READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE. https://nrc-publications.canada.ca/eng/copyright
Vous avez des questions? Nous pouvons vous aider. Pour communiquer directement avec un auteur, consultez la première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées. Si vous n’arrivez pas à les repérer, communiquez avec nous à PublicationsArchive-ArchivesPublications@nrc-cnrc.gc.ca.
Questions? Contact the NRC Publications Archive team at
PublicationsArchive-ArchivesPublications@nrc-cnrc.gc.ca. If you wish to email the authors directly, please see the first page of the publication for their contact information.
NRC Publications Archive
Archives des publications du CNRC
This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.
Access and use of this website and the material on it are subject to the Terms and Conditions set forth at
Moisture content - its significance and interaction in a porous body
Feldman, R. F.; Sereda, P. J.
https://publications-cnrc.canada.ca/fra/droits
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
NRC Publications Record / Notice d'Archives des publications de CNRC:
https://nrc-publications.canada.ca/eng/view/object/?id=17232fee-6973-42a0-b605-7ccfeddd03a2 https://publications-cnrc.canada.ca/fra/voir/objet/?id=17232fee-6973-42a0-b605-7ccfeddd03a228.
Moisture Content-Its Significance and
Interaction in
a
Porous Body*
R.
F. F E L D ~ N AND P.J.
SEREDANational Research Council, Division of Building Research, Ottawa, Canada
ABSTRACT
The meaning of moisture content of a specimen
is
discussed i n terms of the diflerent states of interaction i n which water may exist in the specimen and of the length changes arising from a change in thismoisture content.Sorption isotherms and the associated length changes have been determined for a number of porous systems including molecuEar sieves, porous glass, "Cab-O-Sil" silica, precipitated calcium carbonate, CaSO,. x H , O , and hydrated cement. All these materials, except porous glass were compressed
in
a mold at h i g h pressure to form compacts (rigid porous bodies), and thistechnique
is
discussed.The signiJicance of the sorption isotherms in determining the structure of the adsorbent is discussed, and the added complication of water of structural types (e.g., hydrate, activated adsorp- tion) in the isotherm
is
emphasized. Methods for diflerentiating between the diflerent types are outlined with the aid of theoretical equations applied to the results of the length c h q e and sorption isotherms.The moisture content of a porous body may be defined in a variety of ways, depending on the purpose of the definition and the field of technology in which i t is applied. Despite the diversity of endeavors, however, it must be recognized that water may exist in a porous body in different states as a result of the
*
This paper is a contribution from the Division of Building Research, National Research Council, Canada and is published with the approval of tho Director of the Division.different modes of interaction with the porous body.
It is very difficult to distinguish between the
different states of the water; it is the purpose of this paper to discuss the manner in which length changes arising from a change in the moisture content, together with the sorption isotherms, reflect the state of the water in porous bodies.
THE SIGNIFICANCE OF SORPTION AND
LENGTH CHANGE ISOTHERMS
When a sorption jsotherm is determined z'or
a vapor on a material, the 11 eight gain of the material is measured as a function of the vapor- pressure, a t a constant temperature. Xt each vapor pressure, time is allow-ed for equilibrinn: to be attained between the sorbent and the sorbate.
The molecules of sorbate leave the gas phase and become attached to the surface or penetrate beyond the surface of the sorbent. These are the phenomena of adsorption and absorption, respectively. If the molecules re- main on the surface of the solid sorbent, the attachment may be due to a weak interaction, termed physical adsorption, or a strong one- chemisorption. When the molecules enter the solid, a chemical compound or solid solution is formed. When this occurs the sorbate molecules may penetrate into the field of force
that esists between n~olecules of the sorbent.
