HAL Id: tel-01991183
https://tel.archives-ouvertes.fr/tel-01991183
Submitted on 23 Jan 2019HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires
Syndrome de Usher : Outils innovants pour une
exploration moléculaire exhaustive
Thomas Besnard
To cite this version:
Thomas Besnard. Syndrome de Usher : Outils innovants pour une exploration moléculaire exhaustive. Génétique humaine. Université Montpellier 1, 2012. Français. �tel-01991183�
UNIVERSITÉ MONTPELLIER I
U.F.R. de PHARMACIE
Année 2012THÈSE de DOCTORAT
pour obtenir le grade de
DOCTEUR DE L’UNIVERSITÉ MONTPELLIER I
Discipline : Génétique Moléculaire Formation doctorale : Biologie Santé
École Doctorale : Sciences Chimiques et Biologiques pour la Santé
Syndrome de Usher : Outils innovants pour
une exploration moléculaire exhaustive
présentée et soutenue publiquement par
Thomas BESNARD
Le 5 décembre 2012 à Montpellier, Université Montpellier I, devant la commission d’examen composée de :
Pr Mireille Claustres Pr Patrick Calvas Dr Jean-Michel Rozet Dr Sue Malcolm
Dr Anne-Françoise Roux
PU-PH, Université de Montpellier I
PU-PH, Université Paul Sabatier, Toulouse DR, Inserm U781, Paris
Professeur, University College of London PH, CHU de Montpellier Présidente du jury Rapporteur Rapporteur Examinateur Directeur de thèse
Remerciements
• Je remercie :
Le Professeur Patrick Calvas et le Docteur Jean-Michel Rozet, pour avoir accepté d’être rapporteurs de cette thèse, ainsi que le Professeur Sue Malcolm, pour avoir examiné ce travail.
Le Professeur Mireille Claustres, qui m’a accueilli, soutenu et aiguillé lors de chacune des étapes qui ont ponctué mes années au sein de son laboratoire. Je suis pleinement conscient de la chance que j’ai eue de pouvoir travailler chaque jour à l’interface entre le diagnostic moléculaire et la recherche. Merci de m’avoir permis de réaliser cette expérience si bénéfique.
Le Dr Anne-Françoise Roux, pour m’avoir offert la possibilité d’effectuer ma thèse sous sa direction. Mes années au sein du groupe neurosensoriel ont été extrêmement enrichissantes. Grâce à ton encadrement, j’ai pu à la fois bénéficier de l’autonomie nécessaire à un travail constructif, et de ton soutien dès que cela s’est avéré nécessaire, c’est à dire, fréquemment. Merci !
L’Union Nationale des Aveugles et Déficients Visuels, pour avoir financé une partie de ce travail de thèse.
• Je tiens à remercier l’ensemble des membres du laboratoire pour les cinq années que j’ai passées auprès de vous, et particulièrement :
Le groupe neurosensoriel au complet. Gema avec qui j’ai partagé de nombreuses heures de travail, à la paillasse, devant les séquenceurs, devant Excel! Etre ton voisin de bureau a été un vrai plaisir, enrichissant qui plus est. Je te suis aussi très reconnaissant du soutien que tu m’as apporté tout au long de la rédaction de ce manuscrit.
Christel et David pour leurs corrections et leur relecture du manuscrit, mais aussi pour tout le reste. Christel, pour m’avoir initié à la joie et aux aléas des transcrits ainsi qu’à toutes les perspectives dont découlent leurs études. Merci pour l’ensemble des techniques moléculaires auxquelles tu m’as formé. David, pour tout ce que tu as pu m’apporter au sein
Remerciements
du laboratoire, d’avoir bien voulu essayer de répondre à chacun de mes « pourquoi », qu’ils aient été biologiques, informatiques, sportifs, politiques,! Et puis merci pour tous les à-côtés, les promenades de mi-journée à destination du RU, les footings avec ou sans escaliers, en passant par le volley et les discussions Francisiennes.
Lise pour m’avoir initié à l’étude des gènes USH2 et Val pour m’avoir formé durant de
longues semaines. Votre expérience du secteur diagnostic m’a été extrêmement profitable et a très certainement forgé ma façon de travailler. Susana, pour l’attention particulière que tu m’as apportée et pour ne jamais m’avoir doublé à vélo, pourtant à vive allure ! Vanessa, pour ne jamais avoir râlé sur mon organisation et pour avoir réussi à égayer la Qualité. Enfin, merci à Caroline A et les stagiaires du groupe, particulièrement Alan, Alexandre, Pauline et Claire.
Les responsables des autres équipes du laboratoire : Sylvie, Anne G, Albertina, Magali,
Marie, Marie-Claire et Mireille. Merci pour votre disponibilité et votre aide offertes dès que
cela s’est avéré nécessaire.
Corinne B, pour tes pauses et ton café, mais aussi pour tes bises et ta bonne humeur. Victoria, merci d’avoir été ma collègue de bureau durant tes dernières années de thèse. Je
te suis très reconnaissant de ton soutien et de ta façon d’offrir ton aide presque avant même qu’elle te soit demandée. Enfin, merci pour la relecture de ce manuscrit.
Aliya, pour m’avoir fait une place près de toi durant la rédaction du manuscrit. Corinne T,
pour avoir partagé ces années d’études et de labo avec moi et surtout pour ta façon de toujours prendre les choses par leur bon côté. Julie et Caroline R, pour vos conseils avisés.
Le bureau des étudiants. Jessica pour avoir partagé les joies et aléas du NGS, Anne B, pour ton naturel et ton dynamisme et Alessandro pour avoir apporté, une touche de résultats sportifs au sein du bureau étudiant. Bonne continuation à tous !
A Naima, pour l’énergie que tu émets chaque matin.
Remerciements
• Je souhaite exprimer mes remerciements à mes amis pour tout ce qu’ils m’apportent :
A Guillaume (Morelski), avec qui je me suis initié à la biologie sur les bancs du lycée. A mes collègues de l’IUT : particulièrement Antoine, Anthony, François, Julien et Willy. C’est bon, on est sur nos rails ! Aux montpelliérains : les collègues de l’IUP ; Christophe, pour m’avoir accepté à votre table du RU et pour ne jamais avoir refusé un match de bad ;
Seb pour ta capacité à organiser ; Gaspard, pour m’avoir orienté tout au long de ma thèse
grâce à tes années d’avance. Un merci tout particulier à Amine, pour m’avoir ouvert les portes de Montpellier.
A mes amis kayakistes : l’ensemble de la kermesse yaouank, en particulier à mon Leroux.
Paul, je tiens à te remercier pour la curiosité et l’enthousiasme dont tu as fait part vis à vis de
mes études, durant toutes ces années. De plus, je tiens à adresser mes plus forts sentiments à l’ensemble de la famille Zoungrana pour qui une partie de mes pensées vont chaque jour. Merci Jean, d’avoir aiguillé mon orientation, me menant à ce travail de thèse.
• Je tiens à remercier toute ma famille pour m’avoir soutenu durant ces études :
Mes grands-parents, pour leur ouverture à mon cursus scolaire. A mes parents que je ne
remercierai sans doute jamais assez : Papa, merci « de n’y être pour rien » mais finalement d’être quand même un peu à l’origine de tout ! Maman, merci de m’avoir fait confiance et de m’avoir permis de suivre mon petit chemin, souvent loin de la maison. A mes sœurs, Marion et Alice, pour être des frangines idéales !
• Je tiens à exprimer toute ma reconnaissance à ta famille et tes amis, pour l’accueil et le soutien qu’ils m’ont accordés.
Avant propos
Le syndrome de Usher est une maladie génétique associant surdité congénitale et rétinopathie pigmentaire (RP), auxquelles peuvent s’ajouter des troubles vestibulaires. Les différences exprimées entre ces manifestations définissent une classification en 3 types. Jusqu’au milieu des années 1990, le diagnostic reposait essentiellement sur des bases cliniques. Entre 1995 et 2007, neuf gènes responsables du syndrome de Usher ont été identifiés. Il s’agit des gènes MYO7A, USH1C, CDH23, PCDH15, USH1G impliqués dans le syndrome de Usher de type I, USH2A, GPR98, DFNB31 dans le type II, et CLRN1 dans le type III.
L'assignation de gènes ouvrant de nouvelles perspectives de diagnostic moléculaire, le laboratoire de génétique moléculaire du Centre Hospitalier Universitaire de Montpellier a développé des analyses spécifiques permettant de confirmer et d'affiner le diagnostic du syndrome de Usher. A ce jour, plus de 450 familles ont été étudiées dans notre laboratoire. Le diagnostic moléculaire est un paramètre important pour une meilleure prise en charge des patients, motivant par exemple la mise en place d’une implantation cochléaire précoce ou l’apprentissage de moyens de communication adéquats dès le plus jeune âge. Ce type de diagnostic offre aussi la possibilité d’un conseil génétique adapté en cas de projet parental. A ce jour, même si une correction du déficit auditif peut être apportée, il n’existe pas de possibilité d’amélioration significative du phénotype rétinien.
Identifier précisément les causes moléculaires grâce aux outils d’analyses génétiques permet d’améliorer la compréhension des mécanismes physiopathologiques à l'origine des symptômes du syndrome de Usher. Cette connaissance permettra aussi de susciter de nouvelles pistes thérapeutiques.
