1
The C. elegans class A synthetic multivulva genes inhibit ectopic Ras-mediated vulval development by tightly restricting expression of lin-3 EGF
by
Adam M. Saffer
B.S., Biology and Biochemistry Brandeis University, 2002
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY FEBRUARY 2011
© 2011 Adam M. Saffer. All rights reserved.
The author hereby grants to MIT permission to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole or in part in any medium
now known or hereafter created.
Signature of Author:______________________________________________________ Department of Biology Certified by_____________________________________________________________ H. Robert Horvitz Professor of Biology Thesis Supervisor Accepted by:___________________________________________________________ Stephen P. Bell Professor of Biology Chairman of the Graduate Committee
The C. elegans class A synthetic multivulva genes inhibit ectopic Ras-mediated vulval development by tightly restricting expression of lin-3 EGF
by
Adam M. Saffer
Submitted to the Department of Biology in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
Abstract
The class A and B synthetic multivulva (synMuv) genes of C. elegans
redundantly antagonize an EGF/Ras pathway to prevent ectopic vulval induction. The class B synMuv genes encode many proteins known to remodel chromatin and repress transcription. The class A synMuv genes likely also function in transcription, although their specific molecular functions are unknown.
We have identified a class A synMuv mutation in the promoter of lin-3 EGF, revealing that lin-3 is the key biological target of the class A synMuv genes in vulval development. Using FISH with single mRNA molecule resolution, we found that class AB synMuv double mutants exhibit widespread ectopic lin-3 expression. Our results show that lin-3 EGF is normally expressed in the germline, and many class B synMuv genes have previously been implicated in inhibiting germline fates in somatic cells. We propose that the class A synMuv genes specifically repress ectopic lin-3 EGF
expression through a site in the lin-3 promoter and the class B synMuv genes either directly or indirectly repress lin-3 as a consequence of their role in regulating the germline/soma distinction.
The class A and B synMuv genes had previously been thought of as two parallel pathways, but we have found that each of those pathways is actually composed of multiple parallel pathways. While class AB synMuv double mutants have a strong Muv phenotype, most class AA synMuv double mutants exhibit a weak Muv phenotype, and most pairs of class B synMuv mutants can enhance each other in sensitized
backgrounds, indicating that most genes within a class can function in parallel. We also found that some pairs of synMuv genes cannot act in parallel, indicating that they function together to repress ectopic lin-3 expression.
We also report the molecular characterization of the class A synMuv gene lin-38 and the identification of mcd-1 as a class A synMuv gene. lin-38 and mcd-1 encode paralogous zinc-finger proteins. Unlike previously studied class A synMuv genes that function specifically in vulval development by repressing lin-3, both lin-38 and mcd-1 control multiple aspects of development by regulating target genes other than lin-3.
Thesis Supervisor: H. Robert Horvitz Title: Professor of Biology
3
Acknowledgments
First, I would like to thank my advisor, Bob Horvitz, for giving me the independence to pursue my interests, while also providing advice and encouragement when needed. I would also like to thank my committee members, Chris Kaiser, Richard Hynes, Dennis Kim, Peter Reddien, and Susan Mango.
Iʼd like to thank everyone I have overlapped with in the Horvitz lab (I will not name them individually for fear of accidentally omitting someone). Their advice and assistance over the years has been invaluable. I had the pleasure of working with Erik Andersen and Melissa Harrison in our quest to discover everything there is to know about the C.
elegans vulva. My experience in graduate school would not have been the same
without my baymate Niels Ringstad. His helpful advice and enthusiastic encouragement easily outweighed the inconvenience of losing a few pens a week. I was especially fortunate to have Dave Harris and Erik Andersen as friends and colleagues.
All of the people who accompanied me on the daily trips to Annaʼs, including Melissa Harrison, Johanna Varner, Dan Omura, Dan Denning, Nick Paquin, Mike Hurwitz, and of course Dave Harris, made lunchtimes very enjoyable. I also thank Dan Omura and everyone else I played squash with.
I am grateful for the friendship of many members of Biograd2002, and especially my longtime roommates Rami Rahal, Mauro Calabrese, Lucas Dennis, and Mark Gill. Their friendship made graduate school much more enjoyable.
Finally, I want to thank my parents, Jeff and Paula, and my sister Amy, for their support and all that they have taught me.
Table of Contents
Abstract ...2
Acknowledgments ...3
Table of Contents ...4
Chapter One: Introduction...9
The mammalian EGF signaling pathway...10
EGF signaling in mammalian development ...12
EGF signaling in cancer ...15
C. elegans vulval development...16
EGF/Ras signaling induces vulval fates ...17
Wnt and Notch signaling are also required for normal vulval development...20
Additional roles of EGF signaling in C. elegans ...21
The synMuv genes antagonize EGF signaling ...24
The class A synMuv genes encode putative transcription factors...25
The class B synMuv genes encode transcriptional repressors ...25
synMuv protein complexes ...27
The site of action of the synMuv genes ...28
Relationship between EGF signaling and synMuv genes ...29
The synMuv genes have numerous functions ...29
Conclusion...32
Acknowledgments ...32
Chapter Two: The C. elegans synthetic multivulva genes prevent Ras pathway activation by tightly repressing ectopic expression of lin-3 EGF...33
Summary ...34
Introduction...35
Materials and Methods ...38
Results...40
n4441 causes a dominant class A synMuv phenotype...40
n4441 is an allele of lin-3 ...41
lin-3(n4441) specifically prevents repression of lin-3 ...43
Expression pattern of lin-3 in wild-type animals and synMuv mutants ...44
Discussion ...47
lin-3 is the major target of the class A synMuv genes in vulval development ...47
The class A and B synMuv genes repress lin-3 by two distinct mechanisms...48
The synMuv genes repress lin-3 throughout the animal ...49
Conclusion ...50
Acknowledgments ...51
Table 1: lin-3(n4441) causes a dominant class A synMuv phenotype ...52
Table 2: lin-3 overexpression is enhanced more strongly by a class A synMuv mutation than by a class B synMuv mutation ...53
Figure 1: n4441 is an allele of lin-3...54
Figure 2: The lin-3(n4441) mutation specifically prevents repression of lin-3 ...56 Figure 3: The synMuv genes prevent widespread ectopic expression of lin-3 mRNA. .58
5
Figure 4: The lin-3(n4441) mutation causes widespread ectopic expression of
lin-3 mRNA. ...60
Supplemental Table 1: Oligonucleotides in the lin-3 in situ hybridization probe...62
Chapter Three: Multiple levels of redundant processes inhibit C. elegans vulval cell fates...63
Summary ...64
Introduction...65
Materials and Methods ...68
Results...70
Temperature sensitizes the vulval phenotype...70
Partial loss-of-function mutations in class A or class B synMuv genes can sensitize the vulval phenotype ...71
Most class A-A double mutants have Muv phenotypes ...71
lin-15A and lin-56 act in the same process to inhibit the specification of vulval cell fates...72
Most class B-B double mutants do not have Muv phenotypes ...73
Most class B synMuv genes act redundantly with genes of the same class to inhibit vulval cell fates ...74
Some synMuv genes within the same class function non-redundantly to inhibit vulval cell fates...75
The penetrances of synMuv vulval phenotypes correlate with the level of lin-3 mRNA expression ...77
Discussion ...79
Genetic enhancement tests can distinguish processes that act in series or in parallel...79
The synMuv genes define two distinct classes ...80
Lack of genetic enhancement can identify proteins that function in a complex or a process ...81
Genetic enhancement tests can identify functionally related groups of evolutionarily conserved but uncharacterized genes ...82
Acknowledgments ...83
Table 1: synMuv alleles used in this study. ...84
Table 2: Most class A synMuv double mutants have multivulva phenotypes...85
Table 3: Mutations in most class A synMuv genes enhance the Muv phenotypes caused by mutations in other class A synMuv genes in a class B synMuv mutant background...86
