (g), i=1, 2;
msf-i is the final mass of spiked sample (g), i=1, 2;
mdf-i is the final mass of diluted set of samples (g), i=0, 1, 2;
md0-i is the mass of aliquots of spiked samples for dilution (g), i=0, 1, 2;
md0-f is the final mass of aliquots of spiked samples after dilution (g), i=0, 1, 2;
mx is the mass (g) of the original sample;
mxf is the final mass of the original sample after addition of enriched spikes and 1% HNO3 (g).
In addition, errors-in-variables regression was used to obtain uncertainty [4].
For As speciation, accurately weighted 1 g of sample was placed into 50 mL tube and extracted in 20 mL of trifluoroacetic acid (0.02 M) containing 1% H2O2 following a previously published method [5]. Samples were extracted at 95°C for 1 hours in water bath. Once cooled to room temperature, the samples were centrifuges at 3000 g for 15 minutes and aliquot of 2.5 mL was accurately weighted and transferred into a new vial for standard addition. Three sub-samples of NIST 1568b SRM were treated in the same way and one method blank was used for monitoring As contamination. Standard additions were performed using a traceable arsenic pentoxide as iAs and DMA at concentrations 1x, 2x, and 3x the native sample concentration, based on preliminary testing. The information value of MMA was determined from external calibration using calibration standard prepared from arsenic trioxide. All calibration standards, As(V), As(III) and DMA were cross-calibrated against high purity As standard. Aliquot of each standard was acid digested and analysed in high resolution mode by ICP-MS Element 2.
Chromatographic separation and quantitation of individual As species was achieved by coupling Agilent 1200 Series HPLC (Agilent Technologies, Mississauga, Ontario) to Agilent 8800 ICP-MS (Agilent Technologies, Mississauga, Ontario). Arsenic species were eluted from Hamilton PRP x-100 strong anion exchange column (250 mm x 4.6 mm, 5 µm packing) with guard cartridge (Chromatographic Specialties, Brockville, Ontario) using isocratic elution with 2 mM malonic acid (pH 5.6) and flow rate of 1 mL min-1. Nominal mass values 91 following the mass shift from 75 was monitored by ICP-MS to confirm the presence of As in chromatographic peaks. Additionally, m/z 71 was used as internal standard to monitor
plasma stability during the analysis. Agilent MassHunter Quantitative Analysis software was used for integration of chromatograms.
Moisture content analysis
Three sub-samples of the material (approximately 0.5 to1 g) are weighed into clean glass weighing bottles and placed in the vacuum oven. The room temperature oven was evacuated and maintained below 25 mm Hg to dry the sample to a constant mass. Once dry, the sample weight was recorded to obtain the mass loss. The average result from the individual sub-samples was used.
As species stability testing
Short term isochronous stability study was carried out in accordance of ISO Guide 35 [6]. Briefly, one unit was kept in the freezer at -20 °C as a reference unit. Three units were placed in the refrigerator at 4 °C and three units were place in the oven at 37 °C. After 1 week a unit was moved from the refrigerator and oven to the freezer. This process was repeated after 2 and 4 weeks and all units were analysed at the same time.
Mercury cleaning procedure
Microwave digestion vessels were soaked in 10% HNO3 overnight, followed by overnight heating at 105 °C. All ICP-MS introduction system was rinsed with 5% HNO3 containing 1% 2-mercaptoethanol for 30 min prior to analysis.
Traceability of calibration standards
The certified values are metrologically traceable to the SI through gravimetrically prepared standards of established purity which purities were established by GD-MS at NRC and international measurement intercomparisons. Purity assessment by GD-MS was performed by determining each of the elemental impurity mass fractions (i.e., we
M) presented in the high purity primary standards and subtracting their sum from the ideal value of 1 kg kg-1. With GD-MS analysis, it is possible to provide, with the exception
of H and radioactive elements, full elemental coverage (including C, N and O) with low to sub-ng g-1 detection limits. For those elemental results that cannot be quantitatively determined, a limit of detection (LOD) is reported. More information regarding the use of GD-MS for purity assessment can be found here [7]. For those elements that NRC does not have primary standards assessed by GD-MS, NIST SRMs were used (NIST SRM 3133 Hg for Hg). For traceability of As species, no primary standards traceable to SI is currently available. Thus, commercially available standards were cross-calibrated against primary As standard.