Résumé
Le syndrome de Usher est une maladie génétique associant surdité congénitale et rétinopathie pigmentaire (RP), auxquelles peuvent s’ajouter des troubles vestibulaires. Les différences phénotypiques, distinguées en 3 types cliniques, s’accompagnent d’une hétérogénéité génétique impliquant au moins 10 gènes. Identifier et caractériser les causes moléculaires permet d’améliorer la compréhension des mécanismes physiopathologiques à l'origine des symptômes du syndrome de Usher.
Dans ce cadre, nous nous sommes inscrits dans une recherche d’exhaustivité des études moléculaires. Dans un premier temps, nous avons ainsi mis en place l’analyse et défini le spectre mutationnel des gènes minoritairement impliqués dans le type II (GPR98 et
DFNB31). Nous avons également développé différents outils, notamment pour l’analyse de
variants altérant le mécanisme d’épissage ou touchant les régions promotrices des gènes USH2.
Ces travaux permettent d’obtenir un taux de détection des altérations conduisant au syndrome de Usher type II de 90%. Ce taux est maintenant similaire à celui observé pour le type I, qui constituait jusqu’ici la référence.
Nous avons, dans un second temps, développé le séquençage nouvelle génération (NGS) appliqué à l’exome Usher. L’objectif de cette analyse était de tester la faisabilité et l’efficacité de cette approche, en vue de son éventuelle utilisation en diagnostic moléculaire. La définition des critères de qualité et la mise en place de la priorisation des variants ont été réalisées sur un groupe contrôle. L’étude a ensuite été étendue sur une cohorte de patients de génotype inconnu. Les résultats obtenus montrent qu’une utilisation en diagnostic est possible mais restera dépendante de l’amélioration de la technique du séquençage, de son analyse et des outils bioinformatiques pour interpréter le volume de données ainsi généré.
Mots clés : Syndrome de Usher, génétique, hétérogénéité, analyse moléculaire,
Abstract
Usher syndrome is a genetic disorder combining sensorineural hearing loss (HL) and retinitis pigmentosa (RP). Some patients will also exhibit vestibular areflexia (VA). Clinical and genetic heterogeneity is recognized as the 3 clinical subgroups, defined mainly on the degree of HL and VA, can be caused by mutations in one of the 10 known genes. It is important to use all accessible genetic tools to identify and characterize molecular origin in order to improve the knowledge of the physiopathological mechanisms causing Usher Syndrome.
In this context, we have developed an exhaustive approach. In a first step, we have implemented the analysis and established the mutational spectrum of the 2 minor USH2 genes (GPR98 and DFNB31). In addition, we have developed several tools, in particular to study variants susceptible to alter splicing or lying in the promoter regions of the USH2 genes.
Thanks to this work, the USH2 mutation detection rate has now been raised to 90%, similar to that of USH1.
We have then designed a targeted exome of the Usher genes to be sequenced using the GS Junior system (Roche 454). The aim of the study was to test the feasibility of this new technics for a possible transfer to diagnostic facilities. Quality criteria and variant priorization were set up on a control cohort (previously studied in one of the USH gene). The study has then been extended on a patient cohort. Our results indicate that NGS Usher-exome can be used in molecular diagnostics but improvement of the reliability of the sequencing technology, bioinformatics tools and dedicated databases is essential.
Keywords: Usher Syndrome, genetics, heterogeneity, molecular analysis, next generation
Sommaire ! Remerciements ... 2 ! Avant propos ... 5 ! Résumé ... 6 ! Abstract ... 7 ! Sommaire ... 8 !
Liste des figures 10
!
Liste des tableaux 12
!
Liste des annexes 12
!
Liste des abréviations 13
!
URLs des sites Internet cités 14
! Partie I : Etat des lieux ... 15
! !
I. Généralités : les déficits sensoriels ... 16
! !
A. L’organe de l'audition 16
1.! Les cellules ciliées 17!
2.! La touffe ciliaire 18! 3.! La mécanotransduction 20! ! ! B. L'organe de la vision 20 1.! Anatomie de la rétine 20! 2.! Les photorécepteurs 21! 3.! La phototransduction 21! ! !
II. Le syndrome de Usher ... 23
! !
A. Hétérogénéité clinique et génétique 24
1.! Gènes et protéines USH1 29!
2.! Gènes et protéines USH2 33!
3.! Gène USH3 : CLRN1 36!
4.! Les autres gènes 36!
5.! Gènes Usher et phénotypes non syndromiques 36!
! !
B. Les modèles animaux 37
! !
C. Le rôle des protéines Usher 39
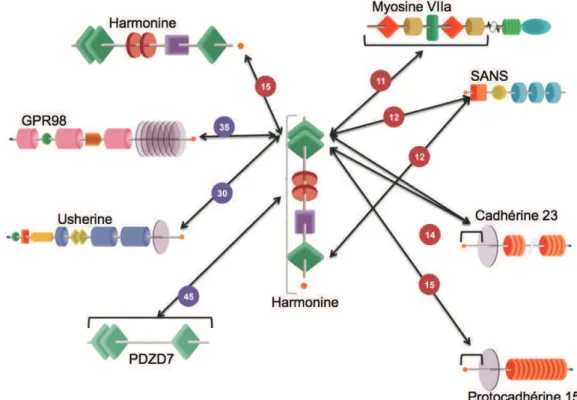
1.! Les interactions entre les protéines Usher 39!
2.! Les autres interactions 42!
3.! Localisation des protéines Usher 43!
4.! Protéines Usher dans les cellules ciliées de l’oreille interne 44!
5.! Protéines Usher dans les photorécepteurs 45!
! !
D. Spectre mutationnel des gènes Usher 46
1.! Hétérogénéité 46!
2.! Effets fondateurs des mutations 47!
3.! Digénisme et oligogénisme 48! ! ! E. Perspectives thérapeutiques 50 1.! Thérapies symptomatiques 51! 2.! Thérapies causales 52! ! !
F. Le diagnostic moléculaire du syndrome de Usher 54
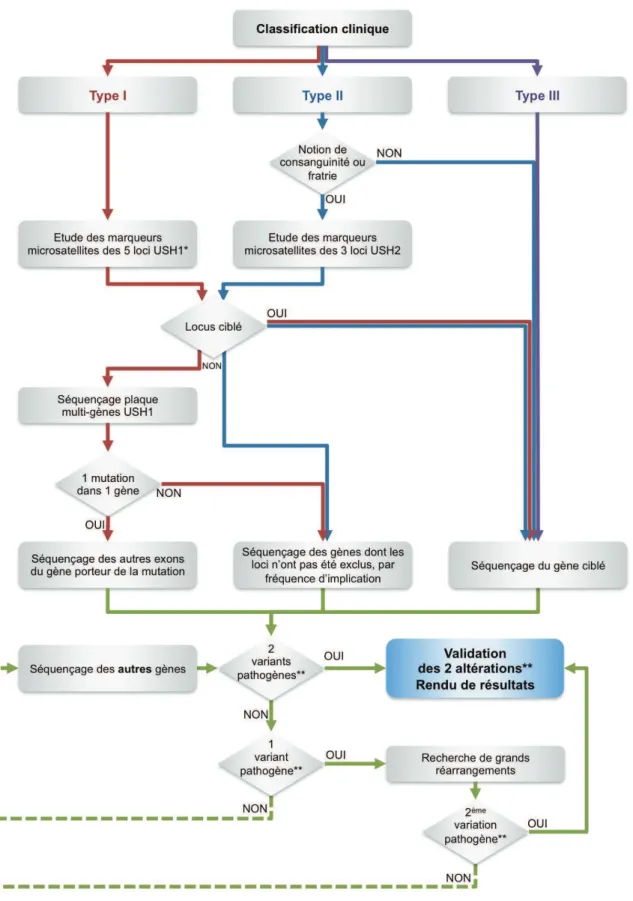
1.! Les stratégies disponibles 54!
2.! La stratégie du laboratoire 54!
! Partie II : Implication des gènes USH2 ... 63
! ! I. Introduction ... 64 ! ! A. Contexte 64 ! ! B. Particularités techniques 65 ! !
II. Article : non-USH2A mutations in USH2 patients ... 66
! !
A. Résumé de l’article 66
! !
B. Article 66
! !
III. Analyses complémentaires ... 79
! !
A. Analyse moléculaire de GPR98 79
! !
B. Analyse de GPR98 dans une cohorte espagnole 81
! !
C. Bilan des altérations du gène GPR98 96
1.! Répartition des altérations 96!
2.! Implication des gènes USH2 98!
! !
D. Mutations localisées en dehors des zones criblées en « routine » 99
1.! Mutations ponctuelles introniques profondes 99!
2.! Altérations localisées dans les régions promotrices 100!
! !
IV. Bilan, discussion et perspectives ... 104
Partie III : Le NGS comme nouvel outil d’analyse des pathologies
! neurosensorielles ... 106
! !
I. Le séquençage nouvelle génération (NGS) ... 107
! !
A. Historique 107
! !
B. Définir les besoins 108
! !
C. Les plateformes disponibles 109
! !
D. Applications cliniques : exomes ciblés et WES 111
! !
II. Etude de l’exome ciblé ... 113
! !
A. Choix des régions capturées 113
! !
B. Capture des séquences 117
! !
C. Séquençage 454 : protocole 117
1.! Capture des séquences (phase I) 118!
2.! PCR en émulsion (Phase II) 120!
3.! Run de séquençage (Phase III) 120!
! !
D. Récupération des données 122
1.! Extraction des données d’intérêt par GSdot 122!