Table 4: Class B synMuv single and class B-B double mutants do not have appreciable Muv phenotypes...87
Table 5: Mutations of most class B and C genes enhance the Muv phenotypes caused by mutations in other class B genes in a class A synMuv mutant background...89
Table 6: Mutations in most class B synMuv genes enhance the Muv phenotypes caused by mutations of other class B synMuv genes in a class A synMuv mutant background...91
Figure 1: Regulation of lin-3 transcription by the synMuv genes...93
Figure 2: Model: The synMuv classes function in two redundant pathways or processes, each composed of separate molecular pathways or processes, to mediate the inhibition of vulval cell fates. ...95
Supplemental Figure 1: Correlation between lin-3 overexpression and the synMuv phenotype ...97
Chapter Four: lin-38 and mcd-1 antagonize the class A synMuv pathway to promote C. elegans vulval development ...98
Summary ...99
Introduction...100
Materials and Methods ...103
Results...108
lin-38 encodes a zinc-finger protein ...108
Characterization of lin-38 class A synMuv alleles...109
Loss of lin-38 causes larval lethality and suppresses the class A synMuv phenotype of lin-38(n751) ...110
Some mcd-1 mutations cause a class A synMuv phenotype...112
mcd-1 and lin-38 class A synMuv alleles exhibit intergenic non-complementation ..113
mcd-1 encodes two distinct transcripts ...114
A subset of mcd-1 mutations cause cell-death and synthetic lethality defects ...116
mcd-1 null phenotype...117
Discussion ...119
LIN-38 and MCD-1 are putative transcription factors...119
lin-38 is required for viability and likely functions as a synMuv suppressor ...120
mcd-1 has two opposing functions in vulval development ...121
mcd-1 controls multiple distinct aspects of development...122
Conclusions ...123
Acknowledgments ...123
Table 1: lin-38 class A synMuv mutations ...124
Table 2: RNAi of lin-38 causes lethality...125
Table 3: Only some mcd-1 mutations cause a class A synMuv phenotype ...126
Table 4: Non-complementation between lin-38 and mcd-1 alleles...128
Table 5: Recombination between lin-38 and mcd-1 alleles ...129
Figure 1: Y48E1B.7 is lin-38...130
Figure 2: LIN-38 is a zinc-finger protein ...132
Figure 3: LIN-38 and MCD-1 are paralogs ...134
Figure 4: mcd-1 gene structure ...136
Figure 5: mcd-1(n4418) does not block programmed cell death ...138
Figure 6: Loss of mcd-1 suppresses the synMuv phenotype ...140
Figure 7: Summary of mcd-1 defects and model for mcd-1 function ...142
Chapter Five: Two putative C. elegans transcription factors, LIN-15A and LIN-56, interact and function redundantly with an Rb pathway to regulate vulval development...144
7
Introduction...146
Materials and Methods ...149
Results...153
lin-56 encodes a putative transcription factor containing a THAP-like domain ...153
LIN-56 is a ubiquitous nuclear protein ...155
LIN-56 protein, but not lin-56 mRNA, is reduced in lin-15A(lf) mutants ...156
LIN-15A protein, but not lin-15A RNA, is reduced in lin-56(lf) mutants ...157
Overexpression of lin-56 does not rescue the lin-15AB(lf) synMuv phenotype, nor does overexpression of lin-15A rescue the lin-56(lf) synMuv phenotype ...157
LIN-56 and LIN-15A physically interact...158
Discussion ...160
LIN-56 and LIN-15A might function as a complex in vivo ...160
The class A synMuv genes likely directly regulate gene expression ...160
Class A synMuv genes might have functions beyond those in vulval development .162 Implications for mammalian tumorigenesis...162
Acknowledgments ...163
Table 1: lin-56 overexpression rescues the lin-56(lf); lin-15B(lf) but not the lin-15AB(lf) synMuv phenotype...164
Table 2: lin-15A overexpression rescues the lin-36(lf); lin-15A(lf) but not the lin-56(lf); lin-36(lf) synMuv phenotype. ...165
Figure 1: Cloning of lin-56. ...166
Figure 2: LIN-56 is broadly expressed and localized to nuclei. ...169
Figure 3: LIN-56 protein but not lin-56 mRNA levels are greatly reduced in lin-15A(lf) mutants. ...173
Figure 4: LIN-15A protein but not lin-15A RNA levels are reduced in lin-56(lf) mutants...176
Figure 5: LIN-15A and LIN-56 interact with each other in the yeast two-hybrid system. ...179
Supplemental Figure 1: Expression of LIN-56 driven by heat-shock promoters ...181
Chapter Six: Future Directions ...182
Where is lin-3 ectopically expressed in each synMuv mutant? ...183
Do any synMuv suppressors promote germline lin-3 expression? ...184
Why is the synMuv phenotype temperature-sensitive? ...185
What transcriptional targets are responsible for the lin-38(null) lethality? ...186
What are the cis-acting synMuv elements in the lin-3 promoter?...187
What proteins physically interact with class A synMuv proteins?...188
What are the molecular functions of the class A synMuv proteins? ...189
What proteins are present at the lin-3 promoter? ...190
Concluding remarks...192
Acknowledgments ...193
Appendix One: Identification of new class A synMuv mutations...194
Materials and Methods ...195
Results...196
Table 2: Class A synMuv screen isolates...199
Appendix Two: The class A synMuv genes lin-15A and lin-56 function in multiple tissues ...200
Introduction...201
Materials and Methods ...203
Results...204
Activity of the dpy-7 and lin-31 promoters...204
lin-15A and lin-56 might function in the Pn.p cells, hyp7, or both ...205
Discussion ...206
Acknowledgments ...207
Table 1: Expression of let-60 cDNA under the control of either dpy-7p or lin-31p rescues the vulvaless phenotype of let-60(n1876) ...208
Table 2: Expression of lin-15A under the control of either dpy-7p or lin-31p rescues the synMuv phenotype...209
Table 3: Expression of lin-56 under the control of either dpy-7p or lin-31p rescues the synMuv phenotype...210
Appendix Three: Progress towards identifying proteins that bind to the lin-3 promoter ...211
Introduction...212
Materials and Methods ...214
Results...218
The effect of the lin-3(4441) mutation on vulval development is not replicated with a repetitive extrachromosomal array ...218
EMSA experiments detect a sequence-specific, lin-3-promoter-binding protein ...219
Affinity purification of lin-3p binding proteins...221
EMSA experiments to test direct interactions between the lin-3 promoter and class A synMuv proteins ...223
Discussion ...224
Acknowledgments ...227
Table 1: lin-3(+) and lin-3(n4441) affect vulval development similarly when present on a repetitive extrachromosomal array ...228
Table 2: Deletion of F13E9.13 does not cause or suppress the synMuv phenotype ...229
Figure 1: Identification of a sequence-specific lin-3p-binding protein...230
Figure 2: Purification of sequence-specific lin-3p binding protein. ...232
Figure 3: EMSA with in vitro transcribed and translated class A synMuv proteins...234
9 Chapter One
Introduction
The Caenorhabditis elegans vulva is an excellent system for studying signal transduction, cell-fate determination, and the role of cell-cell interactions in spatial patterning and development. C. elegans vulval development can be divided into three phases: First, a set of precursor cells with equivalent developmental potential are generated (Kimble, 1981; Sulston and Horvitz, 1977; Sulston and White, 1980). Second, cell-cell interactions specify a subset of those precursor cells to adopt vulval cell fates (Kimble, 1981; Sulston and White, 1980). Finally, the vulval cells divide and their descendants undergo morphogenesis to form the vulva (Sulston and Horvitz,
1977). Extensive screens and genetic analysis have identified a large number of genes that control the different phases of vulval development. Despite containing only 22 nuclei, proper development of the vulva requires a Hox gene, an epidermal growth factor (EGF) and Ras pathway, Notch signaling, and Wnt signaling (Sternberg, 2005). Studies of the C. elegans vulva have also produced insights into cancer biology,
because carcinogenesis involves the misregulation of normal developmental processes. Homologs of many genes required for proper vulval cell-fate specification play a role in cancer, including both oncogenes and tumor suppressor genes. For example, studies of the C. elegans vulva helped elucidate the EGF/Ras signaling pathway (Sternberg, 2006), which is a frequent target of mutations in human cancers (Bos, 1989; Normanno et al., 2006). Because overactivation of the EGF/Ras pathway can promote cancerous growth, it is important that mechanisms exist to negatively regulate EGF/Ras signaling. Therefore, my investigations have focused on the negative regulation of EGF signaling in C. elegans by the synthetic multivulva (synMuv) genes.