Physical adsorption is considered to be the result of the imbalance of forces that an atom in the plane of the surface is subjected to, and the energies inrolved are in the same order of
magnitude as those of condensation. KO satis- dipoles in the first layer of adsorbed nlolecules
factory treatment of surface adsorption \\-hich in turn induce dipoles in the n e s t layer
e s ~ s t c d before 1915 n hen two theories and so on until s e ~ e r a l layers have been built
independent of each other n-ere proposed by up. The forces involved are short-range forces
Polarlr-il and by L a n g ~ n u i r . ~ Polanyl suggest- as opposed to those in the potential theory.
ed that adsarptioil n a s a physical process and This theory was tlie first to give a quantitative tlint the adsorbed phase \\-as inany layers thick, account of the S-shaped isotherm that is 11cld by long range attractive forces from the tjpical of most physical adsorption data, but
surface. Langiiiuir, on the other hand, believed it was shown that tlie binding energy between
that adsorption \$as a chemical process and two adsorbed layers mould be too snlall. The tl:,lt the adsorbed layer was unimolecular. The second tlieorjr, the multimolecular theory of
Lanrnl~iir equation can be applied over the Brunauer. Emnlett and Teller7 is based on t h e
15 hole range of relative pressures, although its assumption that the same forces t h a t produce
uqe is limited to systeins in n hich adsorption condensation are also chiefly responsible for
takes place in a uniniolecular layer. The theory the binding energy of multimolecular adsorp-
is important, lion-ever, because this is the tion and only the first adsorbed layer is
starting point for other equations that fit data attracted strongly by the surface. The second
0.i-er a nider range. layer is adsorbed essentially not by the surface
The capillary condensation theor?, which but by the first adsorbed layer and the adsorp-
originated n-it11 Z ~ i g m o n d y , ~ attributes tion thus propagates from layer to layer. The
adsorption to condensation of the gas in the equation derived on this basis, called the
capillaries of the adsorbent. It was assumed B.E.T. equation, gives a satisfactory explana-
that ill rerv small capillaries, condensation tion of all types of adsorption isotherms;
call take place a t pressures considerably adsorption isotherms have been classified into
lolr er than the normal vapor pressure. The five general types. The B.E.T. theory for the relation betn-een the vapor pressure over the isotherm of a gas adsorbed on a free surface
meniscus of a liquid in a capillary compared to results in the follo~r-ing expression.
tlie bulk hquid is given by the Kelvin equation
-2 y v
plpo =
where
n-here
W
= the weight of gas adsorbed a t vapor
p = the vapor pressure pressure, p
y = the surface tension po = the saturated vapor pressure a t t h e
I' = molar volume of the liquid a t absolute temperature of the adsorbent
temperature, T C = a constant related t o the heat of
l? = the gas constant adsorption
r = the radius of curvature of the liquid in
W ,
= the weight of gas necessary t o formthe capillary. a single layer of molecules over t h e
entire surface. Thus: capillaries of smallest radii fill a t the
lowebt precsures, until finally a t the saturation Although the above equation was derived for
prcs2ure all the pores of the adsorbent will be a flat surface, it is still applicable over a limitcd
filled 1: ith liquid. This theory does not account range for most porous materials. The most
for unin~olecular ad~orption vhich precedes general equation, however, includes the five
capillary condensation and as a rcsult accounts isotherm types for a wide range of relative
for only part of the isotherm. pressures.
Two more tlleorles proposed later were botli Since most solids which are used as catalysts
ba-ed on multimolecular adsorption. The first: or adsorbents in many industrial applications
referred to as the polarization theory5, and also materials used in build~ng are of a
esplains the adcorption of nolipolar molecules porous nature, an adcquate measure of t h e
on ionic adsorbents by assuming t h a t the surface area must include the aren of t h e
28. MOISTURE CONTENT I N
A
POROUS BODY 235angstroms in diameter. The molecules of a gas provide a suitable instrument for measuring the total area since the area covered by a single molecule can be determined from- known physical constants of the gas. As was shown above, the B.E.T. equation, applied to a n adsorption isotherm, enables the determina- tion of the weight of gas necessary t o form a single layer over the entire surface; this makes possible the measurement of the total area. Information pertaining to the physical structure of a porous body, the pore size and pore-size distribution may also be obtained from the adsorption-desorption isotherm by application of the Kelvin equation; the
hysteresis loops which
.