2.! Critères de conservation des variants 124!
! !
E. Cohortes étudiées 126
1.! Groupe « contrôle » 126!
2.! Groupe « patients Usher » 128!
3.! Groupe « patients surdités » 128!
! !
III. Résultats ... 129
! !
A. Groupe « contrôle » 129
1.! Données brutes et critères de qualité 129!
2.! Identification des variants 135!
! !
B. Groupe « patients Usher » 144
! !
C. Groupe « patients surdités » : étude pilote 146
! !
IV. Discussion et perspectives ... 148
! Conclusion ... 154
Liste des figures
Figure 1 : Organe de l'audition 17!
Figure 2 : Rangées de cellules ciliées d’un organe de Corti humain 17!
Figure 3 : Liens des stéréocils 19!
Figure 4 : Coupe transversale d’un œil et détail des couches rétiniennes 21!
Figure 5 : Photoexcitation des photorécepteurs 22!
Figure 6 : Chronologie d'identification des loci et des gènes Usher 26!
Figure 7 : Répartition clinique des patients mutés dans un des gènes USH1 28!
Figure 8 : Répartition clinique des patients mutés dans un des gènes USH2 28!
Figure 9 : Principales isoformes des protéines USH1 31!
Figure 10 : Pourcentage d'implication des gènes chez les patients USH1 32!
Figure 11 : Principales isoformes des protéines USH2 et USH3 35!
Figure 12 : Interactions entre les différentes protéines USH 39!
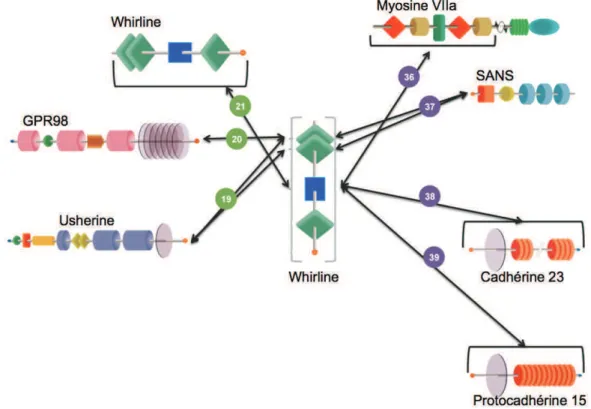
Figure 13 : Représentation des interactions entre les protéines USH et harmonine 40!
Figure 14 : Représentation des interactions entre les protéines USH et whirline 41!
Figure 15 : Localisation spatiale des protéines Usher dans les cellules cibles 43!
Figure 16 : Représentation des interactomes Usher situés au niveau des stéréocils des
cellules ciliées de l'oreille interne 44!
Figure 17 : Interactome Usher situé au niveau du cil connecteur des photorécepteurs 45!
Figure 18 : Implant cochléaire 51!
Figure 19 : Logigramme de la stratégie de diagnostic moléculaire du laboratoire 55!
Figure 20 : Diagramme de classification des variants non décrits (VSCIs) 57!
Figure 21 : Diagramme de Venn des propriétés des acides aminés 60!
Figure 22 : Représentation schématique d’une étude en minigène 61!
Figure 23 : Répartition de l’ensemble des variants GPR98 considérés comme pathogènes 97!
Figure 24 : Pourcentage d'implication des gènes chez les patients USH2 98!
Figure 25 : Grandes classes de promoteurs chez les mammifères 100!
Figure 26 : Identification des îlots CpG dans GPR98 et USH2A 101!
Figure 27 : Activité luciférase des régions promotrices de USH2A et GPR98 102!
Figure 28 : Etude de la boite TATA identifiée dans la région d'intérêt USH2A 103!
Figure 29 : Tableau récapitulatif des promoteurs USH2A et GPR98 103!
Figure 30 : Variables du NGS 108!
Figure 31 : Techniques d'amplification et d'isolement des séquences des 3 séquenceurs
Figure 32 : Représentation des sondes de capture pour un exon donné 117!
Figure 33 : Diagramme du séquençage de l'exome NS. 119!
Figure 34 : Représentation du pyroséquençage 454 GS 121!
Figure 35 : Diagrammes générés par GSdot 123!
Figure 36 : répartition des patients du groupe contrôle 126!
Figure 37 : Données brutes moyennes par run de séquençage 130!
Figure 38 : Illustration schématique du pourcentage moyen de séquences par run pour une
région d’intérêt 131!
Figure 39 : Profondeur moyenne de couverture par gène 132!
Figure 40 : Dispersion des régions en fonction de leur profondeur de couverture à l’issue du
séquençage et de leur teneur en GC 133!
Figure 41 : Dispersion des régions en fonction de leur profondeur de couverture à l’issue du
séquençage et du nombre moyen de sondes par nucléotide capturé 134!
Figure 42 : Ratio profondeur moyenne autosomes / profondeur moyenne Chr X 135!
Figure 43 : Nombre de variations selon filtration 136!
Figure 44 : Répartition des variants précédemment identifiés en Sanger 137!
Figure 45 : Filtre de priorisation des variants issus du groupe contrôle 138!
Figure 46 : Electrophérogramme de confirmation de la variation c.166_167insC dans PDZD7
pour U329 par séquençage Sanger 141!
Figure 47 : Priorisation de l'analyse dans le groupe de patients Usher 144!
Figure 48 : Filtre de priorisation des variants du groupe patients surdité 146!
Liste des tableaux
Tableau 1 : Prévalence du syndrome de Usher 23!
Tableau 2 : Loci et gènes impliqués dans le syndrome de Usher 26!
Tableau 3 : Références des gènes Usher 29!
Tableau 4 : Pathologies non-syndromiques impliquant les gènes Usher 37!
Tableau 5 : Les modèles animaux 38!
Tableau 6 : Génotypes des familles rapportées dans l’étude de Zheng et al, 2005 49!
Tableau 7 : Analyse des gènes USH2 80!
Tableau 8 : Répartition des variants GPR98 par type et par classe de pathogénicité 81!
Tableau 9 : Publications des mutations GPR98 96!
Tableau 10 : Comparatif des principales caractéristiques des séquenceurs nouvelle
génération dédiés au diagnostic. Adapté de (Loman et al., 2012) 109!
Tableau 11 : Références NCBI des gènes Usher capturés 115!
Tableau 12 : Références NCBI des autres gènes capturés 116!
Tableau 13 : Patients du groupe contrôle et gènes analysés 127!
Tableau 14 : Nombre moyen de séquences obtenues par run 130!
Tableau 15 : Répartition des régions par gène, en fonction de leur couverture 132!
Tableau 16 : Patients du groupe contrôle porteurs de 1 ou 2 mutations 139!
Tableau 17 : Variants d'intérêt du groupe « Patients Usher » 145!
Tableau 18 : Variants d'intérêt du groupe surdité 147!
Liste des annexes
Annexe 1 : Répartition des patients recensés dans LOVD USHBases 174!
Annexe 2 : Usher syndrome type II caused by activation of an USH2A pseudoexon:
implications for diagnosis and therapy. Vaché et al., 2012 176!
Annexe 3 : Nasal epithelial cells are a reliable source to study splicing variants in Usher
syndrome. Vaché et al., 2010 184!
Liste des abréviations
7TM : Domaine transmembranaire à 7 hélices aCGH : array comparative genomic hybridization BBS : Syndrome de Bardet-Biedl
CCE – OHC : Cellules ciliées externes CCI – IHC : Cellules ciliées internes
DFNA : Surdité non syndromique à transmission autosomique dominante DFNB : Surdité non syndromique à transmission autosomique récessive EAR : Epilepsy associated repeat
(c)GMP : (cyclic) Guanosine monophosphate GDP : Guanosine diphosphate
GTP : Guanosine triphosphate kb : Kilobase
LSF : Langue des signes française PBM : PDZ-binding motif
PDZ : Post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), Zonula occludens-1 protein (zo-1)
PST-rich region : Proline serine threonine rich region RCPG : Récepteurs couplés aux protéines G
RP : Rétinite pigmentaire
RPNS : Rétinite pigmentaire non syndromique TBP : Tata Binding Protein
TSS : Site d’initiation de la transcription SFF : Standard Flowgram Format USH : Usher
USMA : Usher Syndrome Missense Analysis UTR : Région non traduite
VSCI : Variation de Signification Clinique Inconnue WES : Whole Exome Sequencing
URLs des sites Internet cités
1000Genomes : http://browser.1000genomes.org/index.html ConSite : http://asp.ii.uib.no:8090/cgi-bin/CONSITE/consite CpG island Searcher : http://cpgislands.usc.edu/
Deafness Variation Database : http://deafnessvariationdatabase.org/ Ensembl : http://www.ensembl.org/index.html
Exome Variant Server : http://evs.gs.washington.edu/EVS/ Galaxy : https://main.g2.bx.psu.edu/ GSdot : https://neuro-2.iurc.montp.inserm.fr/454/ HSF : http://www.umd.be/HSF/ Jaspar : http://jaspar.genereg.net/ Mutalyzer : https://mutalyzer.nl primer3plus : http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi Seq Answers : http://seqanswers.com/wiki/Software
TFSearch : http://www.cbrc.jp/research/db/TFSEARCH UniProt : http://www.uniprot.org/
UCSC : http://genome.ucsc.edu/
USMA : https://neuro-2.iurc.montp.inserm.fr/USMA/
Généralités : les déficits sensoriels L’organe de l'audition
Le syndrome de Usher a été décrit pour la première fois par l’ophtalmologiste allemand Albrecht Von Graefe en 1858. Le caractère héréditaire sera mise en évidence, en 1914, par l'ophtalmologiste britannique Charles Usher, suite à l’étude de 69 patients atteints de RP (Usher, 1914). A ce jour, plus d'une cinquantaine de syndromes associent des pertes visuelles et auditives. Le syndrome de Usher est le plus commun d’entre eux et est impliqué pour la moitié des patients adultes atteints de surdi-cécité (Saihan et al., 2009). Ces troubles sensoriels résultent de l’atteinte simultanée des organes auditifs et visuels. Dans certaines formes du syndrome, l’atteinte de l’organe auditif conduit aussi à des troubles vestibulaires.