The mammalian EGF signaling pathway
In mammals, signaling by EGF-like ligands controls many aspects of
development and cell-proliferation. EGF is the founding member of the EGF-like family of peptide growth factors (Cohen, 1962; Cohen and Elliott, 1963). Many other EGF-like growth factors have since been identified, including transforming growth factor ! (TGF-!), amphiregulin, heparin-binding EGF (HB-EGF), betacellulin, epiregulin, epigen, and the neuregulins (Busfield et al., 1997; Carraway et al., 1997; Chang et al., 1997;
11
Derynck et al., 1984; Higashiyama et al., 1992; Holmes et al., 1992; Lee et al., 1985; Riese et al., 1995; Shing et al., 1993; Shoyab et al., 1989; Strachan et al., 2001; Toyoda et al., 1995). Each EGF-like growth factor has one or more EGF domains, a motif of approximately 40 amino acids containing six cysteines with stereotypical spacing (Schneider and Wolf, 2009). EGF-like growth factors are synthesized as large
transmembrane proteins (Derynck et al., 1984; Gray et al., 1983; Higashiyama et al., 1992), and the membrane-bound growth factors can signal to adjacent cells in a
process known as juxtacrine signaling (Harris et al., 2003). The extracellular domains of EGF-like growth factors are also released by proteolytic cleavage, allowing them to signal to the cells that released them (autocrine signaling), or to diffuse and signal to neighboring or more distant cells (paracrine signaling) (Singh and Harris, 2005).
Regulation of proteolytic cleavage of EGF-like ligands is necessary for proper signaling. For example, mice in which HB-EGF is replaced by an uncleavable HB-EGF show heart defects similar to loss of HB-EGF, while replacement of HB-EGF with a constitutively soluble form of HB-EGF causes severe hyperplasia defects, perhaps as a result of excessive HB-EGF signaling (Yamazaki et al., 2003).
EGF binds to the EGF receptor (EGFR), also called ErbB1 or HER1. There are three other related receptors, ErbB2/Neu/HER2, ErbB3/HER3, and ErbB4/HER4. Hereafter I refer to this receptor family as the ErbB receptors. All ErbB receptors are transmembrane receptor tyrosine kinases (RTKs) with an extracellular ligand-binding domain and a cytoplasmic protein tyrosine kinase domain. The binding of EGF-like ligands to ErbB receptors leads to either homodimerization or heterodimerization of the receptors, and possibly to oligomerization of larger numbers of receptor molecules (Schlessinger, 2000). Some ligands, such as EGF, TGF-!, and amphiregulin bind specifically to EGFR, while other ligands, including HB-EGF, bind to either EGFR or ErbB4, and the neuregulin ligands bind to ErbB3 and ErbB4 (Carraway et al., 1994; Carraway et al., 1997; Olayioye et al., 2000). There is no known ligand for ErbB2, but ErbB2 is the preferred heterodimerization partner for the other ErbB family members, underscoring the importance of heterodimerization (Graus-Porta et al., 1997; Tzahar et al., 1996). Furthermore, at least eight of the ten possible homodimers and
heterodimers of ErbB receptors can be formed in response to ligand binding (Tzahar et al., 1996).
Upon binding of an EGF-like ligand to an ErbB receptor, the receptor dimerizes, activating its protein tyrosine kinase activity and autophosphorylating the receptor dimer. The phosphorylated tyrosine residues serve as docking sites for proteins with Src
homology 2 (SH2) and phosphotyrosine binding (PTB) domains, leading to the
activation of various signal transduction pathways (Schlessinger, 2000). For example, phosphorylated ErbB receptors activate phosolipase C gamma (PLC"), which in turn generates the second messengers inositol triphosphate (IP3) and diacylglylcerol (DAG) (Rhee and Bae, 1997). Perhaps the best known pathway downstream of EGF signaling is the Ras and mitogen activated protein kinase (MAPK) signaling cascade. Ras is a small guanine nucleotide binding protein that is active when bound to GTP, but is inactive when bound to GDP. Ras possesses intrinsic GTPase activity that converts GTP to GDP, inactivating the Ras protein (Sweet et al., 1984). Upon EGF receptor activation, the Grb2 adaptor protein binds to the phosphorylated receptor and recruits the guanine nucleotide exchange factor SOS, which switches Ras proteins from an inactive GDP-bound state to an active GTP-bound state (Buday and Downward, 1993; Chardin et al., 1993; Egan et al., 1993; Gale et al., 1993; Li et al., 1993; Olivier et al., 1993; Rozakis-Adcock et al., 1993; Simon et al., 1993). Activated Ras can
subsequently activate multiple downstream targets, including the protein kinase Raf (Vojtek et al., 1993; Warne et al., 1993; Zhang et al., 1993). Raf phosphorylates MEK (MAPK/Erk kinase), which phosphorylates MAPKs, which then translocate to the
nucleus and phosphorylate targets including transcription factors. Ras/MAPK signaling controls many processes and is best known for promoting cell proliferation.
EGF signaling in mammalian development
EGF and most EGF-like ligands were identified by their ability to promote growth and proliferation in cell culture or in vivo, and mutant mice lacking various ErbB
receptors indicate that EGF signaling has many roles in normal development. The different ErbB receptor knockouts have a combination of overlapping and distinct
13
defects, as might be expected given that the receptors function in multiple homo- and hetero-dimeric combinations. Mice lacking EGFR grow slowly and exhibit numerous developmental defects (Miettinen et al., 1995; Sibilia and Wagner, 1995; Threadgill et al., 1995). The mice die at various stages depending on the background and exhibit abnormal epithelial development, including thin skin and hair growth defects. Targeted expression of a dominant negative EGFR, as well as transplantation experiments, also implicate EGFR in mammary duct development (Wiesen et al., 1999; Xie et al., 1997). Mutant mice lacking ErbB2 die as a result of cardiac defects, and animals in which the cardiac defect is rescued show that ErbB2 also promotes the proliferation and survival of sensory neurons, motor neurons, and Schwann cells (Lee et al., 1995; Morris et al., 1999). ErbB3 knockout mice have abnormal development of neurons, heart, and other organs (Erickson et al., 1997; Riethmacher et al., 1997). Mice lacking ErbB4 exhibit lethal cardiac development defects and axon guidance defects, and a neuron-specific ErbB4 knockout has some behavioral abnormalities (Gassmann et al., 1995; Golub et al., 2004). Overall, the ErbB family knockouts implicate EGF signaling in promoting proliferation and survival of many cell-types in vivo.
Mutant mice lacking EGF-like ligands exhibit far fewer abnormalities than
receptor mutants, suggesting that there is substantial redundancy amongst the EGF-like ligands. The only known essential EGF-like ligands are HB-EGF and neuregulin-1. Mice lacking neuregulin-1 die during embryogenesis with heart defects and the absence of Schwann cell precursors and some neurons (Meyer and Birchmeier, 1995). Most mice without HB-EGF also die during embryogenesis and exhibit heart defects (Iwamoto et al., 2003; Jackson et al., 2003). Mutant mice lacking TGF-! are fertile and healthy, although they do have wavy hair and whiskers as a result of disorganized hair follicles as well as slight defects in neuronal proliferation (Blum, 1998; Luetteke et al., 1993; Mann et al., 1993; Tropepe et al., 1997). EGF, betacellulin, and amphiregulin knockouts are viable and fertile, although amphiregulin mutants have mammary duct development defects and betacellulin mutants enhance the phenotype of HB-EGF mutants (Jackson et al., 2003; Luetteke et al., 1999). Furthermore, a triple mutant between EGF, TGF-!, and amphiregulin is slow-growing but viable (Luetteke et al., 1999; Troyer et al., 2001).
Many of the phenotypes caused by loss of EGF-like ligands are similar to defects
caused by ErbB receptors, as might be expected. The relatively mild defects caused by loss of most EGF-like ligands suggests that multiple EGF-like ligands have overlapping roles in vivo.
Because EGF-like ligands often act over short distances, EGF signaling can be controlled by limiting the spatial and temporal expression of EGF-like ligands. Some EGF-like ligands are widely expressed, while others are expressed in more restricted patterns. For example, neuregulin-1 is expressed in many mesenchymal and neuronal cells, neuregulin-2 is expressed specifically in the nervous system, and neuregulin-3 is expressed highly only in the pancreas and muscle (Harari et al., 1999; Meyer and Birchmeier, 1995; Zhang et al., 1997). Epiregulin is primarily expressed specifically in the placenta and in certain macrophages (Toyoda et al., 1997). Perhaps the most striking example of restricted EGF-like ligand expression is HB-EGF expression in the uterus: HB-EGF is expressed 6-7 hours before implantation precisely at the site where the blastocyst attaches (Das et al., 1994).