often appear inphysical adsorption isotherms are character- ized by the fact that a given amount of vapor remains adsorbed a t a lower pressure on the desorption branch of the loop than on the adsorption branch. The "delayed meniscus" theory, proposed independently by CohanB and C ~ e l i n g h , ~ considers a capillary open a t both ends; adsorption will occur on a n
annular ring on the ~vall of the pore, but the
desorption occurs from the surface of a normal meniscus in a completely filled pore so t h a t the normal Kelvin equation will describe the vapor pressure over the meniscus. Calcula- tions of surface area from the pore-size dis- tribution have been made, and good agree- ment with the result obtained from the B.E.T. method has been a c l ~ i e v e d . l ~ - ~ ~
Some adsorbents are capable of adsorbing large amounts of sorbate, and in some cases i t is not easy to tell whether the sorbate is
attached physically or otllern~ise; naturally in
these cases application of theory derived for physical adsorbed water must be done with caution. The sorption of water on a hydrated sorbent represents an example of this latter case, and here it is sometimes difficult t o separ- ate the hydrate n-ater from the adsorbed xater. Early efforts to study the state of the sorbate in porous materials by measuring length changes during the isotherm were nlade by
J. IT7. R 1 ~ B a i n . l " ~ ~ He pointed out that accord-
ing to capillary condensation theory tllc
adsorbate should cause a contraction of the adsorbent ratlier than an espansion. He considered that since, a t that time, all investigators found espansion for charcoal and not contraction, the capillary condensation
theory has been disproved. Since that time, however, other ~ o r k e r s l ~ , ~ ~ found marked contractions in the length change isotherms, and the discrepancy was attributed t o the difference in the degree of charring with the
other charcoals. A study of adsorption
hysteresis by means of the length change isotherm of porous glasslo provided a sound basis for the capillary condensation theory in the high relative pressure region of the isotherm.
It has been recognized t h a t adsorption is due to the imbalance of forces on the surface of the solid adsorbent, and because of this, the adsorbent should not be insensitive t o the process of adsorption; a study of the length changes of a porous body should provide information on the process of adsorption and the effect of the adsorbate on the body and its position or function within the body. BanghamlB and his collaborators made the most extensive study of the expansion of charcoal due to adsorption of vapors. They noted t h a t their results over certain ranges of concentrations could be expressed by a n equation that assumed direct proportionalit:- between the surface pressure
A
F and the l i n ~ a respansion
AZIZ.
Thuswhere
X
is a collstant. Further work byBangham and Razoukl9 showed that the surface pressure or decrease in surface free energy call be evaluated fronl the adsorption isotherm by integrating the Gibbs eq11atioi1'O
where s is the surface coneeiltration of the
adsorbate in g-nloles/cm2. Data fro111 Bond,
Griffith and JIaggs" indicated a linear
relation betncen the espansion and surface free energy decrease for the adsorption of water on coal. Tliis has also been sllown for
n-ater on porous g1n~s.l0 Bangllan~ and his
c o - , ~ o r ~ e r s ~ - ~ ~ esplflilled tlle process of espansion during adsorption in the follon.ing n a y : a porous nlatcrial may be considered to be corilposed of snlall subunits; each unit,
maintains its size and shape by n bnl;lnce
bctnwn two opposing sets offorces. the surface forces nhicll tend to aggregate tllc mntcrial
236 INTERACTION OF IIIOISTURE A N D MATERIALS impart rigidity to the bloclr. The adsorption of
vapor causes a reduction in the surface forces so t h a t the unit expands. The rcsult nlay be considered as a force applied tangentially to the total area of the porous body. Because the surface of the porous body is high in many cases, the force described above and thus the expansion can be considerable.
USE OF COMPACTS AS SAMPLES To study the relationships of length change associated with sorption of vapors, i t is dcsir- able to have rigid porous bodies of a variety of materials 11-hich may be defined and classified into general systems. The technique for producing such samples must allow for control of composition, purity, surface area, and void fraction; the porous bodies must also be reproducible.
These requirements were met in large measure by compacts, produced by compress- ing powdered materials (particle size 10 to 0.01 p) in a mold a t pressures in the range of 5000 to 150,000 psi.25 Some materials formed
satisfactory rigid bodics over a wide mnge of pressures, giving a mnge of void fractions.
Otl~ers could be forincd only in a narrow range
of press~~res. I11 gencral, most systems yieldcd
a body that had a unique straight-line relation between the void fraction and the logarithm of the con~pactiilg pressure. The conlpacts that were prepared were 1.25 in. in diameter and 0.015 to 0.11 in. thick. For this range of thiclrness, the density did not vary with thickness a t a constant pressure of compaction.