I. Généralités : les déficits sensoriels
A. L’organe de l'audition
L’oreille des mammifères est organisée en trois compartiments : l’oreille externe, moyenne et interne (Figure 1-a, page 17). L'oreille externe, constituée du pavillon et du conduit auditif externe, capte les ondes sonores, qui font vibrer le tympan et sont transmises à l'oreille interne par l'intermédiaire des trois osselets de l'oreille moyenne (marteau, enclume, étrier). L'oreille interne transforme le message sonore en influx électrique (processus de transduction mécano-électrique) conduit par le nerf auditif jusqu'au cerveau où l'information sonore est décodée.
Au sein de la cochlée (Figure 1-b, page 17), se situe l’organe de Corti, qui contient les cellules ciliées responsables de la transformation du signal sonore en signal électrique interprétable par le cerveau. La cochlée humaine permet la perception de fréquences sonores comprises entre 20 Hz et 20 kHz (pour revue El-Amraoui et al. 2010).
Chez l’homme, l’atteinte auditive est l’un des troubles neurosensoriels les plus répandus et peut être engendrée par de nombreux facteurs extrinsèques tels que la surexposition au bruit ou la prise de molécules ototoxiques (comme les aminoglycosides), mais peut aussi avoir une origine génétique (pour revue Smith et al., 2012). On estime d’ailleurs que l’implication génétique est supérieure à 50% pour les troubles auditifs congénitaux ou pré-linguaux. Parmi les causes génétiques, on compte 77% de troubles transmis sur le mode autosomique récessif, 22% sur le mode autosomique dominant et 1% lié à l’X (Petit and Richardson, 2009). Récemment, l’équipe de Lin a étudié en séquençage nouvelle génération, plus de 150 gènes impliqués dans les surdités génétiques englobant les formes syndromiques et non syndromiques (Lin et al., 2012), démontrant l’importante hétérogénéité génétique de ces affections.
Généralités : les déficits sensoriels L’organe de l'audition
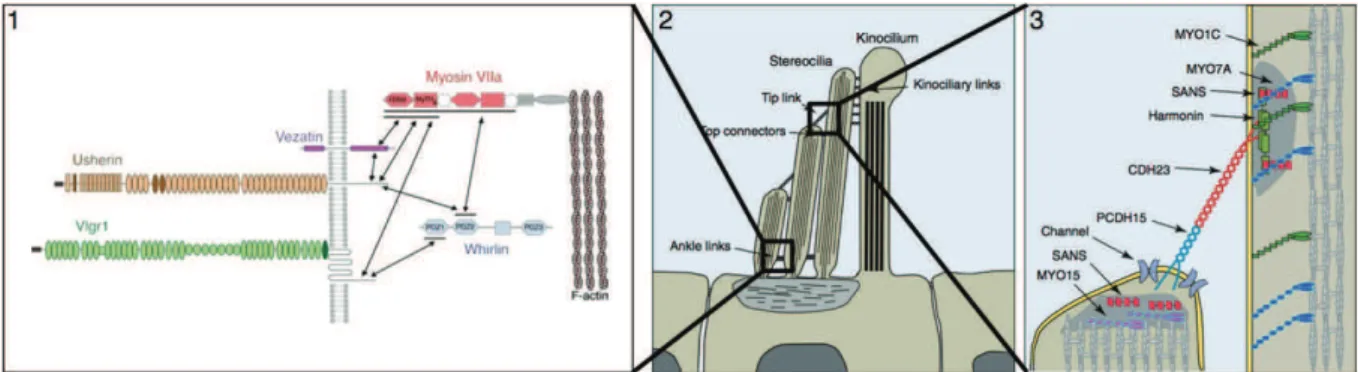
Figure 1 : Organe de l'audition
Adaptée de (Frolenkov et al., 2004). a- Représentation schématique de l’oreille et de ses trois parties distinctes (externe, moyenne et interne). b- Schéma d’une coupe horizontale de cochlée. c- Schéma de la partie mécanosensible de la cochlée : l’organe de Corti.
1. Les cellules ciliées
Les cellules ciliées de l’oreille interne sont présentes chez tous les mammifères. Leur nombre est fixe puisque les mammifères ont perdu la capacité à régénérer ces cellules (pour revue El-Amraoui et al. 2010). Chaque cochlée contient entre 15000 et 30000 cellules ciliées. Elles sont organisées en quatre rangées (Figure 2, page 17) : trois alignements de cellules ciliées externes (CCE–OHC : outer hair cells) responsables de l’amplification du signal sonore, et une rangée de cellules ciliées internes (CCI–IHC : inner hair cells), actrices de la transduction mécano-électrique du signal sonore et de sa transmission (Figure 1-c, page 17). Il existe un gradient morphologique des CCE entre la base et l’apex de la cochlée. Les cellules ciliées externes à la base, plus petites et rigides sont spécialisées dans l’amplification des fréquences élevées tandis que les cellules à l’apex de la cochlée, plus longues et plus souples, sont spécialisées dans l’analyse des fréquences basses (Legendre et al., 2009).
Sur leur face apicale, chacune de ces cellules (CCE et CCI) possède une touffe ciliaire composée de stéréocils.
Figure 2 : Rangées de cellules ciliées d’un organe de Corti humain
Généralités : les déficits sensoriels L’organe de l'audition
Des cellules ciliées sont aussi présentes dans le vestibule. Leur organisation est très semblable à celle des cellules ciliées de la cochlée. Elles possèdent sur leur face apicale, une touffe ciliaire ayant aussi un rôle de mécano-transduction sensible aux mouvements. Cet organe participe notamment à la fonction de l’équilibre en agissant comme un accéléromètre sensible aux déplacements angulaires (mouvements de rotation de la tête) et linéaires (Purves et al., 2001).
2. La touffe ciliaire
La touffe ciliaire est extrêmement bien organisée et forme un V sur la face apicale des cellules ciliées externes tandis que leur organisation est plus longiligne sur la face apicale des cellules ciliées internes. (Figure 2, page 17). En réalité, les stéréocils la composant sont des microvillosités de la paroi des cellules maintenues par une structure filamenteuse d’actine. Ils sont alignés en rang de taille décroissante, les plus grands faisant partie des rangées dont le nombre de stéréocils est le plus faible. Ils constituent les organites de la mécano-transduction. Seul un véritable cil, le kinocil, est présent sur la face apicale de ces cellules ciliées durant leur maturation (Nayak et al., 2007). Il est à l’origine de l’organisation très caractéristique de cet assemblage. Nayak décrit le développement de la touffe ciliaire en 3 étapes : lors de la première phase, des microvillosités se forment autour du kinocil. Chez la souris, cette apparition de microvillosités semble se réaliser entre E13 et E16 (13 à 16 jours de vie embryonnaire) (Lefèvre et al., 2008). Durant cette période, des liens latéraux transitoires sont présents entre les stéréocils mais aussi entre les stéréocils et le kinocil central. Ensuite, entre E17,5 et E18,5, le kinocil migre vers la périphérie. Sa position déterminera l’orientation de la touffe ciliaire. Les stéréocils les plus proches du kinocil initient alors leur étape d’élongation, qui sera suivie successivement par l’élongation des autres stéréocils, dans les rangées de plus en plus éloignées. La seconde phase de maturation consiste en l’élargissement des stéréocils avant la dernière phase terminant leur élongation et aboutissant à cette structure en escalier. L’élongation se déroule chez la souris, en post-natal, entre P1 et P5. Au cours de la mise en place de la touffe ciliaire, différents liens apparaissent. Certains d’entre eux sont transitoires car ils ne semblent pas être présents au niveau des stéréocils des touffes ciliaires arrivées à maturation. Sans compter les liens latéraux transitoires présents au moment de l’élongation des microvillosités en stéréocils, les liens inter-stéréocils chez la souris sont au nombre de 4 : le lien apical (en anglais : tip-links), 2 types de liens latéraux (en anglais : top-links et shaft connectors) et le lien d’ancrage (en anglais : ankle links). La Figure 3 (page 19) représente ces différents liens ainsi que leur stade d’apparition et de disparition durant la maturation des stéréocils et du développement
Généralités : les déficits sensoriels L’organe de l'audition
de la touffe ciliaire. Les tip-links et les top-links sont les seuls liens qui semblent persister au niveau des stéréocils des cellules ciliées matures chez la souris (Goodyear et al., 2005).
Les liens apicaux et les shaft connectors sont les premiers à être mis en place entre les stéréocils. Le composant principal des shaft connectors est la protéine codée par le gène
PTPRQ. Celle-ci est requise pour la maturation des stéréocils mais sa fonction précise est
actuellement inconnue (Goodyear et al., 2012).