Consistent with the restricted expression of many EGF-like ligands,
overexpression, ectopic expression, or exogenous application of EGF-like ligands can disrupt normal development. For example, EGF was first identified based on its ability to cause premature eyelid opening and incisor eruption in newborn mice when
exogenously applied (Cohen, 1962). In contrast to the relatively normal phenotype of mice lacking TGF-!, TGF-! overexpression causes substantial defects, including hyperplasia in the epithelia of many organs and excessive proliferation of multiple cell types in the pancreas (Sandgren et al., 1990). Epigen-overexpressing mice have enlarged sebaceous glands, and transgenic mice expressing amphiregulin exhibit a psoriasis-like phenotype, perhaps both reflecting overproliferation (Cook et al., 1997; Dahlhoff et al., 2010). Furthermore, while betacellulin knockout mice are relatively normal, mice overexpressing betacellulin have alterations in skull shape, multiple
defects in eye development, and severe lung abnormalities that lead to reduced lifespan (Schneider et al., 2005). Taken together, these experiments show that controlled
15 EGF signaling in cancer
Many pathways that control development are co-opted by tumor cells, and EGF signaling is a frequent target of misregulation in cancers. Normal cells depend on continuously receiving growth signals such as EGF-like peptides to proliferate. By contrast, tumor cells develop independence from external growth factor signals, allowing them to proliferate excessively. One mechanism by which cancer cells can overcome their dependence on external growth factor signals is by producing those growth factors themselves and promoting their own growth via autocrine signaling. TGF-! is
expressed in many cell lines derived from tumors and was initially identified as a
substance secreted by tumor cells that was capable of transforming other cells in culture (Todaro et al., 1980). In vivo, overexpression of TGF-! causes carcinomas (Sandgren et al., 1990).
ErbB receptors are also misregulated in cancer cells. Mutations can allow ErbB receptors to signal independently of ligand binding, or the receptors can be amplified so as to respond more strongly to existing ligand concentrations (Sharma et al., 2007). As a result, drugs that target EGFR are in clinical use. These include anti-EGFR
monoclonal antibodies that compete with EGF ligands for binding sites and small molecule inhibitors that compete with ATP-binding and thereby inhibit ErbB receptor tyrosine kinase activity (Ciardiello and Tortora, 2008). Tumor cells can also acquire mutations in signaling molecules that transduce the growth signals downstream of ErbB receptors, causing constitutively active signaling. For example, approximately 20% of human cancers have an activating mutation in a Ras gene (Bos, 1989).
Because inappropriate activation of EGF and Ras signaling can promote cancerous growth, genes that negatively regulate EGF/Ras signaling are likely
candidates to function as tumor suppressor genes. One such tumor suppressor is the neurofibromatosis type 1 (NF1) gene, which encodes a GTPase activating protein and negatively regulates Ras by promoting the intrinsic ability of Ras to inactivate itself by hydrolyzing GTP to GDP (Weiss et al., 1999). The NF1 disorder, which is inherited in a dominant fashion, is caused by a loss-of-function mutation of one allele of NF1. When
the second allele is compromised by loss of heterozygosity, malignant tumors appear, presumably as a result of EGF/Ras pathway overactivation (Weiss et al., 1999).
C. elegans vulval development
Much of the current knowledge of EGF signaling comes from studies of vulval development in C. elegans. The six cells P(3-8).p, also called Pn.p cells, are each innately capable of adopting either a vulval or a non-vulval cell fate. In wild-type animals, an inductive signal from the anchor cell of the somatic gonad causes three of the cells, P(5-7).p, to adopt vulval cell fates at the beginning of the third larval stage. P(5-7).p then divide several times and their 22 descendents form the vulva (Sulston and Horvitz, 1977). The other three cells, P(3,4,8).p, do not receive sufficient inductive signal to adopt vulval cell fates. Therefore, P(3,4,8).p adopt non-vulval cell fates, divide once, and their progeny fuse with the hypodermis.
A series of physical perturbations showed that Pn.p cells are each capable of adopting either vulval or non-vulval fates, and that the anchor cell induces vulval fates through a diffusible signal. If the anchor cell is removed by laser-ablating either the anchor cell precursors or the entire gonad, then no vulval induction occurs and all six Pn.p cells adopt non-vulval fates (Kimble, 1981; Sulston and White, 1980). Conversely, laser ablation of every cell in the gonad except the anchor cell does not prevent wild-type vulval development (Kimble, 1981). In wild-wild-type animals, the anchor cell is closest to P6.p, and P(5-7).p are induced to adopt vulval cell fates. In dig-1 mutants, the gonad, including the anchor cell, is frequently displaced. This leads to a corresponding shift in the specific Pn.p cells that are induced to adopt vulval cell fates. For example, when the anchor cell is closest to P5.p, P(4-6).p adopt vulval fates and P(3,7,8).p adopt non-vulval fates (Thomas et al., 1990). In wild-type animals, only P(5-7).p adopt non-vulval fates, but if P(5-7).p are ablated then P(3,4,8).p will adopt vulval fates, proving that each Pn.p cell is capable of adopting a vulval cell fate (Sulston and White, 1980). Ablation of the gonad and some Pn.p cells showed that the default state of Pn.p cells in the absence of these cell-cell interactions is a non-vulval fate (Sternberg and Horvitz, 1986). Taken together, these experiments prove that each Pn.p cell is capable of adopting either a
17
vulval or a non-vulval cell fate, and that a signal from the anchor cell induces the closest Pn.p cells to adopt vulval cell fates.
In wild-type animals, the anchor cell is located in close proximity to P6.p along the ventral side of the animal. In some dig-1 mutant animals, the gonad, and hence the anchor cell, is displaced to the dorsal side of the animal (Thomas et al., 1990). Despite being located up to 45 µm away from the nearest Pn.p cell, the anchor cell signal can still induce vulval cell fates. Therefore, the inductive signal from the anchor cell can act at a significant distance, and is likely to be a secreted and diffusible factor.
EGF/Ras signaling induces vulval fates
Many mutants with abnormal vulval development have been identified (e.g. Ferguson and Horvitz, 1985). Animals in which none of the six Pn.p cells adopt a vulval cell fate have a vulvaless (Vul) phenotype. Animals in which more than three cells adopt vulval cell fates have a multivulva (Muv) phenotype, with the extra vulval cells forming ectopic pseudovulvae on the ventral side of the animal. Studies of mutants with multivulva and vulvaless phenotypes led to the discovery of an EGF/Ras pathway that induces vulval cell fates. Loss-of-function mutations in components of the EGF/Ras pathway prevent vulval induction, causing a vulvaless phenotype (Aroian et al., 1990; Beitel et al., 1990; Han and Sternberg, 1990; Hill and Sternberg, 1992). Conversely, overactivation of the EGF/Ras pathway causes a multivulva phenotype (Beitel et al., 1990; Han and Sternberg, 1990; Hill and Sternberg, 1992; Katz et al., 1996). Studies of EGF/Ras signaling in C. elegans vulval development, along with studies of the
Drosophila melanogastar eye and mammalian cell culture, helped elucidate the
components of this pathway and provided genetic evidence for the order of action of the different components (Sternberg, 2006). C. elegans mutants with abnormal vulval development have allowed the study of core component of EGF/Ras signaling as well as multiple regulatory inputs.
lin-3 encodes a membrane-spanning protein with homology to EGF-like growth
factors, and mutations in lin-3 cause a vulvaless phenotype (Hill and Sternberg, 1992). Unlike mammals, which possess numerous partially redundant EGF-like ligands, lin-3 is
the sole C. elegans EGF-like ligand (www.wormbase.org WS190). lin-3 is expressed in the anchor cell, where it acts as the inductive signal that controls vulval development (Hill and Sternberg, 1992). There are both membrane-bound and secreted forms of LIN-3 (Dutt et al., 2004). During vulval development, the anchor cell is in close proximity to P6.p, so the membrane bound form of LIN-3 might be used. The anchor cell can also induce vulval development at a distance (Thomas et al., 1990), likely using secreted LIN-3.
let-23 encodes an EGF receptor subfamily receptor tyrosine kinase (Aroian et al.,
1990). LIN-3 EGF expressed from the anchor cell is thought to bind to LET-23 EGFR in the Pn.p cells, and mosaic analysis indicates that let-23 acts cell-autonomously in Pn.p cells to specify vulval fates (Koga and Ohshima, 1995; Simske and Kim, 1995).