Determination of trace metals by Lab 2
Samples were digested using AOAC 2015.01 [8]. In short, 0.25-0.5 g of sample was digested using a mixture of concentrated HNO3 and 30% H2O2 in CEM Mars Express microwave. The digested samples were brought up to volume with DIW and analyzed using an Agilent 8800 ICP-QQQ-MS for As, Cd, and Pb and by Brooks Rand Merx-T for Hg.
Inorganic As was extracted from the samples using the FDA Elemental Analysis Manual Method 4.11 [9] with minor modifications. Approximately 0.5-1 g of sample was extracted with dilute HNO3 and H2O2 on a hot block (Environmental Express) at 90°C for 2 hours. The extracts were brought up to volume with DIW, filtered through a 0.45 μm filter and then analyzed by IC-ICP-MS on an Agilent 7700 ICP-MS.
Traceability of calibration standards
All standards used were NIST traceable. Arsenic speciation standards were purchased from Spex, Inorganic Ventures and High Purity Standards (AsIII, AsV, DMA and MMA). After receiving the standards, their purity was confirmed by analyzing 3 different preparations in triplicate (n=9). Every analytical batch is verified by standards from a second source (NIST Traceable) as well as reference materials (NIST 1640a + TMDA 70.2 and USGS T221) to validate our calibration.
Determination of trace metals by Lab 3
For determination of total element concentration, 0.2 g sample was measured gravimetrically and microwave digested in 2 mL of concentrated HNO3 and 2 mL of 30% w/w H2O2 using open vessel
digestion in a CEM Mars microwave system. The samples were diluted to a final mass of 50 g with DIW. Calibration standards for element analysis were prepared by appropriate dilution of stock multi-standard solution 71A (Inorganic Ventures) in the same solvent as sample extraction. Agilent 7900 ICP-MS was used for multi-element analysis optimised and operated in hydrogen collision cell mode. Online addition of Ge internal standard at approximately 10 μg L-1 was implemented to correct for ICP-MS signal-drift.
Arsenic species were extracted from samples (0.1 g) using microwave extraction in 10 mL of 1% HNO3 and 2% H2O2 (5 min 50 °C, 5 min 75 °C, 30 min 95°C). All samples were centrifuged at 13 000 rpm for 10 min prior to analysis with HG-ICPMS or HPLC−ICPMS [10]. Calibration standards for HG-ICP-MS were prepared by appropriate dilutions of As(V) solution (Spex CLAS2-2Y) in the same solvent as used for the extraction. Standards for HPLC-ICPMS were prepared from sodium cacodylate (Sigma, 98%), also in matrix-matched solvent. Speciation was carried out on an Agilent 1100 HPLC system connected directly to an Agilent 8800 ICP-QQQ-MS in O2 mode. PRPX-100 Hamilton anion exchange column (10 μm, 4.6 mm × 250 mm) with a flow rate of 1 mL min−1 was used. Mobile phase was 25 mM ammonium carbonate (pH 8.5) for HPLC−ICPMS and 10 μg L-1 Ge-solution was continuously added as internal standard via a T-piece before the nebulizer.
An Agilent Hydride Generation Accessory for ICP-MS was also used. For that, the ICP-QQQ-MS was used for As detection in He mode as described in detail elsewhere [10]. Briefly, the samples were injected using autosampler and transported to the hydride generator (0.5 mL min-1) where the sample mixed with HCl (5M, 2.5 mL min-1) and NaBH4 (2% (w/v) in antifoam, 0.5 mL min-1) in a mixing coil before entering the gas–liquid separator (GLS). The gaseous sample was then transported to the ICP-MS with an argon gas flow (0.3 L min-1) using the make-up gas line of the ICP-MS, separating online the iAs from DMA. To this, an argon flow (0.85–0.95 L min-1) carrying a nebulized solution of the IS using the peristaltic pump of the ICP-MS was added creating wet plasma conditions.