Experinlental details for the procedures used in the determinations of length and sorption isotherills have been d e s c r i b ~ d . ~ ~
CLASSIFICATION OF THE POROUS SYSTEMS STUDIED
The samples repor'tcd on by the a ~ t h o r s ~ ~ - ~ ~ can be classified as follows:
(1) A system composed of large particles
t h a t yield a low surface area and have no internal micropore structure. This type was represented by compacts of precipitated
J R E L A T I V E HUMIDITY, ' l o W E I G H T CHANGE, O/o
(b)
L E N G T H CHANGE( c )
L E N G T H CHANGE ASISOTHERM R E L A T E D TO
W E I G H T CHANGE
28. HOISTURE CONTENT IN A POROUS BODY
R E L A T I V E H U M I D I T Y , '/a W E I G H T C H A N G E , "/a
Fro. 2. System 11 compact of silica diluted with calcium carbonate.
calcium carbonate of 1- t o 5-p particle size.
(2) A system composed of very small
particles yielding a large surface area that have no internal micropore structure. This mas represented by silica ("Cab-0-Sil" supplied by
G. L. Cabot Inc.) with particle sizes of 0.01 to
0.02 p.
(3) A system composed of large particles
with an internal micropore system. This produced a pore-system similar to Type 1 modified by its own micropore system.
Molecular sieves (Linde 4A-supplied by
Linde Company) a n aluminum silicate with a
particle size of 2 to 5 p and a crystal structure
consisting of 11.4-A cavities separated by 4.2-A openings represented this system.
(4) A system similar to Type 1 but conl-
posed of a hydrated material, in this case calcium sulfate hemihydrate of about 1-p particle size.
( 5 ) Asjrstem similar to TJ-pe 3 but composed of a hydrated material. in this case hydrated cemcnt of about 1-p particle size.
I n all these cases, n7ater 1-apor as sorbate n as utilized in the reported work.
A number of the compacts used n-ere immersed in kerosene and carbon tetrachloride and did not show any length change. This was interpreted as an absence of residual strain from the result o f t he compression of particles.
When exposed to high 11unlidit~- or saturated
with water, howeyer, most con~pacts sho~ved
a degree of relaxation or irrercrsible l e l ~ g t l ~ change especially when the alternate condi- tions of n e t and dry n ere cycled a nnn;ber of times. Some colnpacts tended to disintegrate
~vllen saturated with n ater. T11cs~ tendel~cies
placed certain limits on tlle use of con~pacts in
studying the length changesarising fro111 v a t e r sorption.
DISCUSSION OF RELATIONSHIPS
The results of the measurenlc~~ts of sorptior~
and the associated length changcs for the five distinct systenls arc summarized in Fipa. 1 to 5 .
The al~alyscs of thcsc rcaults indicntcrl certz~in
conlinoll rCl,~tionships 11 hich may bc descr~bed
n ith rcfrxrc~~cc to the systems st~~clicd.
I L 7 T E R A C T I O N OF ,JlOISTURE AhrD M A T E R I A L S
WEIGHT CHANGE, ' l o
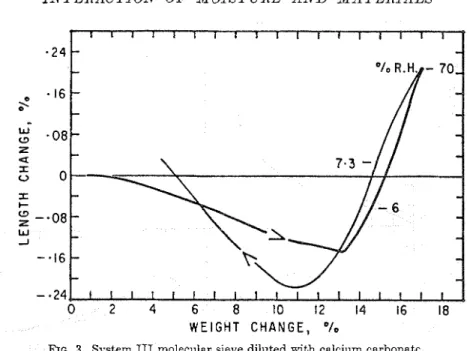
RG. 3. System I11 molecular sieve diluted with calcium carbonatr.
internal surface area of the porous body has a with system 1, sho~vn in Fig. 1, having a
direct influence upon the total length change surface area 5.6 m2/g (as calculated by the
associated with the sorption of water from the B.E.T. method from the sorption isotherm),
dry state to near saturation. The expansion of illustrates this point when compared with that
0.03per cent a t 50 per cent humidity obtained of 0.07 per cent obtained with System 2 as
R E L A T I'JE H U M I D I T Y , '10 W E I G H T C H A N G E , "10
28. MOISTURE CONTENT I N A POROUS B O D Y 239
R E L A T I V E H U M I D I T Y , % WEIGHT CHANGE, 7,
Fro. 5. System V compact of hydrated cement.
shown in Fig. 2 and having a surface area of
36.1 m2/g.