Mutations in lin-2, lin-7, and lin-10 cause a vulvaless phenotype as a result of the subcellular mislocalization of LET-23. lin-2 encodes a membrane-associated guanylate kinase (MAGUK) protein, and lin-7 and lin-10 encode PDZ domain-containing proteins (Hoskins et al., 1996; Simske et al., 1996; Whitfield et al., 1999). LET-23 is specifically localized to the basolateral side of cell junctions in Pn.p cells in wild-type animals, but in
lin-2, lin-7, and lin-10 mutants LET-23 is broadly localized along the entire cell
membrane (Simske et al., 1996; Whitfield et al., 1999). LIN-2, LIN-7, LIN-10, and LET-23 form a complex required for proper LET-23 localization (Kaech et al., 1998; Simske et al., 1996), which is in turn required for LET-23 to receive the LIN-3 inductive signal from the anchor cell. Unlike other components of EGF signaling, lin-2, lin-7, and
lin-10 are involved only in vulval development, and null mutations do not exhibit other
pleiotropic defects exhibited by most components of EGF signaling (see below) (Ferguson and Horvitz, 1985; Hoskins et al., 1996).
EGF signaling in vulval development acts through a Ras/MAPK signaling cascade. Upon binding of LIN-3 to LET-23, LET-23 is likely to dimerize and
autophosphorylate. The phosphorylated LET-23 then binds to SEM-5, an adaptor protein with one SH2 and two SH3 domains (Clark et al., 1992a). sem-5 was originally studied in C. elegans for its role in the Ras pathway in vulval development and sex myoblast migration (Clark et al., 1992a), and the sem-5 homolog Grb2 was later
19
identified as a component of the EGF/Ras pathway in mammals (Lowenstein et al., 1992). The guanine nucleotide exchange factor SOS-1/LET-341 is recruited, and subsequently activates LET-60 Ras (Chang et al., 2000). Both sem-5 and sos-1 are required for normal vulval development, and genetic evidence suggests that sem-5 and
sos-1 act downstream of let-23 EGFR and upstream of let-60 Ras (Chang et al., 2000;
Clark et al., 1992a, b).
Unlike most genes in the C. elegans EGF/Ras pathway that were identified as Vul mutants, both Vul and Muv alleles of the Ras homolog let-60 were isolated in screens. Gain-of-function alleles or overexpression of let-60 cause a Muv phenotype, while loss-of-function alleles cause a Vul phenotype (Beitel et al., 1990; Ferguson and Horvitz, 1985; Han et al., 1990; Han and Sternberg, 1990). Many of the gain-of-function mutations in let-60 result in a glycine-to-glutamic acid change at codon 13 (Beitel et al., 1990). Mutations in that same codon of human Ras genes have been found in cancers (Bos et al., 1985).
Activation of LET-60 Ras initiates a kinase signaling cascade. The RAF family serine/threonine kinase LIN-45, the MEK serine/threonine and tyrosine kinase homolog MEK-2, and the MAP kinase homolog MPK-1/SUR-1 are all required for vulval
development and function downstream of LET-60 (Han et al., 1993; Kornfeld et al., 1995a; Lackner et al., 1994; Wu and Han, 1994; Wu et al., 1995). Biochemical evidence shows that MEK-2 functions between LIN-45 and MPK-1/SUR-1 in a kinase cascade (Wu et al., 1995). The KSR-1 and KSR-2 proteins are similar to Raf and are thought to function as scaffolds that promote Raf/MEK/MAPK signaling, and animals lacking both ksr-1 and ksr-2 have a phenotype similar to strong EGF/Ras pathway loss-of-function mutants (Kornfeld et al., 1995b; Ohmachi et al., 2002; Sundaram and Han, 1995).
Two transcription factors function directly downstream of the EGF/Ras pathway. The ETS-domain transcription factor LIN-1 negatively regulates vulval cell fates, and loss-of-function mutations of lin-1 cause a Muv phenotype (Beitel et al., 1995). The winged helix transcription factor LIN-31 both positively and negatively regulates vulval cell fates, and lin-31 loss-of-function leads to a mix of Muv and Vul phenotypes (Miller et
al., 1993). LIN-1 and LIN-31 form a complex, and their binding is disrupted by
phosphorylation of LIN-1 and LIN-31 by MPK-1/SUR-1 (Tan et al., 1998). In addition to the core elements of the Ras pathway described here, many additional positive and negative regulators promote or inhibit Ras signaling in C. elegans (Sundaram, 2006).
Wnt and Notch signaling are also required for normal vulval development Besides EGF/Ras signaling, multiple other signaling pathways are required to form a wild-type vulva. Wnt signaling prevents the Pn.p cells from prematurely fusing with the hypodermis, which would render them unable to receive and respond to the inductive signal from the anchor cell (Eisenmann and Kim, 2000; Eisenmann et al., 1998). Wnt signaling also promotes the adoption of vulval cell fates by Pn.p cells (Eisenmann and Kim, 2000; Gleason et al., 2002). The role of Wnt signaling in vulval development is somewhat obscured by the substantial redundancy in the Wnt pathway, as there are multiple Wnt ligands and receptors involved (Gleason et al., 2006).
In wild-type animals, P6.p adopts a primary vulval cell fate and produces eight descendants, and P5.p and P7.p adopt secondary vulval cell fates and produce seven descendants each (Sulston and Horvitz, 1977). Additional differences between the progeny produced by primary and secondary Pn.p cells can be distinguished by gene expression reporters (Inoue et al., 2002). The adoption of secondary vulval cell fates and the precise patterning of primary and secondary fates are regulated by a Notch pathway that signals between the Pn.p cells (Greenwald et al., 1983; Sternberg, 1988; Yochem et al., 1988). Patterning the vulva requires crosstalk between the EGF/Ras and Notch pathways to ensure that the correct cell adopts the primary vulval cell fate (Chen and Greenwald, 2004; Shaye and Greenwald, 2002; Yoo et al., 2004). In wild-type animals, primary vulval cell fates are specified by EGF/Ras signaling and secondary vulval cell fates are induced in P5.p and P7.p by a combination of Notch and EGF signaling (Katz et al., 1995). P6.p is closest to the anchor cell, so it receives the strongest inductive signal and adopts the primary fate. P6.p subsequently signals to the adjacent Pn.p cells to adopt secondary fates. In mosaic animals in which P6.p has wild-type let-23 but P5.p and P7.p have both lost let-23, P5.p and P7.p can still properly
21
adopt secondary vulval cell fates, indicating that Notch signaling alone in the absence of EGF/Ras signaling can specify secondary fates (Koga and Ohshima, 1995; Simske and Kim, 1995). In Muv animals resulting from EGF pathway overactivation, Notch signaling is still present, typically causing an alternating pattern of primary and secondary vulval cell fates among the six Pn.p cells (Sternberg, 1988).
Additional roles of EGF signaling in C. elegans
In addition to its role in vulval cell-fate specification, EGF signaling also controls many other aspects of C. elegans development and behavior. Similarly to vulval development, several cell fates require EGF signaling transduced by a Ras/MAPK signaling cascade. Null mutations in lin-3 EGF, let-23 EGFR, let-60 Ras, and other components of the Ras/MAPK pathway cause lethality (Beitel et al., 1990; Ferguson and Horvitz, 1985; Han et al., 1990). Specifically, animals die during the first larval stage and exhibit a fluid-filled appearance known as rod-like lethality. Most alleles of genes in the EGF/Ras pathway identified in screens for abnormal vulval development are partial loss-of-function alleles that impair vulval development but do not cause lethality. This rod-like lethality could reflect a general requirement for EGF/Ras
signaling to promote growth of the animal, or it could be caused by a relatively specific cell-fate defect. Mosaic experiments showed that loss of let-60 in large portions of the animal does not cause lethality, and that lethality is caused specifically by loss of let-60 in the duct cell (Yochem et al., 1997). The duct cell is required for osmoregulation, and ablation of the duct cell also causes lethality (Nelson and Riddle, 1984).