Traceability of calibration standards
Standard used for total metals quantitation (Inorganic Venture 71A) and inorganic As speciation standards (Inorganic Ventures (AsIII and AsV) are traceable to NIST. Cacodylic acid (Argos Organic) used for quantitation of DMA is not NIST traceable.
Determination of trace metals Lab 4
About 1 g of samples were digested with 5 mL concentrated HNO3 and 1 mL H2O2 using hot block at 180 O
C for 3 h. Samples were then diluted to 10 mL with DIW and analysed for total As using ICP-MS with an external calibration.
For As speciation, approximately 0.5 g samples were extracted in 0.15 M HNO3 on a hot block at 100 OC for 2 h. After extraction, the samples were diluted to 10 g with DIW, centrifuged at 3000 rpm and then filtered through 0.45 µm membrane. LC-ICP-MS separation was carried out on ODS column (4.6 mm i.d. x 250 mm) and mobile phase consisting of 10 mM sodium 1-butanesulfonate, 4 mM malonic acid, 4 mM Tetramethyl-ammonium hydroxide and 0.05 % methanol (pH 3.0) at flow rate 0.75 ml min-1 at room temperature.
Traceability of calibration standards
Inorganic As speciation standards (Inorganic Ventures (AsIII and AsV) used are traceable to NIST and NMIJ 7913-a was used for quantitation of DMA.
Determination of trace metals by Lab 5
Total As was determined using the FDA EAM method §4.7 [11]. In summary, samples were subjected to microwave-assisted digestion (Mars Xpress, CEM) with 8 mL of concentrated HNO3 and 1 mL of H2O2, following dilution to approximately 200 g with DIW, and analyzed by ICP-MS (Agilent 7900). Analysis was done in He mode with an online internal standard (Rh) and quantitation was performed by external calibration.
Arsenic speciation was performed using the FDA EAM method §4.11 [9]. In summary, samples were extract with 0.28M HNO3 at 95°C for 90 minutes in a hot block digestion system (DigiPREP MS, SCP Science). The extract was diluted with DIW, then neutralized for a final dilution factor of ca. 50X. The extract was then filtered using 0.45 μm nylon syringe filter and analyzed by HPLC-ICP-MS. The HPLC used was an Agilent 1260 and the ICP-MS was an Agilent 8800, with He as a collision gas. The column used was a Hamilton PRP-X100 anion exchange column (4.1 × 250 mm, 10 μm) with matching guard column (Hamilton, Reno, NV). The chromatographic conditions include a mobile phase of 10 mM ammonium phosphate dibasic in water with the pH adjusted to 8.25 using NH4OH, mobile phase flow rate of 1.0 mL min−1 and an injection volume of 100 μL. The column effluent was connected directly to
the nebulizer of the detector with a post-column marker (instrument drift standard) against appropriate external calibration standards including AsIII, DMA, MMA, and AsV. The iAs was reported as the sum of AsIII and AsV.
Traceability of calibration standards
Standards for total metals analysis were prepared from NIST traceable stock solutions (Inorganic Venture). Speciation standards were analyzed for total As content versus NIST traceable As standard. Additionally, the purity of each standard was checked by analyzing using HPLC-ICPMS, if the primary peak was >98%, then the impurities were ignored and the total As concentration was applied in calculations. For those that were <98%, the composition was adjusted – this occurred for MMA with 7% AsV.
Calculation of expanded uncertainty
The expanded uncertainty (U) is equal to U = kuc where uc is the combined standard uncertainty calculated according to the JCGM Guide [12, 13] and k is the coverage factor. A coverage factor of two (2) was applied. It is intended that UCRM accounts for every aspect that reasonably contributes to the uncertainty of the measurement.
The standard combined uncertainty, uc, was determined using the following equation:
u2c = u2char + u2hom + u2method
where:
uchar is the batch characterization,
uhom uncertainties related to possible between-bottle variation
umethod uncertainties related to inconsistency between the various measurement methods.
