A Rapid and Inexpensive Method for the Purification of DNA from Lichens and their Symbionts
8
0
0
Texte intégral
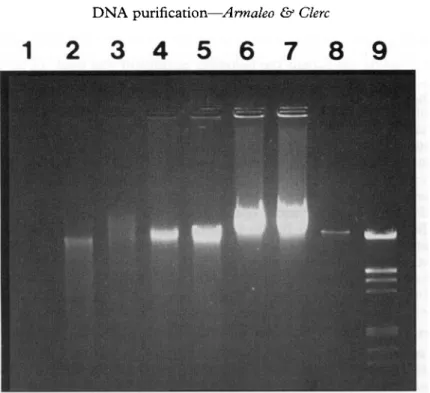
Figure
Documents relatifs