EGF signaling also regulates the fate of the P11 and P12 ventral cord precursor cells (Aroian and Sternberg, 1991; Fixsen et al., 1985; Jiang and Sternberg, 1998). If either P11 or P12 is ablated in a wild-type animal the other cell adopts a P12 fate,
indicating that P12 is the primary fate (Sulston and White, 1980). Loss of EGF signaling causes P12 to transform to a P11 fate (Jiang and Sternberg, 1998). EGF signaling in P11 and P12 acts through let-60 Ras, as a let-60 loss-of-function mutation also causes a P12-to-P11 transformation (Jiang and Sternberg, 1998). Conversely, expression of a
fate (Jiang and Sternberg, 1998). Therefore, EGF signaling in P11 and P12 promotes the adoption of the P12 fate.
EGF signaling also controls development of certain cells in the male tail during spicule development. Partial loss-of-function mutations in lin-3 EGF, let-23 EGFR,
let-60 Ras, and other components of the EGF/Ras signaling pathway prevent the
adoption of certain anterior cell fates (Chamberlin and Sternberg, 1994). Expression of
lin-3 from a ubiquitous heat-shock promoter causes the ectopic adoption of those fates
(Chamberlin and Sternberg, 1994).
After the vulva is formed a connection must be established between the vulva and the uterus, and this connection also requires EGF signaling. lin-3 is expressed in specific descendants of the Pn.p cells in the vulva, and let-23 is expressed in the ventral-most cells of the uterus (Chang et al., 1999). Partial loss-of-function mutations in let-23 or let-60 prevent the adoption of the uterine uv1 cell fate necessary to make the vulval-uterine connection (Chang et al., 1999).
EGF signaling has additional roles outside of cell-fate specification, generally acting through pathways other than Ras/MAPK signaling. A strong non-null lin-3 allele and certain let-23 alleles cause hermaphrodite sterility as the result of a defect in ovulation (Aroian et al., 1990; Ferguson and Horvitz, 1985). The somatic gonad appears grossly normal and oocytes mature normally, but oocytes do not enter the spermatheca where they are fertilized in wild-type animals (Clandinin et al., 1998).
let-60 Ras is not involved in this process, and instead let-23 signals through inositol
triphosphate, possibly regulating calcium levels in the spermatheca (Clandinin et al., 1998).
EGF signaling also regulates behavioral quiescence in C. elegans. Expression of
lin-3 from a ubiquitous heat-shock promoter causes animals to cease pharyngeal
pumping and grow more slowly (Van Buskirk and Sternberg, 2007). The growth and pumping defects caused by lin-3 overexpression act through phospholipase C-" and diacylglycerol but not through let-60 Ras (Van Buskirk and Sternberg, 2007). The phenotype caused by heat-shock lin-3 expression mimics the lethargus period which precedes each larval molt in C. elegans, and let-23 activation in a specific neuron
23
induces quiescence (Van Buskirk and Sternberg, 2007). In summary, lin-3 has multiple Ras-independent roles in behavioral regulation, in addition to the multiple
Ras-dependent roles of EGF signaling in cell-fate specification in C. elegans.
A common theme among all of the roles of EGF signaling in C. elegans is that misexpression of lin-3 causes numerous developmental and behavioral defects.
Consistent with the adverse effects of lin-3 overexpression and ectopic expression, lin-3 expression is normally tightly restricted to specific cells and tissues that correspond to the different roles of EGF signaling. At the late L2 and early L3 stage when vulval induction occurs a lin-3::lacZ fusion is expressed specifically in the anchor cell and not in any other cells near the developing vulva (Hill and Sternberg, 1992). lin-3 is also expressed in a temporally regulated manner in the male tail, spermatheca, and in specific vulval cells, likely corresponding to the roles of EGF signaling in male tail development, ovulation, and uterine development, respectively (Chang et al., 1999; Hwang and Sternberg, 2004). Additionally, lin-3 expression is seen in the pharynx
throughout development, perhaps to regulate quiescence (Hwang and Sternberg, 2004). Because lin-3 is expressed in distinct tissues and cells at different times, there are likely to be multiple cis-regulatory elements in the lin-3 locus controlling the different aspects of lin-3 expression. Supporting this theory, lin-3::lacZ and lin-3::gfp fusions with different extents of 5ʼ non-coding regions have differing expression patterns, with more extensive 5ʼ regions conferring expression in additional tissues (Chang et al., 1999; Hill and Sternberg, 1992; Hwang and Sternberg, 2004). The only aspect of lin-3 expression for which the basis of spatial specificity has been studied is the anchor cell. The
lin-3(e1417) mutation causes a vulvaless phenotype, but does not result in any of the
other defects associated with lin-3 loss-of-function (Liu et al., 1999). lin-3(e1417) affects a conserved 59 bp element located in a lin-3 intron that is both necessary and sufficient to drive expression in the anchor cell (Hwang and Sternberg, 2004). This element contains consensus binding sites for the bHLH protein HLH-2 and the nuclear hormone receptor NHR-25. Both NHR-25 and HLH-2 bind to this element in vitro, and RNAi of
hlh-2 at the appropriate stage eliminates lin-3 expression in the anchor cell (Hwang and
restricted manner. The different sources of lin-3 control a variety of developmental and behavioral processes, and ectopic expression of lin-3 can disrupt the precise regulation of these processes.
The synMuv genes antagonize EGF signaling
Because excessive or misplaced EGF signaling can disrupt normal development, it is important for C. elegans to properly restrict EGF signaling. The synthetic multivulva (synMuv) genes oppose EGF signaling in vulval development. The synMuv genes are grouped into two redundant classes, A and B. Animals carrying a mutation in a single synMuv gene have essentially wild-type vulval development, but animals carrying mutations in both a class A and a class B synMuv gene exhibit a Muv phenotype
(Ferguson and Horvitz, 1989). Animals with two class A synMuv mutations or two class B synMuv mutations have mostly normal vulval development. Therefore, the synMuv genes form two redundant pathways that prevent ectopic vulval development. When either pathway is mutated, the other pathway is still active and prevents excess vulval development, but when both pathways are mutated extra Pn.p cells adopt vulval cell fates. Genes with redundant functions can be difficult to identify by genetic screens. However, the synMuv genes were found in four distinct ways. The first two synMuv genes were discovered when a Muv strain was fortuitously isolated with two unlinked mutations that were both required to cause a Muv phenotype (Horvitz and Sulston, 1980). Second, a number of synMuv mutations were discovered when a strain carrying a background mutation in a class A synMuv gene was mutagenized, leading to many Muv strains that carried two synMuv mutations (Ferguson and Horvitz, 1989). Third, the class A and B synMuv genes lin-15A and lin-15B are located adjacent to each other in an operon, and mutations that eliminate both 15A and 15B (called 15 or
lin-15AB mutations) were isolated in screens (Clark et al., 1994; Ferguson and Horvitz,
1989; Huang et al., 1994). Finally, screens have been performed using animals carrying one synMuv mutation to identify synMuv mutations in the other class. These approaches, as well as directly testing candidate genes, have led to the identification of four class A synMuv genes and approximately 25 class B synMuv genes.
25
The class A synMuv genes encode putative transcription factors
Four class A synMuv genes have been discovered: lin-8, lin-15A, lin-38, and
lin-56. lin-8 encodes a novel acidic protein that is the founding member of a family of C. elegans proteins (Davison et al., 2005). lin-15A and lin-56 encode THAP domain
proteins (Clark et al., 1994; Huang et al., 1994; Davison, 2003). The THAP domain is a zinc finger-like motif present in many organisms including C. elegans, Drosophila
melanogastar, and humans, and can exhibit sequence-specific DNA-binding activity
(Bessiere et al., 2008; Clouaire et al., 2005; Liew et al., 2007; Roussigne et al., 2003; Sabogal et al., 2009). THAP domain proteins repress transcription of many genes, possibly by recruiting corepressor proteins (Cayrol et al., 2007; Dejosez et al., 2008; Macfarlan et al., 2005). LIN-15A and LIN-56 interact in vitro and are required for each otherʼs stability in vivo, suggesting that they function in a complex together (Davison, 2003). LIN-8, LIN-15A, and LIN-56 are all expressed broadly throughout the animal and are primarily localized to the nucleus (Davison, 2003; Davison et al., 2005). Based on their domains and nuclear localization, the class A synMuv genes might be
transcriptional regulators. However, as none of the class A synMuv proteins has extensive homology to well-characterized proteins, the specific molecular functions of the class A synMuv genes are still unclear.