The most useful relation derived from the results \rras that between length change and weight change (moisture content change)
during adsorption. It had been found by
earlier investigators1° t h a t for porous glass, the length change vs weight change was a linear relation from zero up to about 60 per cent humidity. This relation has been confirmed by results for Systenls 1 and 2, and i t serves as a tool to be applied to sj7steins such as 4 and 5 nhcre hydrate water conlplicates the analyses of the sorption process.
An exception to this general behavior was encountered with System 3 \\-here the material eshibits a unique sorption length change rela- tion as shown in Fig. 3. This co~lsisted of an initial contraction on adsorption a t relative pressures )selo\v 10 per cent humidity follon.ed
b y an expansion. An eren greater contractiou occurred during desorption. This unusual result n-as considered to be related to the specific dimensions of the micropore system n~hich \\-as inlposed by the lattice of the molecular sieves. I t n-as postulated t h a t nlolecular bridging in the 4 . 2 - 1 openings caused contractioil as first proposed bj- Lakhanpal and Flood30 for adsorptio~l on charcoal. The larger contraction on the desorption cycle contributing to the observed hysteresis \\as esplainecl by tension ill the vater present in the ll.L'--< cnl-itit.s. Because tliis contraction occurrcd bclou. 10 p1.r cent humidity, representing an cqui\-nlent tcnsion far in excess of what a colunin of n'tter in n
en\-it>- could withstand, the uatcr \ \ a s
considered to be in a nonequilibrimn state.
\Yatcr in this systc~n. tllcrcfixt,. OCCLIIIIC:: a unique positioil as indicated by the intcrc~ction
240 INTERACTIOS OF VOISTURE AND HATERIALS with the sorbents, and the characteristics, if
attributed to sorbcd water, nil1 lead to mis- leading conclusions.
For systems such as 1 and 2 (where length
change vs weight change showed a linear
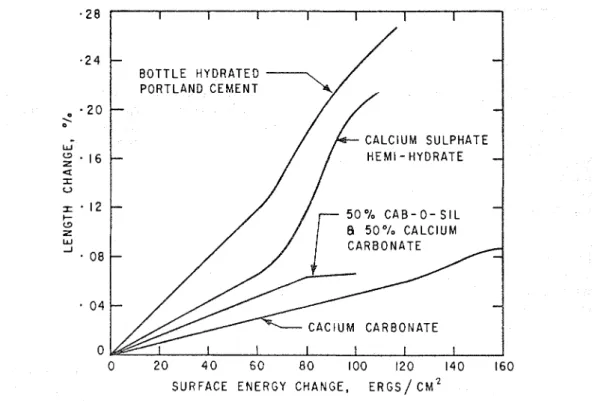
relation with adsorption as described earlier), the plot of length change as a function of the free energy change (as calculated from the sorption isotherm by the Gibbs equation) also gare a straight linen hich passed tllrough zero, thus obeying the Bangham equation. Such plots are presented in Fig. 6 . This relation gives the second tool to be applied to systems such as 4 and 5 t o enable the differentiation of the hydrate n-ater from sorbed water.
I n any hydrate system such as calcium sulfate, n-here 11)-dration or dehydration can occur simultaneously n-it11 the adsorption or deqorption process, i t has been difficult t o interpret the results of sorption nleasurements and. as a result, difficult to use these for deter- mination of the surface area. I n these systems, the anlount of n-ater taken up for hydration often does not correspoild to the theoretical anlount and is dependent on the history of the sample.
The conlmon relationships obtained for
Systems 1 and 2 were applied to Systems 4
represented by CaS0,-CaSO,.%H,O for
~vhich the sorption and length c l ~ a n ~ ~ r e s u l t s are shown in Fig. 4. For this system, the relationsllip of length change to weight change for sorption of water on anhydrous CaSO, can be divided into three regions: OA representing the region of hydration to form CaS0,-xH,O together with some adsorption is a region of low length change for a relatively large weight change; region AB representing
adsorption and characterized by the linear relation of length change to weight change from which it n-as concluded t h a t the point representing zero adsorbed water should be located on the projection of this straight line; and region BC representingsorption considered partly due to activated or chemical adsorption. This conclusion as discussed later was based on the discontinuity and secondary hysteresis observed in the isotherms and the plot of
surface energy decrease vs length change in
this region (Fig. 6).