En ce qui concerne le lien apical, il établit une jonction entre un stéréocil et les stéréocils adjacents des rangées inférieures. Il est formé par l’interaction entre la cadhérine 23 (partie haute du lien apical, sur le stéréocil le plus grand) et la protocadhérine 15 sur le stéréocil le plus petit (Kazmierczak et al., 2007). Ce lien joue un rôle majeur dans la mécano-transduction. Le lien d’ancrage semble se mettre en place plus tardivement. Goodyear a uniquement pu l’identifier en post-natal, par microscopie électronique (Goodyear et al., 2005). Ce lien est issu de l’interaction entre les protéines codées notamment par les gènes USH2 : usherine, GPR98 et whirline (Michalski et al., 2007). Les parties extracellulaires de usherine et GPR98 interagissent et permettent le maintien des stéréocils en touffe, à la surface des cellules ciliées.
Généralités : les déficits sensoriels L'organe de la vision
3. La mécanotransduction
La mécanotransduction permet la transformation du signal sonore en signal électrique. Ce dernier est ensuite interprété par le système nerveux central. Cette transformation s’effectue au niveau des cellules ciliées internes de l’organe de Corti situées au sein de la cochlée. Les stéréocils réagissent aux ondes sonores en s’inclinant en fonction de l’intensité de ces ondes. La sensibilité de ce mécanisme est extrêmement élevée puisque l’inclinaison maximale des stéréocils avoisine une angulation de l’ordre du degré (Corey and Hudspeth, 1983). Elle suffit pourtant à déclencher la mécanotransduction du signal initial. Cette inclinaison des stéréocils entraîne alors un étirement du lien apical créant une tension suffisante pour permettre l’ouverture d’un canal encore non défini au niveau du stéréocil le plus petit du lien, sur son extrémité apicale. C’est ce canal qui permet le passage des
cations, particulièrement les ions Ca2+, entraînant alors une dépolarisation de la cellule ciliée
et activant l’émission de neurotransmetteurs au niveau de la base de ces cellules en direction du système nerveux central. Le temps de réaction, de l’ordre de la microseconde,
conforte l’hypothèse de l’absence d’intermédiaire enzymatique qui ralentirait
considérablement ce temps de réponse (Corey and Hudspeth, 1983).
B. L'organe de la vision
1. Anatomie de la rétine
Avec la sclérotique et la cornée, la rétine est l’une des trois couches composant le globe oculaire (Figure 4, page 21). La rétine est en quelque sorte le capteur photosensible de l’œil et le foyer de la phototransduction qui convertit une source lumineuse en signal électrique. Elle recouvre environ 75% du globe oculaire. La rétine est composée de 8 sous-couches présentées dans la Figure 4 (page 21). Les sous-couches 6 et 7 forment la couche des photorécepteurs. Cette couche est reliée à la couche granuleuse interne (4) par une zone d’articulation interneuronale, la couche plexiforme externe (5) (Rozet and Kaplan, 2005). La couche granuleuse interne contient 3 types de neurones, les cellules amacrines (D), les cellules horizontales (F) et les cellules bipolaires (E). Enfin la couche granuleuse interne est reliée à la couche des cellules ganglionnaires par une seconde zone d’articulation interneuronale, la couche plexiforme interne (3). La couche ganglionnaire (1 et 2) est composée de cellules ganglionnaires dont le faisceau d’axones forme le nerf optique. La couche 8 représente l’épithélium pigmentaire. Celui-ci couvre la totalité de la surface rétinienne. A sa surface, chaque cellule épithéliale est constituée de microvillosités en
Généralités : les déficits sensoriels L'organe de la vision
contact avec les photorécepteurs. Dans la rétine périphérique chaque cellule est ainsi en contact avec 22 segments externes de bâtonnets et dans la région maculaire avec 30 segments externes de cônes (Rozet and Kaplan, 2005).
Figure 4 : Coupe transversale d’un œil et détail des couches rétiniennes
Adaptée de (Livesey and Cepko, 2001).
2. Les photorécepteurs
Deux types de photorécepteurs se distinguent : les cônes et les bâtonnets. Ces derniers, nettement plus nombreux (120 millions contre 6,5 millions de cônes) sont hautement sensibles à l’intensité lumineuse et permettent de ce fait, la vision en condition d’obscurité. Le pigment visuel des bâtonnets est la rhodopsine. Les cônes sont responsables de la vision en couleurs et en condition lumineuse. On distingue alors trois types de pigments visuels appelés photopsines (ou iodopsines), chacun étant excité par des plages de longueurs d’ondes différentes.
3. La phototransduction
La phototransduction rassemble l’ensemble des réactions enzymatiques qui se déroulent au niveau du segment externe de chaque photorécepteur (Kennan et al., 2005). La Figure 5 (page 22) illustre cette cascade au niveau des bâtonnets. En absence de lumière, le GMPc (Guanosine-3'-5'-monophosphate cyclique) est fixé aux canaux à ions Ca2+/Na+. Cette
Généralités : les déficits sensoriels L'organe de la vision
La photoexcitation permet la conversion de la rhodopsine en meta-II-rhodopsine (Rho*) en présence d’une source lumineuse. Rho* catalyse alors l’activation par le GTP de centaines de molécules de transducine (T) qui, à leur tour, activent, ou plutôt désinhibent, en se liant à leur inhibiteur, les sous-unités catalytiques des phosphodiestérases (PDE). Ces enzymes hydrolytiques extrêmement efficaces peuvent alors en quelques dixièmes de secondes éliminer du cytoplasme environnant des centaines de milliers de molécules de GMPc, ce qui stoppe l’influx ionique (Kennan et al., 2005; Yau and Hardie, 2009). Le changement de polarité consécutif est à l’origine du signal électrique qui sera alors acheminé vers le cerveau
via le nerf optique.
Figure 5 : Photoexcitation des photorécepteurs
Le syndrome de Usher
II. Le syndrome de Usher
Ce syndrome est hétérogène d’un point de vue clinique et génétique. Alors que les gènes n’étaient pas encore connus, des études épidémiologiques ont permis d'estimer une prévalence moyenne en Europe et en Amérique à 5 cas pour 100 000 naissances (Tableau 1, page 23). En France, la fréquence est de l'ordre de 3 à 5/100 000. Certaines études à l’échelle de villes comme à Birmingham en Angleterre ou Heidelberg et Mannheim en Allemagne établissent une prévalence un peu supérieure avec 6,2 cas pour 100000 individus. D’autres publications ciblées sur des populations d’Oregon et d’Iowa aux Etats-Unis, ou encore de la péninsule de Macano au Venezuela, font état de prévalences respectivement de 17 et de 76 pour 100000 mais elles ne reflètent probablement pas la prévalence globale.
Tableau 1 : Prévalence du syndrome de Usher
Prévalence Auteur Année Pays Région / Province
3 / 100 000 (Hallgren, 1959) Suède et Norvège 4,4 / 100 000 (Boughman et al., 1983) Etats Unis 3,6 / 100 000 (Grøndahl, 1987) Norvège 3,2 / 100 000 (Tamayo et al., 1991) Colombie
5 / 100 000 (Rosenberg et al., 1997) Danemark
6,2 / 100 000 (Hope et al., 1997) Angleterre Birmingham 4,2 / 100 000 (Espinós et al., 1998) Espagne Valencia
6,2 / 100 000 (Spandau and Rohrschneider, 2002) Allemagne Heidelberg, Mannheim 76 / 100 000 (Keogh et al., 2004) Venezuela Péninsule de Macano
(pop Latino-Américaine) 1 / 6000
(17 / 100 000) (Kimberling et al., 2010) Etats-Unis Oregon et Iowa 3 / 100 000
(20% surdi-cécités) Wittich et al., 2012 Canada Montréal 3,5 / 100 000 (Västinsalo et al., 2012) Finlande
Globalement, on admet une prévalence de 1 sur 25000 à 30000, mais ce chiffre pourrait être affiné par des analyses moléculaires sur des recrutements exhaustifs à plus grande échelle.
Le syndrome de Usher Hétérogénéité clinique et génétique
A. Hétérogénéité clinique et génétique
Le syndrome de Usher associe une surdité congénitale et une rétinite pigmentaire,
auxquelles peuvent s’adjoindre des troubles vestibulaires. Il existe 3 types cliniques distincts de syndrome de Usher : type I (USH1), type II (USH2) et type III (USH3). Ils se différencient selon le degré de sévérité de l'atteinte auditive, son évolution, ainsi que la présence ou non de troubles vestibulaires.
La surdité se définit en fonction de la perte auditive d’après la norme ISO389 (ISO/TC, 1996) : • Moyenne : 26-40 dB • Modérée : 41-55 dB • Modérément sévère : 56-70 dB • Sévère : 71-90 dB • Profonde : 91-120 dB
Il s'agit de surdité neurosensorielle, due à une altération de l’oreille interne (à opposer aux surdités de conduction engendrées par des troubles de l’acheminement de l’onde sonore au niveau de l’oreille externe, du tympan ou de l’oreille moyenne).
L’atteinte vestibulaire entraîne notamment des troubles de l’équilibre. Ils peuvent être à l’origine d’un retard d’acquisition de la marche.