The class B synMuv genes encode transcriptional repressors
Many class B synMuv genes are homologous to genes known to be involved in chromatin remodeling and transcriptional repression. Nucleosomes, composed of approximately 146 base pairs of DNA wrapped around an octamer consisting of two H2A, two H2B, two H3, and two H4 histones, are the core unit of chromatin. These nucleosomes then form higher order structures that can compact the chromatin,
typically rendering it less accessible to polymerases. Many proteins can affect the state and structure of chromatin and thereby regulate transcription.
Histone tails extend from the nucleosome and are subjected to many covalent modifications, including acetylation, methylation, phosphorylation,ubiquitylation, and sumoylation. Modifications can either promote or repress transcription, depending on
both the specific covalent modification and the residue that it is applied to. For example, methylation of lysine 9 of histone H3 typically leads to a repressive chromatin state that inhibits transcription, but methylation of lysine 4 of histone H3 typically promotes
transcription (Kouzarides, 2007). The class B synMuv genes include both the histone deacetlyase HDA-1 and the histone acetylase MYS-1 (Ceol and Horvitz, 2004; Lu and Horvitz, 1998). It is not known if HDA-1 and MYS-1 affect different residues of histones at the same target genes, or if they affect two separate target genes that have opposing effects on vulval development. Two histone lysine methyltransferases have been
identified as class B synMuv genes, MET-1 and MET-2 (Andersen and Horvitz, 2007; Poulin et al., 2005). MET-1 is homologous to histone H3 lysine 36 histone
methyltransferases, and MET-2 is homologous to histone H3 lysine 9 histone methyltransferases. Covalent modifications are often “read” by proteins that bind
specific covalently-modified histones to regulate transcription. For example, methylated lysine 9 of histone H3 serves as a binding site for chromodomain proteins such as HP1, which represses transcription (Bannister et al., 2001; Lachner et al., 2001; Nakayama et al., 2001). The C. elegans HP1 homolog hpl-2 is a class B synMuv gene (Couteau et al., 2002).
ATP-dependent chromatin remodeling enzymes use energy from the hydrolysis of ATP to remodel chromatin by either sliding nucleosomes along the DNA or by
displacing nucleosomes from the DNA. These actions can alter the accessibility of the DNA and thereby affect transcription (Saha et al., 2006). One such enzyme is Mi-2, a component of the nucleosome remodeling and histone deacetlyase (NuRD) complex (Tong et al., 1998; Wade et al., 1998; Zhang et al., 1998). The C. elegans Mi-2
homolog LET-418 and homologs of several other components of the NuRD complex are class B synMuv proteins (Lu and Horvitz, 1998; von Zelewsky et al., 2000).
Other class B synMuv genes encode transcription factors that can bind to specific DNA regulatory sequences and regulate expression of adjacent genes. The class B synMuv genes lin-35, dpl-1, and efl-1 encode the C. elegans homologs of the retinoblastoma gene (Rb), DP, and E2F, respectively (Ceol and Horvitz, 2001; Lu and Horvitz, 1998). Rb is a tumor suppressor, and either Rb or other components of the Rb
27
pathway are inactivated in the majority of human cancers (Hanahan and Weinberg, 2000). E2F and DP proteins form heterodimeric transcription factors that bind DNA in a sequence-specific manner. Rb can bind to the DP/E2F heterodimer and modulate its effects on transcription. Rb, DP, and E2F have been extensively studied for their roles in regulating cell-cycle progression (Dyson, 1998). Additionally, Rb controls the
expression of many genes involved in development and differentiation in Drosophila (Dimova et al., 2003).
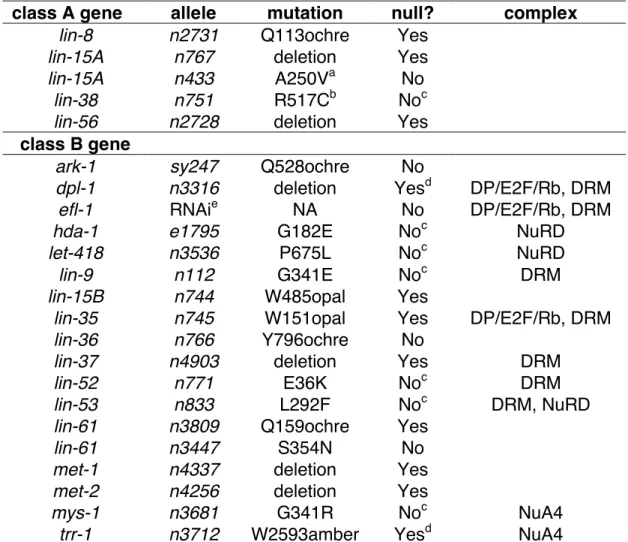
Some class B synMuv genes do not have known functions, and many of these genes are conserved in humans. For example, LIN-9, LIN-37, LIN-52, and LIN-54 are conserved proteins, and their specific molecular functions are not well understood (Beitel et al., 2000; Harrison et al., 2006; Thomas et al., 2003). Other class B synMuv genes such as lin-13, lin-15B, and lin-36 do not have obvious homologs in mammals, but do encode proteins with specific domains that are present in many animals, including humans (Clark et al., 1994; Huang et al., 1994; Melendez and Greenwald, 2000; Reddy and Villeneuve, 2004; Thomas and Horvitz, 1999).
synMuv protein complexes
Based on interaction studies in C. elegans and homology to well-studied yeast and mammalian complexes, the synMuv genes are thought to form a number of distinct complexes. The class A synMuv proteins LIN-15A and LIN-56 form a complex
(Davison, 2003). The class B synMuv proteins HDA-1, LIN-53, and LET-418 are the C.
elegans homologs of members of the NuRD complex (Harrison et al., 2006; Lu and
Horvitz, 1998; von Zelewsky et al., 2000). The NuRD complex is a corepressor that combines histone deacetlyase and ATP-dependent chromatin remodeling activities. Eight other class B synMuv proteins, including LIN-35 and DPL-1, are present in the DRM complex, which is distinct from the C. elegans NuRD-like complex (Harrison et al., 2006). The DRM complex is highly similar to the Drosophila dREAM and Myb-MuvB complexes that repress the transcription of many E2F target genes (Korenjak et al., 2004; Lewis et al., 2004). Another group of class B synMuv genes, previously referred to as the class C synMuv genes (see Chapter Three for explanation), are predicted to
form a C. elegans Tip60/NuA4-like histone acetyltranferase complex (Ceol and Horvitz, 2004). The class B synMuv genes hpl-2 and lin-13 both exhibit a Muv phenotype at high temperatures as single mutants in addition to their class B synMuv phenotype, and they form a complex in vivo (Coustham et al., 2006; Couteau et al., 2002; Melendez and Greenwald, 2000). Some synMuv proteins might be members of multiple complexes that control vulval development and other processes. For example, the RbAp48 homolog LIN-53 is present in both the DRM and NuRD complexes (Harrison et al., 2006; Lu and Horvitz, 1998). Surprisingly, yeast two-hybrid and in vitro binding assays indicate that the class A synMuv protein LIN-8 and the class B synMuv protein LIN-35 physically interact (Davison et al., 2005). Given the different roles of lin-8 and lin-35 in vulval development, this interaction might not be relevant to vulval development but might instead be important for a non-vulval function of these two genes.