To verify t h a t region AB represents adsorbed water and to determine its absolute
- 2 8 - 2 4 B O T T L E H Y D R A T E D P O R T L A N D C E M E N T
.
. 2 0 \ W C A L C I U M S U L P H A T E g.16 H E M I-
H Y D R A T E 4 I U x-
1 2 t- 0 % C A B - 0 - S I L w z 5 0 % C A L C I U M W -I 0 8 0 4 C A C l U M C A R B O N A T E 0 0 2 0 4 0 6 0 8 0 100 1 2 0 1 4 0 160 S U R F A C E E N E R G Y C H A N G E , E R G S / CM'28. MOISTURE CONTENT I N A POROUS BODY 241
quantity, a method based on the Gibbs and Banghain equations has bcen \ilorkcd
I n this procedure, a l~ypotl~ctical case is talcen where i t was assumed that all tlle adsorbed water could be removcd without decomposition of the hemihydrate; the11 the point for zero adsorbed water would lie on the extrapolatcd straight line BA. When a point was selected representing the zero for adsorbed water, then the surface area could be calculated (using the B.E.T. method) and the free energy change could be determined (using the Gibbs equation). By a trial and error proccdure involving the selection of a number of such points, a series of plots of length change vs free
energy change were co~lstructed and found to
be linear; the selected point that gave a line passing through the origin was considered as defining the correctly selected point for zero adsorbed water. Thus the region AB was shown to obey the Gibbs and Bangham equations and verified the initial hypothesis that this water
was physically adsorbed. It also provided a
method to distinguish between the hydrate and adsorbed water and to estimate them quantitatively thus enabling t l ~ e calculation of the B.E.T. area. When the plot of free energy decrease vs Al/1 was extended to include the region where the discontinuity occurred, a surprising increase in the slope of this curve mas observed immediately after the point equiv- alent to the discontinuity. This was believed to be explained by regarding the discontinuity as a n example of activated adsorption. I n this case, as the adsorbate went on the surface, changes to the adsorbent occurred; i t is ionic in nature and by no means inert to the adsorption process, as the length change isotherm showed. I n effect, more sites were made available for adsorption, generated by the initial coverage of the adsorbent by tohe adsorbate. Because of its nature, this type of sorption cannot be reversible, and the esplana- tion of the discontinuity would nccessarily in- clude an irreversible isotherm; this accounted for the secondary hysteresis in the iso- therms
It has been found that this ~ l l e t l ~ o d for differentiating between hydratc and sorbed water and n~hich was applied ill the case of System 4 (one component) can also be applied to Systenl 5 ~ i ~ h i c h represents a multi-
component hydrate system of great
c ~ m p l e x i t y . ~ ~ The basic relationships obtained by sorption and length change measurements for this system are shown ill Fig. 5. The length change us neight change relation below 25 per cent humidity shows two distinct linear
portions with an abrupt transition. It was
logical to consider the point of intersection of these lines (urhich occurred a t pressure between 10 and 30 p ) as the datum point and t h a t represented the zero point for adsorbed
waterz1 [see B and G, Figure 5(c)]. A surface
area calculation by the B.E.T. metllod using this datum point for zero adsorbed water yielded a value of 80.6 m2/g. By using the modi- ficd form of the Gibbs adsorption equation and
the method described above for System 4, i t
mas possible to calculate the position of the point representing zero adsorbed water. This point coincided with the experimental datum point and gave a value for surface area by B.E.T. method of 73.1 m2/g. This close agree- ment added support to the idea that the volume change and sorption characteristics of the hydrated cement could be described by the Gibbs adsorption and Bangham equations.
On desorption from the saturated or near- saturated state most materials show a primary hysteresis region xvhere large contractions are observed (mainly due to contractive force of the menisci) and followed by a region of no
change as shown by EF in Figure 5(c). This
effect was clearly demonstrated in System 2.