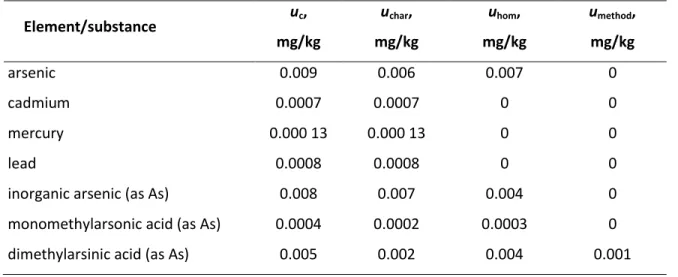
Table S2. Uncertainty components for BARI-1 Element/substance uc, mg/kg uchar, mg/kg uhom, mg/kg umethod, mg/kg arsenic 0.009 0.006 0.007 0 cadmium 0.0007 0.0007 0 0 mercury 0.000 13 0.000 13 0 0 lead 0.0008 0.0008 0 0
inorganic arsenic (as As) 0.008 0.007 0.004 0 monomethylarsonic acid (as As) 0.0004 0.0002 0.0003 0 dimethylarsinic acid (as As) 0.005 0.002 0.004 0.001
As it could be seen from Table S2, the uchar , except for arsenic, and its species, is the dominant source of uncertainty, which is expected due to the low-level of those measurands in the proposed CRM. For Arsenic and its species, uhom also contributes to the overall uncertainty.
References
1. Yang, L. and R.E. Sturgeon, High accuracy and precision isotope dilution mass spectrometry: An application to the determination of Mo in seawater. J. Anal. At. Spectrom., 2009. 24: p. 1327-1335.
2. Gao, Y., Sturgeon RE, Mester Z, Hou X, Zheng C, Yang L, Direct Determination of Trace Antimony in Natural Waters by Photochemical Vapor Generation ICPMS: Method Optimization and Comparison of Quantitation Strategies. Anal. Chem., 2015. 87: p. 7996-8004.
3. Meija, J., E. Pagliano, and Z. Mester, Coordinate Swapping in Standard Addition Graphs for Analytical Chemistry: A Simplified Path for Uncertainty Calculation in Linear and Nonlinear Plots. Anal. Chem., 2014. 86: p. 8563-8567.
4. Meija, J. and M.M. Chartrand, Uncertainty evaluation in normalization of isotope delta measurement results against international reference materials. Anal Bioanal Chem, 2018.
410(3): p. 1061-1069.
5. Raber, G., Stock N, Hanel P, Murko M, Navratilova J, Francesconi KA, An improved HPLC–ICPMS method for determining inorganic arsenic in food: application to rice, wheat and tuna fish. Food Chem, 2012. 134(1): p. 524-532.
6. ISO, International Organization for Standardization (ISO), ISO Guide 34, General requirements for the competence of reference material producers. 2009: Geneva, Switzerland.
7. Sturgeon, R.E., Methven B, Willie SN, Grinberg P, Assignment of purity to primary metal calibrants using pin-cell VG 9000 glow discharge mass spectrometry: a primary method with direct traceability to the SI international system of units? Metrologia, 2014. 51: p. 410-422. 8. AOAC SMPR 2012.007. Standard method performance requirements for determination of heavy
metals in a variety of foods and beverages. J AOAC Int, 2013. 96(4): p. 704.
9. Kubachka, K.M., Shockey, N. V., Hanley, T. A., Conklin, S. D., Heitkemper, D. T., 4.11 Arsenic Speciation in Rice and Rice Products Using High Performance Liquid Chromatography-Inductively Coupled Plasma-Mass Spectrometric Determination. 2012.
10. Musil S, Pétursdóttir ÁH, Raab A, Gunnlaugsdóttir H, Krupp E, Feldmann J,Speciation without chromatography using selective hydride generation: inorganic arsenic in rice and samples of marine origin et al.,. Anal Chem, 2014. 86(2): p. 993-9.
11. Gray, P.J., Mindak, W. R., Cheng, J., 4.7 Inductively Coupled Plasma-Mass Spectrometric Determination of Arsenic, Cadmium, Chromium, Lead, Mercury, and Other Elements in Food Using Microwave Assisted Digestion. 2015.
12. JCGM, Evaluation of measurement data — Guide to the expression of uncertainty in measurement. 2008.
13. DerSimonian R, Kacker, R, Random-effects model for meta-analysis of clinical trials: an update. Contemporary clinical trials, 2007. 28(2): p. 105-114.