The site of action of the synMuv genes
The site of action of components of the EGF signaling pathway in vulval induction is clear: lin-3 EGF is expressed in the anchor cell, and let-23 EGFR and all downstream signaling components including the transcription factor lin-31 function cell-autonomously in the Pn.p cells (Hill and Sternberg, 1992; Koga and Ohshima, 1995; Lackner et al., 1994; Miller et al., 1996; Simske and Kim, 1995; Yochem et al., 1997). The site-of-action of the synMuv genes is less clear. The synMuv phenotype is epistatic to anchor cell ablation, indicating that the synMuv genes function outside of the anchor cell (Ferguson et al., 1987; Sternberg and Horvitz, 1989). Mosaic analyses of the synMuv genes lin-15 and lin-37 clearly showed that they do not act cell autonomously in the Pn.p cells (Hedgecock and Herman, 1995; Herman and Hedgecock, 1990). However, mosaic analyses did not clearly implicate any one cell or tissue as the site-of-action of the synMuv genes (Hedgecock and Herman, 1995; Herman and Hedgecock, 1990; Thomas and Horvitz, 1999). More recently, Myers and Greenwald (2005) reported that expression of a lin-35 cDNA driven specifically in the hyp7 hypodermal syncytium using the dpy-7 promoter was sufficient to rescue the lin-35 synMuv phenotype. However, the rescue was not complete and I have found that the expression pattern of the dpy-7
29
promoter is broader than was previously thought (Appendix Two), so it is possible that other cells in addition to hyp7 contribute to the synMuv phenotype.
Relationship between EGF signaling and synMuv genes
The relationship between the EGF/Ras pathway and the synMuv genes in vulval development has been a longstanding question. Double mutant experiments indicated that the synMuv phenotype was epistatic to the Vul phenotype caused by a reduction of
lin-3 EGF function, but the Vul phenotype caused by loss-of-function mutations in let-23
EGFR or let-60 Ras was epistatic to the synMuv phenotype (Ceol and Horvitz, 2001, 2004; Huang et al., 1994; Lu and Horvitz, 1998). This observation led to the hypothesis that LET-23 may have some basal signaling activity in the absence of LIN-3 binding (Ceol and Horvitz, 2001). However, these epistasis experiments were confounded because non-null alleles of lin-3 were used, since null alleles of lin-3 cause lethality that precludes assaying vulval development. The first hint that the synMuv phenotype might not be epistatic to lin-3 loss-of-function was that a partial loss-of-function mutation of
lin-3 in trans to a lin-3 null mutation partially suppresses a weak synMuv phenotype
(Ferguson et al., 1987). Later, it was found that RNAi directed against lin-3 could efficiently suppress the synMuv phenotype of many synMuv double mutants (Cui et al., 2006a). Because the synMuv phenotype requires EGF signaling, EGF signaling must act either downstream of or in parallel to the synMuv genes. lin-3 mRNA levels are grossly wild-type in synMuv single mutants, but lin-3 is overexpressed in synMuv double mutants (Andersen and Horvitz, 2007; Cui et al., 2006a). Thus, the class A and B synMuv genes redundantly repress lin-3 expression, and lin-3 is likely to be a direct or indirect target of the synMuv genes in vulval development.
The synMuv genes have numerous functions
synMuv single mutants typically do not have major defects in vulval development, and most class A synMuv single mutants have a superficially wild-type phenotype. However, most class B synMuv single mutants exhibit numerous abnormalities.
and efl-1 identified a large number of transcripts with altered expression (Chi and
Reinke, 2006; Kirienko and Fay, 2007), and many defects other than those in vulval development have been identified in class B synMuv single mutants. Some of these defects are present in many synMuv mutants, while other defects are specific to much smaller subsets of the synMuv genes. These various processes involve many different groups of synMuv genes. Most of the defects caused by synMuv mutations do not resemble the defects that result from either loss-of-function or overexpression of lin-3, indicating that they result from effects on other target genes.
The initial sign that the synMuv genes might be involved in processes other than vulval development came from studies of lin-9, the first class B synMuv gene identified. The first allele of lin-9 was a partial loss-of-function allele, and a non-complementation screen isolated strong lin-9 loss-of-function mutations that cause sterility (Ferguson and Horvitz, 1989). Subsequently, many class B synMuv mutations were found to cause sterility (Ceol and Horvitz, 2001; Ceol et al., 2006; von Zelewsky et al., 2000). The sterility of these mutants might not indicate a specific role in fertility for these genes. Rather, these genes could be absolutely essential for all aspects of growth and viability, and defects are seen in vulval development and fertility because the maternal
contribution of the gene has been exhausted by the time those processes occur. The divisions of the Pn.p cells are some of the latest divisions in the animal (Sulston and Horvitz, 1977), which may be why defects are seen in vulval development and not in other aspects of development.
The Rb gene has an important role in mammals controlling progression through the cell cycle. Likewise, the C. elegans Rb homolog lin-35 is a negative regulator of the G1/S progression (Boxem and van den Heuvel, 2001). Many other synMuv genes, including efl-1, dpl-1, lin-9, lin-15B, and lin-36 also function in cell cycle progression (Boxem and van den Heuvel, 2002). Screens for mutants synthetically lethal with lin-35 loss-of-function implicated lin-35 Rb and other class B synMuv genes in pharyngeal development, gonadal development, and general growth and viability (Bender et al., 2007; Bender et al., 2004; Cui et al., 2004; Fay et al., 2003; Fay et al., 2004).
31
Therefore, lin-35 Rb is required for multiple development processes where its role is masked by redundancy, much like the function of lin-35 in vulval development.
Many class B synMuv mutants exhibit a set of abnormalities that can be attributed to an underlying defect in maintaining the distinction between germline and soma. In these mutants, somatic cells adopt a more germline-like appearance and inappropriately express PGL-1, a component of P granules that is normally expressed only in the germline (Kawasaki et al., 1998; Unhavaithaya et al., 2002; Wang et al., 2005). Transgenes in C. elegans created by injecting DNA into the gonad are large, multicopy, and repetitive arrays (Mello et al., 1991; Stinchcomb et al., 1985). These transgenes are expressed in somatic cells, but are typically silenced in the germline (Kelly et al., 1997). In some class B synMuv mutant backgrounds, repetitive transgenes are also silenced in somatic cells, perhaps reflecting the adoption of germline
characteristics that include transgene silencing (Hsieh et al., 1999). Many of these class B synMuv mutants are also hypersensitive to RNAi, possibly as a result of
misexpression of the germline-specific RNA-dependent RNA polymerase EGO-1 (Wang et al., 2005). Some class B synMuv genes, such as the histone methyltransferases
met-1 and met-2, do not affect the distinction between germline and soma (Andersen
and Horvitz, 2007). Other synMuv mutants exhibit only some aspects of the soma to germline transformation. For example, mutations in the class B synMuv genes tam-1 and lex-1 cause silencing of repetitive transgenes but do not affect expression of the germline marker PGL-1 (Hsieh et al., 1999; Tseng et al., 2007). It is also possible that
tam-1 and lex-1 do not play any role in distinguishing germline and somatic fates, and
that their effects on transgene silencing are caused by other mechanisms. Furthermore, it is not known if the class B synMuv genes impart somatic identity by directly repressing many or all germline-specific genes, or if the class B synMuv genes control a small number of master regulators that specify germline and somatic fates and thereby indirectly repress the expression of germline genes.
Conclusion
The C. elegans vulva has been an informative system for the study of EGF/Ras signaling. The synMuv genes, which oppose EGF signaling in vulval development, function in chromatin remodeling and transcriptional repression to inhibit the ectopic adoption of vulval cell fates. My thesis work has focused on identifying synMuv genes, determining how the synMuv genes interact with each other, and elucidating the relationship between the synMuv genes and the EGF/Ras pathway. I have specifically focused on the class A synMuv genes, as they have been studied much less extensively than the class B synMuv genes. In Chapter Two I show that the class A synMuv genes antagonize vulval development primarily by repressing lin-3, and that the class A and B synMuv genes function globally to prevent ectopic lin-3 expression. In Chapter Three I show that the class A synMuv genes function in multiple parallel pathways to repress
lin-3 expression. In Chapter Four I report the identification of class A synMuv alleles of mcd-1 and the cloning and characterization of lin-38. Finally, I discuss what future
approaches might be taken to study further the synMuv genes and their role in repressing lin-3 EGF expression.
Acknowledgments
33 Chapter Two
The C. elegans synthetic multivulva genes prevent Ras pathway activation by tightly repressing ectopic expression of lin-3 EGF
Adam M. Saffer, Dong hyun Kim, Alexander van Oudenaarden, and H. Robert Horvitz
Chapter Two: The C. elegans synthetic multivulva genes prevent Ras pathway activation by tightly repressing ectopic expression of lin-3 EGF
I fixed animals for in situ hybridizations, Dong hyun Kim performed the hybridizations and collected the images, and I analyzed the images. I performed all other experiments in this chapter and wrote the manuscript.