Here, for the adsorption part of the loop, the length change isotherm shoxi-ed a flattening of
the curve a t about 10 per cent humidity and a
contraction following this. This effect was also exhibited in the desorption part of the cycle where first a n expansion folio\\-ed by a contrac- tion was observed. This is one of the very few examples of expansion followed by contrac- tion during the adsorption part of the cycle. The dl/!-AP plot (Fig. 6) sho\vs clearly where
menisci formation had occurred. It is signifi-
cant that this primary hysteresis region in Systenls 2 and 5 extended do\vn t o approxi- mately 30 per cent humidity and confirined the presence of menisci in t h a t low humidity region. The dcsorption isotherm in the primary liysteresis region (System 5) \illen used for the calculation of surfacc area as described above yields a value of 101 ~ n ~ / g m , nhich agrees fairly well with the other methods of calcula- tion.
212 I N T E R A C T I O N OF H O I S T U R E AhrD M A T E R I A L S CONCLUSION
It has been sho~\-n hen. measurement of
sorption isothemls and the associated length change isotherms not only gix-e illfornlation regarding the state of the water held in a rigid porous body b u t also reflect sonle of t h e ph-aical and chemical properties of t h e material and the geometry of its pore system. Thus the term moisture content of a sample may represent water in different states because of the different physical and chenlical inter- actions with the material and also because of the geometric collfiguration of the space i t m a y occupy. The chemical and pllr-sical interaction results in certain basic relationships of volume changes associated wit11 the moisture content changes in the region of d r y t o partial saturation. These relationships have been discussed. I n the region of higher humidity, in n-hich primary hysteresis occurs, the relation- ship of moisture content to volume change is m a i n l - influenced by the geometry of tlle pore sj-stem; and contraction due t o menisci forces occurs.
I t is commonly assumed t h a t moisture content refers t o a definite and implicitly defined quantity of moisture present in a material. This is not necessarily the case. Much careful study may be necessary i n order t o be able t o define moisture c o n t e ~ l t usefully for a n y given purpose. or t o interpret the results obtnined from a particular method of measure- ment. K h e n it is desired t o study effects such as dimensional change, hysteresis and hydra- tion. elaborate nletllods including those used in this xtork may be required in order t o be able t o differentiate betxeen amounts of water held in different ways.
References
1. Polanyi, >I., "Adsorption von Gasen (Dunpfen dureh ein Pastes nicht Pluchtiges Adsorbens),"
I'erhundl. Deut. P h y s i k , 16, S>60 (1916). 2. Langmuir, I., "Chemical Reactions a t Low
Pressures," J. B 7 n . Cheri. Soc., 37, 1139 (1915).
3. Lan,puir, I., "The Adsorption of Gases on Plane Surfaces of Giass, Mica, and Platinum,"
J . A m . Cttcm. Soc., 4 0 , 1361 (1916).
4. Zsigmr~ndy, I?., "Structure of Gelatinous Silicic Acid. Theory of Dehydration," 2. Anorg. Crhem., 71, 356 ( 1 9 1 1 ) .
5 . de Boer, J. H., and Zwikker, C., "Adsorption as a Result of Polarization. The Adsorption Iso- therm," 2. Phyaik. C h m . B - 3 , 407 (1929).
6. Bradley, R. S., "Pol~molecular Adsorbed Films,"
J . Chetn. Soc., 139, 1467-1474 (1936). 7. Brunauer, S., Emmet, P. H., and Teller, W. E.,
"Adsorption of Gases in Multi-molecular Layers," J. rim. Chem. Soc., 60, 309 (1936). 8. Cohan, L. >I., "Sorption Hysteresis and the
Vapor Pressure of Concave Surfaces," J. A m .
Clienl. Soc., 60, 443 (1938).
9. Coelingh, 11. B., "Optical Invest,igations of the Liquid-vapor Equilibriuin in Capillary Sys- tems," Kolloid Z., 87, 251 (1939).
10. Amberg, C. H., and RIcIntosh, R., "A Study of Adsorption Hysteresis by Means of Length Changes of a Rod of Porous Glass," C a n . J.
Cllon., 30, 1012 (1952).
11. Icistler, S. S., Fischer, E. A., a.nd Freeman, I. R., "Sorption and Surface Area in Silica Aerogel,"
Am. Chem. Soc., 65, 1909 (1943).