La rétinite pigmentaire (RP) est à l’origine de la déficience visuelle des patients Usher. Que la RP soit syndromique ou non, son évolution est lente chez la plupart des patients et peut se dérouler sur plusieurs décennies. L’âge d’apparition est très variable entre les individus atteints. Les RP se caractérisent par un dépôt de pigment rétinien, initialement à la périphérie de la rétine, ainsi que par une dégénérescence initiale des bâtonnets suivie de celle des cônes (Hamel, 2006). Le terme anglais est rod-cone dystrophy. L’héméralopie (perte de la capacité à voir dans la pénombre) est d’ailleurs très souvent le premier signe clinique diagnostiqué chez les patients atteints de RP. Au fur et à mesure que les dépôts pigmentaires gagnent du terrain vers le centre de la rétine, le champ visuel poursuit son rétrécissement. La rétinite conduit à une perte totale de la vision, mais en général, les patients ont encore la capacité de percevoir la lumière, notamment au niveau périphérique (Hamel, 2006).
Le syndrome de Usher Hétérogénéité clinique et génétique
Le syndrome de Usher de type I est le plus sévère. Il associe une surdité congénitale profonde et des troubles vestibulaires à une RP progressive pouvant apparaître dès l’enfance (Kimberling et al., 1991). Il peut persister toutefois une audition résiduelle dans les basses fréquences.
Le syndrome de Usher de type II est le plus fréquent. Il se caractérise par une surdité pré-linguale modérée à sévère, des fonctions vestibulaires normales (Abadie et al., 2011) ainsi qu’une RP variable d’apparition généralement plus tardive (Kimberling et al., 1991).
Enfin, le syndrome de Usher de type III est le plus rare avec une implication de moins de 5% sur l’ensemble des patients Usher. En revanche, il est plus fréquent dans les populations finlandaises et juives Ashkénazes, et représente 40% des cas dans ces populations pour lesquelles un effet fondateur est reconnu. Cet aspect sera détaillé dans le chapitre II.D.2 (page 47).
Une importante hétérogénéité génétique caractérise le syndrome de Usher. La découverte des gènes impliqués est récente puisque le premier locus, USH2A, a été identifié en 1990 et le clonage du gène correspondant publié en 1998 (Figure 6, page 26). Sur les 17 loci initialement candidats (Tableau 2, page 26), l’implication de deux d’entre eux, USH1A et USH2B, a été réfutée. Par ailleurs, aucun gène n’a pu être encore localisé dans les régions USH1E, USH1H et USH1K. Au total, neuf gènes sont reconnus par de nombreux travaux comme étant impliqués dans le syndrome de Usher : MYO7A, USH1C, CDH23, PCDH15 et
SANS dans le type I, USH2A, GPR98 et DFNB31 dans le type II, et CLRN dans le type III
(Tableau 2, page 26).
Récemment, l’implication de trois autres gènes a été suggérée. Ebermann montre un effet possible du gène PDZD7, soit en digénisme avec GPR98 dans une famille, soit en tant que modificateur aggravant le phénotype rétinien dans une fratrie (Ebermann et al., 2010). Une variation faux-sens à l’état homozygote dans le gène HARS serait à l’origine d’un syndrome de Usher de type IIIB chez deux patients issus de la communauté Amish (Puffenberger et al., 2012). Enfin, le gène CIB2 a été impliqué dans les surdités non syndromiques (DFNB48) et dans le syndrome de Usher de type IJ. A priori, dans une famille pakistanaise, le syndrome de Usher de type I coségrège avec la variation CIB2 c.192G>C (p.Glu64Asp) à l’état homozygote (Riazuddin et al., 2012).
Le syndrome de Usher Hétérogénéité clinique et génétique
Figure 6 : Chronologie d'identification des loci et des gènes Usher
Chaque rectangle correspond à l’intervalle entre la description du locus et la publication du gène responsable.
Tableau 2 : Loci et gènes impliqués dans le syndrome de Usher
Locus Auteur identification Gène Auteur identification
USH1A (Chr14q) (Kaplan et al., 1992) Réfuté (Gerber et al., 2006)
USH1B (Chr11q) (Kimberling et al., 1992) MYO7A (Weil et al., 1995)
USH1C (Chr11p) (Smith et al., 1992) USH1C (Verpy et al., 2000; Bitner-Glindzicz
et al., 2000)
USH1D (Chr10q) (Gerber et al., 1996; Wayne et al., 1996) CDH23 (Bolz et al., 2001; Bork et al., 2001)
USH1E (Chr21q) (Chaïb et al., 1997)
USH1F (Chr10q) (Wayne, 1997. abstract ASHG) PCDH15 (Alagramam et al., 2001; Ahmed et
al., 2001)
USH1G (Chr17q) (Mustapha et al., 2002a) SANS (Weil et al., 2003)
USH1H (Chr15q) (Ahmed et al., 2009)
USH1J (Chr15q) (Riazuddin et al., 2012) CIB2 (Riazuddin et al., 2012)
USH1K (Chr10p) (Jaworek et al., 2012)
USH2A (Chr1q) (Kimberling et al., 1990) USH2A (Eudy et al., 1998)
USH2B (Chr3p) (Hmani et al., 1999) Réfuté
USH2C (Chr5q) (Pieke-Dahl et al., 2000) GPR98 (Weston et al., 2004)
USH2D (Chr9q) (Mustapha et al., 2002b) DFNB31 (Ebermann et al., 2007b)
(Chr10q) PDZD7 (Ebermann et al., 2010)
USH3A (Chr3q) (Sankila et al., 1995) CLRN1 (Joensuu et al., 2001)
Le syndrome de Usher Hétérogénéité clinique et génétique
De plus, il existe aussi, au sein d’un même type clinique, une hétérogénéité des symptômes et de leur évolution. Il est donc difficile d’établir des corrélations fines entre les génotypes et les phénotypes. Une étude visant à définir de façon précise l’atteinte auditive a été conduite sur 100 patients USH2 dont les génotypes étaient connus (Abadie et al., 2011). Nous avons observé une tendance à une perte auditive plus sévère chez les patients mutés dans le gène
GPR98 par rapport aux patients mutés dans le gène USH2A. En revanche, cette étude n’a
pas établi de corrélation entre les différents types de mutations et la nature de l’atteinte auditive. Il est probable que les facteurs environnementaux soient trop influents pour permettre ce type d’analyse.
Malgré cette hétérogénéité, la caractérisation clinique des patients permet, dans la majorité des cas, de les classer dans l'un des trois types cliniques. Et très généralement, lorsque le diagnostic moléculaire peut être établi, le gène impliqué corrèle avec ce type. Dans les rares cas contraires, on parle de patients Usher atypiques.
A ce niveau, il est important de noter que sont également considérés comme des patients Usher dits « atypiques », des patients dont le génotype est encore inconnu, et dont le phénotype ne permet pas de les classer dans l'un des trois types définis.
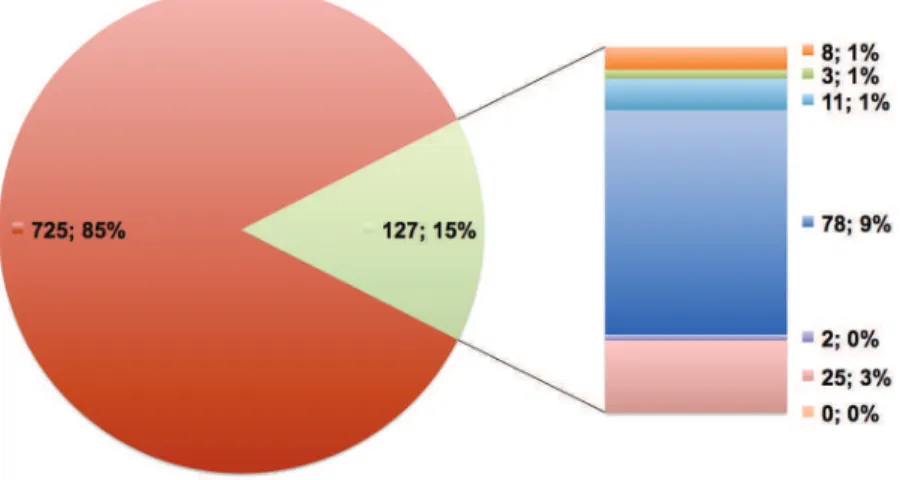
En utilisant LOVD USHbases, banque de données de type locus spécifique dédiée aux variations génomiques des gènes responsables du syndrome de Usher publiées dans la littérature, nous avons pu étudier le phénotype des patients publiés en fonction de leur génotype pathogène. A ce jour, on observe que 85% des patients mutés dans un des gènes USH1, ont bien un phénotype USH1 (Figure 7, page 28). Parmi les 15% restants, 2/3 des patients présentent une surdité non syndromique (voir chapitre II.A.5, page 36) et 1/3 ont un phénotype Usher différent de celui attendu (donc, définis comme Usher atypiques ; 2% classés en USH2 ou USH3 et 3% autres).
De même, 85% des patients mutés dans un des gènes USH2 ont un phénotype USH2 (Figure 8, page 28). Parmi les autres 15%, 33% n’ont pas de phénotype associé et 47% présentent une forme non syndromique de RP (voir chapitre II.A.5, page 36). Finalement, 20% des patients mutés dans un gène USH2 ont un phénotype différent de celui attendu (2% associés à un autre phénotype Usher, USH1 ou USH3 et 1% autre).
Le syndrome de Usher Hétérogénéité clinique et génétique
Figure 7 : Répartition clinique des patients mutés dans un des gènes USH1
Basée sur les informations relatives aux 852 patients mutés dans un des gènes USH1 et répertoriés dans LOVD USHBases. USH1, 2 ou 3 : Phénotype Usher respectivement de type I, II ou III ; DFNA/B : surdité non syndromique de transmission autosomique dominante/récessive; ARRP : Rétinite pigmentaire non syndromique à transmission autosomique récessive ; Autres : Phénotype différent des précédents ; NC : phénotype non communiqué
Figure 8 : Répartition clinique des patients mutés dans un des gènes USH2
Basée sur les informations relatives aux 1000 patients mutés dans un des gènes USH2 et répertoriés dans LOVD USHBases. USH1, 2 ou 3 : Phénotype Usher respectivement de type I, II ou III ; DFNA/B : surdité non syndromique de transmission autosomique dominante/récessive; ARRP : Rétinite pigmentaire non syndromique à transmission autosomique récessive ; Autres : Phénotype différent des précédents ; NC : phénotype non communiqué
Ces chiffres, représentant une méta-analyse de la littérature internationale sur le syndrome de Usher, indiquent clairement que la classification clinique en trois types reste pertinente en 2012.
Hétérogénéité clinique et génétique Gènes et protéines USH1
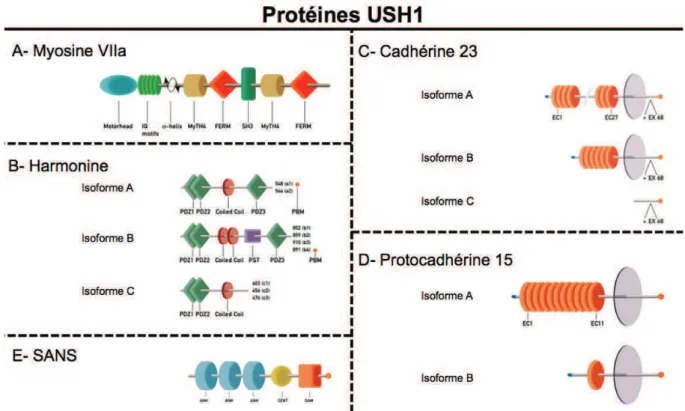
1. Gènes et protéines USH1
A ce jour, 6 gènes ont été identifiés comme responsables du syndrome de Usher de type I (Tableau 3, page 29). Ils codent pour des protéines dont les fonctions sont hétérogènes (Figure 9, page 31) Le sixième gène, CIB2, a été caractérisé par Riazuddin et al. 2012 durant la rédaction de ce manuscrit. De ce fait, il n’a pas été étudié durant ce travail de thèse.
Tableau 3 : Références des gènes Usher
Locus Symbole HGNC HGNC ID Gene MIM Phenotype MIM Entrez ID RefSeqGene NCBI Ensembl Gene ID USH1 11q13 MYO7A 7606 *276903 #276900 4647 NG_009086.1 ENSG00000137474 11p14 USH1C 12597 *605242 #276904 10083 NG_011883.1 ENSG00000006611 10q22 CDH23 13733 *605516 #601067 64072 NG_008835.1 ENSG00000107736 10q21 PCDH15 14674 *605514 #602083 65217 NG_009191.1 ENSG00000150275 17q25 USH1G 16356 *607696 #606943 124590 NG_007882.1 ENSG00000182040 15q24 CIB2 10518 *605564 - 24579 - ENSG00000136425 USH2 1q41 USH2A 12601 *608400 #276901 7399 NG_009497.1 ENSG00000042781 5q13 GPR98 17416 *602851 #605472 84059 NG_007083.1 ENSG00000164199 9q32 DFNB31 16361 *607928 #611383 25861 NG_016700.1 ENSG00000095397 USH3 3q25 CLRN1 12605 *606397 #276902 7401 NG_009168.1 ENSG00000163646
HGNC : Human Genome Organisation (HUGO) Gene Nomenclature Committee ; HGNC ID : identifiant HGNC ; MIM : Mendelian Inheritance in Man ; Entrez ID : numéro d’identification du gène au NCBI (National Center for Biotechnology Information, USA) ; NCBI RefSeqGene : Numero d’accession de la séquence du gène au NCBI ; Ensembl Gene ID : Identifiant dans la base de données Ensembl.
a. MYO7A
C’est en 1992 qu’a pu être identifié le locus USH1B sur le chromosome 11q (Kimberling et al., 1992). Le gène MYO7A fut mis en évidence en 1995 (Weil et al., 1995). Il code pour la myosine VIIa (Figure 9-A, page 31). Considérée comme une myosine non-conventionnelle, myo7a possède les caractéristiques des protéines à tête motrice. L’isoforme majeure contient 2215 acides aminés (NP_000251.3 ; ENSP00000386331) codée par la transcription de 49 exons (NM_000260.3 ; ENST00000409709). La large extrémité C-terminale jouerait
Hétérogénéité clinique et génétique Gènes et protéines USH1
b. USH1C
En 1992, Smith cartographie un autre locus, USH1C, sur le bras court du chromosome 11 (Smith et al., 1992) dans une famille acadienne (région francophone nord-américaine entourant le Nouveau-Brunswick). En 2000, deux équipes identifient le gène USH1C comme responsable du syndrome de Usher de type I dans plusieurs familles (Bitner-Glindzicz et al., 2000; Verpy et al., 2000). Verpy et al., montrèrent également que ce gène peut être impliqué dans les surdités non syndromiques à transmission autosomique récessive (Verpy et al., 2000). USH1C code pour l’harmonine (Figure 9-B, page 31), une protéine d’échafaudage (« scaffold protein ») dont les nombreuses isoformes peuvent être classées en 3 sous-types (a, b et c). Le transcrit codant pour la plus grande isoforme (NP_710142.1 ; ENSP00000005226) est issu de 27 exons (NM_153676.3 ; ENST00000005226).
c. CDH23
Le locus USH1D fut identifié en 1996 (Gerber et al., 1996; Wayne et al., 1996) sur le bras long du chromosome 10. En 2001, Bolz et Bork identifièrent indépendamment le gène codant pour la cadhérine 23 (Figure 9-C, page 31) en étudiant son isoforme de 3354 acides aminés (Bolz et al. 2001; Bork et al. 2001). Il occupe une région chromosomique d’environ 420 Kb.
L’isoforme A (la plus grande) de la cadhérine 23 (NP_071407.4 ; ENSP00000224721) est le produit d’un transcrit de 69 exons (NM_022124.5 ; ENST00000224721)
d. PCDH15
En 1997, le locus USH1F situé sur le bras long du chromosome 10q fit l’objet d’une communication à l’ASHG (Wayne, 1997. abstract ASHG) et le gène PCDH15 fut identifié en 2001 par 2 équipes en parallèle (Alagramam et al. 2001; Ahmed et al. 2001). Le gène s’étend sur près de 1 Mb en 10q21.
De nombreux transcrits ont été observés pour ce gène (Ahmed et al., 2008), regroupés en trois classes principales dénommées CD-1, 2 ou 3. Ces classes diffèrent essentiellement par la partie 3’ des transcrits, et notamment par la nature de l’exon final qui encode entre 184 et 500 acides aminés.
Le transcrit historiquement le plus étudié (NM_033056.3 ; ENST00000320301) est composé de 33 exons codant une protéine de 1955 acides aminés (NP_001136236.1 ; ENSP00000354950), la protocadhérine 15 (Figure 9-D, page 31)
Hétérogénéité clinique et génétique Gènes et protéines USH1
e. USH1G
Le locus USH1G fut identifié en 2002 (Mustapha et al., 2002a), puis le gène SANS, d’environ 7 kb fut décrit l’année suivante (Weil et al., 2003, 200).
Seul un transcrit, issu de 3 exons et composé de 3561 nucléotides est actuellement connu (NM_173477.2 ; ENST00000319642). Il code pour une protéine de 461 acides aminés (NP_775748.2. ; ENSP00000320076), SANS (Figure 9-E, page 31).
Figure 9 : Principales isoformes des protéines USH1
Adaptée de (Kremer et al., 2006). A- Myosine VIIa se compose d’un domaine moteur de myosine (myosin motorhead), de 5 motifs de fixation de calmoduline (IQ motifs, pour isoleucine (I), glutamine (Q)), 2 répétitions d’un domaine Myosin Tail Homology 4 (MyTH4) suivi d’un domaine FERM séparées par un domaine Src homology 3 (SH3). B- Les différentes isoformes d’harmonine sont composées de 2 ou 3 domaines PDZ (post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1)), d’1 ou 2 domaines coiled-coil (CC) et potentiellement d’un motif PBM (PDZ binding motif) et/ou d’une région riche en proline, sérine et thréonine (PST). C- Cadhérine 23 est composée d’un motif PBM et potentiellement, d’un domaine transmembranaire (TM) et de répétitions de domaines extracellulaires de cadhérine (jusqu’à 27 domaines) permettant la fixation des ions Ca2+. D- Protocadhérine 15 présente une organisation similaire à cadhérine 23 avec jusqu’à 11 domaines EC. E- Sans est constituée de 2 domaines ankyrines (ANK), d’une région centrale (CENT), d’un motif stérile alpha (SAM) ainsi que d’un motif PBM.
Hétérogénéité clinique et génétique Gènes et protéines USH1
f. Implication des gènes USH1
Récemment, deux études sur de grandes cohortes de patients USH1 ont été réalisées. Pour la première, les patients sont majoritairement français (Roux et al., 2011) tandis que la seconde étude recense en grande majorité des patients USH du Royaume-Uni (Le Quesne Stabej et al., 2012). L’implication des différents gènes USH1 dans ces études est représentée par la Figure 10 (page 32). Pour chaque gène, la variation d’implication n’excède pas 13 points entre les deux études.