12. Barrett, E. P., J o p e r , L. G., and Halenda, P. P., "The Determination of Pore Volume and Area Distribution in Porous Substances. I Computa- tions froin Nitrogen Isotherms," J. A m . Chem.
Soc., 73, 373 (1951).
13. Pierce, C., "Comput'ation of Pore Size from Physical Adsorption Data," J. Phys. Chsm.,
57, 149-52 (1953).
14. 3lcBain, J . W., "The Sorption of Gases and Vapors by Solids," p. 148, London, Rutledge,
1932.
16. NcBain, J . IY., Porter, J. L., and Sessions, R. F., "The Nature of the Sorption of Water by Charcoal," J. d m . Chem. Soc., 55, 2294 (1933). 16. Haines, R. S., and 3lacIntosh, R., "Length
Changes of Activated Carbon Rods Caused by Adsorption of Vapours," J. Chena. Phys., 15, 26 (1947).
17. Wiig, E . 0.: and Juhola, A. J., "The Adsorption of Water Vapour on -Activated Charcoal," J.
A m . C h o n . Soc., 71, 861-568 (February 1949). 18. Bangham, D. H., Fal~houry, N., and Mohamed, A. F., "The Swelling of Charcoal. I1 Some Factors Controlling the Espansion Caused by Water Benzene and Pyridine Vapors," PTOC.
R o y . Soc., Ser. A , 138, 162 (1932).
19. Bangha~n, D. H., and liazouk, R . I., "Adsorption and the Wettability of Solid Surfaces," Trans.
Faraday Soc., 33, 1463 (1937).
20. F'reundlich, H., "Colloid and Capillary Chemistry," p. 46, London, Rlethuen, 1926. 21. Bond, R . L., Griffith, M., and Maggs, F. A. P.,
"Some Properties of Water Adsorbed in the Capillary Structure of Coal," Discussions
Faraduy S o c . , 3, 29 (1946).
22. Bangham, D. H., "Some Physical Aspects of Coal and Coke Structure. Proceedings of a Con- ference on the Ultrafine Structure of Coals and Cokes," p. 18, British Con1 Utilization Research Association, 1944.
23. Bangham, D. H., and AIaggs, F. -4. P., "The Strength and Elastic Constants of Coals in Relation to Their Ultrafine Structure," Pro- ceedings of a Conference on the Ultrafine Structure of Coals and Colres, p. 118, British Coal Utilization Research Association, 1944.
28. MOISTURE C O N T E N T I N A POROGS B O D Y 213
24. Bangham, D. E., "The Swelling and Shrinkage of Porous Materials and the Role of Surface Forccs in Determining Technical Strength," Sym- posium organized by t h e Society of The Chemical Industry in conjunction with tho hllnistry of Works, London, May 8, 1946. 25. Sereda, P. J., and Feldman, R . F., "Compacts of
Powdered Materials as Porous Bodies for Use in Sorption Studies," J. A p p l . Chern., 13, 150 (1963).
26. Feldman, R . F., and Soreda, P. J., "The Use of Compacts to Study the Sorption Characteristics of Powdered Plaster of Paris," J. A p p l . Chem.,
13, 158 (1963).
27. Feldman, R. F., and Sereda, P. J., "A Datum Point for Estimating t h e Adsorbed Water in
Hydrated Portland Cement," J. A p p l . Chem.,
13, 375 (1903).
28. Feldman, R. F., and Seredn, P . J., "The Sorption of JVater on Compacts of Bottle-hytlratcd Cement. I The Sorption and length Change Isotherms," J. Appl. Chem.. 1 4 , 87-93 (19641. 29. Feldman, R . F., and Scrcda, P . J., "The Sorption
of Water on Compacts of Bottle-hydrated Cement. I1 Thermodynamic Considerations and Theory of V o l u n ~ e Change," J. A p p l .
Chem., 1 4 , 93-104 ( 1 9 6 4 ) .
30. Lakhanpal, M. L., and Flood, E. A., "Stresses and Strains in Adsorbate-adsorbent Systems. I V Contractions of Activcltcd Carbon on Adsorp- tion of Gases and 'iTapours of Low Initiai Pressures," Can. J. Chem., 35, 887 (1957j.