Figure 10 : Pourcentage d'implication des gènes chez les patients USH1
Cette analyse prend uniquement en compte les patients pour lesquels un génotype pathogène a été identifié.
Comme l’avait mis en évidence une précédente étude (Weil et al., 1995), MYO7A est le gène le plus impliqué dans le syndrome de Usher de type I et représente entre plus de 60% des cas. La Figure 10 montre une nette différence en ce qui concerne l’implication du gène
USH1C, beaucoup plus présent au Royaume-Uni. Cette différence peut s’expliquer par la
présence d’une mutation fréquente dans cette population (c.496+1G>A), jamais retrouvée jusqu’ici, en France (données issues de LOVD USHBases). Les deux populations diffèrent beaucoup moins concernant les gènes CDH23 et PCDH15.
Enfin, aucun patient porteur de mutations dans USH1G n’a été décrit au cours de ces deux études. Cependant, notre équipe a identifié depuis, des mutations dans ce gène chez deux patients (données non publiées).
0 10 20 30 40 50 60 70
MYO7A CDH23 PCDH15 USH1C USH1G
63,3 20 12,2 4,5 0 61,0 12,2 9,7 17,1 0,0 Po u rc e n ta g e d 'i m p li c a ti o n Roux et al. 2011 Le Quesne et al. 2012
Hétérogénéité clinique et génétique Gènes et protéines USH2
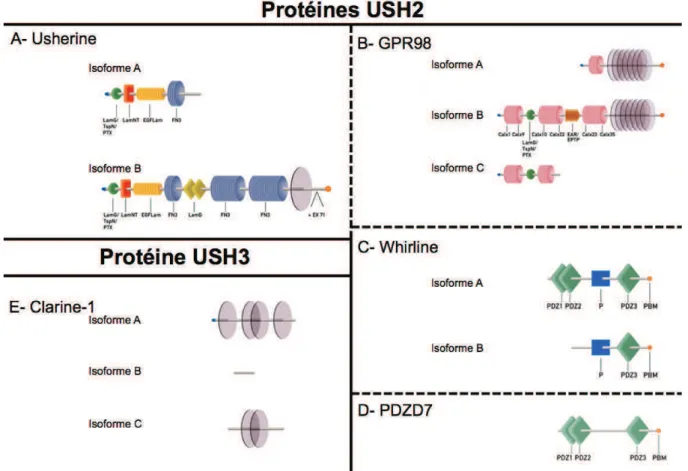
2. Gènes et protéines USH2
Trois gènes ont été identifiés comme responsables du syndrome de Usher de type II (Tableau 3, page 29). Les protéines codées par ces gènes sont représentées Figure 11 (page 35).
a. USH2A
En 1990, suite à l’étude de marqueurs ADN chez huit familles atteintes d’un syndrome de Usher de type II, Kimberling a cartographié le locus USH2A sur le chromosome 1q (Kimberling et al., 1990). Il affina la position en 1995 en le situant en 1q41 (Kimberling et al., 1995). En 1998, une partie du gène fut cloné. Elle représentait 21 exons (NM_007123.5 ;
ENST00000366942) codant pour la petite isoforme usherine (NP_009054.5 ;
ENSP00000355909) et couvrant 250 kb (Eudy et al., 1998). En criblant ces exons, seulement 63% des mutations attendues dans USH2A étaient identifiées et il fallut attendre 2004 et les travaux de van Wijk pour que 51 nouveaux exons soient identifiés (van Wijk et al., 2004). Le gène USH2A couvre une région chromosomique d’environ 800 kb. Il est composé de 72 exons (NM_206933.2 ; ENST00000307340) codant pour une protéine de 5202 acides aminés qui comporte notamment une large partie extracellulaire (NP_775748.2 ; ENSP00000320076).
D’après UniProt (http://www.uniprot.org/), usherine comporte 3 parties : une partie cytoplasmique possédant en son extrémité C-terminale un motif PDZ-binding (PBM), un domaine transmembranaire et une imposante partie extracellulaire (Figure 11-A, page 35). Celle-ci possède un domaine Laminin N-terminal, 10 domaines Laminin EGF-like, 2 domaines Laminin G-like ainsi que 35 domaines Fibronectine type-III (van Wijk et al., 2004).
b. GPR98
En 2000, soit 10 ans après l’identification du locus USH2A, Pieke-Dahl et al. ont mis en évidence un nouveau locus, USH2C, situé sur le chromosome 5q entre 5q14.3 et 5q21.1 (Pieke-Dahl et al., 2000). Un gène se distingua rapidement comme probablement responsable du syndrome de Usher, de par son expression chez le fœtus humain, au niveau de la cochlée et de la rétine (Weston et al., 2004) : il s’agissait du gène GPR98, codant « the
very large G-coupled receptor » ou VLGR1, désormais connue sous le nom de G
protein-coupled receptor 98 (GPR98) (Figure 11-B, page 35). Comme les autres protéines transmembranaires, elle est composée de 3 régions distinctes. C’est sa structure en sept hélices hydrophobes transmembranaires (7TM) séparant son domaine cytoplasmique de son
Hétérogénéité clinique et génétique Gènes et protéines USH2
des récepteurs couplés aux protéines G (RCPG). Ce gène s’étend sur plus de 600 kb, se compose de 90 exons pour l’isoforme la plus longue, GPR98 (NM_032119.3 ; ENST00000405460), et code pour l’une des plus grandes protéines décrites (6306 acides aminés ; NP_115495.3 ; ENSP00000384582).
Trois isoformes (initialement VLGR1a, b et c) générées par épissage alternatif ont été mises en évidence (McMillan et al., 2002). La partie cytoplasmique des isoformes GPR98 A et B (Figure 11-B, page 35) possède en C-terminal et au même titre que usherine, un motif PBM, tandis que sa large partie extracellulaire contient un domaine GPS (G-proteolytic site) proche du domaine 7TM, 35 domaines Calx-β (homologues aux domaines spécifiques des protéines
régulant les échanges NA+/Ca2+), ainsi que 7 domaines EAR (epilepsy associated repeat)
(Weston et al., 2004). Ces derniers doivent leur nom à l’association initiale d’une partie de ce gène (à l’époque appelé MASS1) à une pathologie autosomique dominante dont les symptômes sont des crises convulsives hyperthermiques. Mais en 2006, cette association fut rejetée par les travaux de Deprez associant ces troubles au locus FEB4 situé entre 5q14.3 et 5q23.1 (Deprez et al., 2006).
Aujourd’hui encore GPR98 fait partie de la famille des récepteurs couplés au protéines G, dits « orphelins » car le ligand reste inconnu et la fonction n’est pas véritablement établie (Foord et al., 2005). Toutefois, elle semble être impliquée dans le maintien de l’intégrité de la touffe ciliaire des cellules ciliées de l’oreille interne (McGee et al., 2006; Michalski et al., 2007), le développement du système nerveux central et la fonction des photorécepteurs (McMillan et al., 2002; Schwartz et al., 2005).
c. DFNB31
Le locus DNFB31 a été mis en évidence en 2002 dans la région 9q32-9q34, chez une famille atteinte de surdité congénitale (Mustapha et al., 2002b). Bien que les premières mutations aient été identifiées chez des patients souffrant de surdité non syndromique (Mburu et al., 2003), son implication dans le syndrome de Usher de type II fut démontrée en 2007 (Ebermann et al., 2007b).
D’après Uniprot, l’isoforme la plus longue (Figure 11-C, page 35) composée de 907 acides aminés (NP_056219.3 ; ENSP00000354623) est issue d’un transcrit composé de 12 exons (NM_015404.3 ; ENST00000362057). Trois domaines PDZ sont présents et font de la whirline une protéine d'échafaudage.
Hétérogénéité clinique et génétique Gènes et protéines USH2
Figure 11 : Principales isoformes des protéines USH2 et USH3 Adaptée de (Kremer et al., 2006).
A- L’isoforme A d’usherine contient un domaine thrombospondin/pentaxin/laminin G-like (LamG/TspN/PTX), un domain Laminin-N-terminal (LamNT), 10 domaines laminine EGF-like (EGF Lam) et 4 domaines fibronectine 3 (FN3) tandis que l’isoforme B possède en plus, 2 domaines laminine G (LamG), 23 domaines FN3, un domaine transmembranaire (TM) ainsi qu’un motif PDZ binding motif (PBM). B- L’isoforme la plus grande de GPR98 (isoforme B) contient 35 domaines Calx-beta entre lesquels s’intercalent un domaine LamG/TspN/PTX ainsi qu’une région composée de 7 domaines EAR (epilepsy associated repeat), une région à 7 TM et un motif PBM. C- l’isoforme A de la whirline contient 3 domaines PDZ, une région riche en proline (P) ainsi qu’un domaine PBM. D- PDZD7 contient 3 domaines PDZ et un motif PBM. E- Clarine contient seulement 4 domaines TP dans son isoforme la plus grande.
d. Implication des gènes USH2
Fin 2008, date à laquelle j’ai débuté mon travail de thèse, seul le gène USH2A était analysé dans les laboratoires et représentait 80% des cas USH2 (Baux et al., 2007). L’implication des autres gènes USH2 a été définie au cours des 4 dernières années. Ce travail faisant l’objet de la première partie de ma thèse, il sera développé dans la Partie II.