Development of BMP type I receptor kinase inhibitors for the
treatment of fibrodysplasia ossificans progressiva and the
study of the BMP signaling pathway.
by
Agustin Humberto Mohedas
B.S. Biomedical Engineering Texas A&M University, 2007
SUBMITTED TO THE HARVARD-MIT DIVISION OF HEALTH SCIENCES AND TECHNOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE
DEGREE OF
DOCTOR OF PHILOSOPHY IN MEDICAL ENGINEERING AND MEDICAL PHYSICS AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
JUNE 2014
C 2014 Massachusetts Institute of Technology. All rights reserve A4C T TS INS E OF TECHOLOGY
UN 18 2014
Signature redacted
BRARIE o
S u Harvard-MIT Division of Health Sciences and TechnologyMay 19, 2014
Certified by:
Signature redacted
6-'
Accepted by:Paul B. Yu, MD, PhD Assistant Professor of Medicine
Signature red acted
IThesis Supervisor
Emery N. Brown, MD, PhD Director Harvard-MIT Program in Health Sciences and Technology Professor of Computational Neuroscience and Health Sciences and Technology
Development of BMP type I receptor kinase inhibitors for the
treatment of fibrodysplasia ossificans progressiva and the
study of the BMP signaling pathway.
by
Agustin Humberto Mohedas
Submitted to the Harvard-MIT Division of Health Sciences and Technology on May 19, 2014 in Partial Fulfillment of the
Requirements for the Degree of Doctor of Philosophy in Medical Engineering and Medical Physics
Abstract
The BMP signaling pathway is essential for embryonic development and the maintenance of tissue homeostasis. Dysregulated BMP signaling, both loss and gain-of-function, has been demonstrated in the pathogenesis of diseases including cancer, atherosclerosis, anemia and particularly hereditary disorders such as pulmonary arterial hypertension, hereditary hemorrhagic telangiectasia, and fibrodysplasia ossificans progressiva (FOP). FOP is a rare and disabling condition caused by a highly recurrent mutation in the ACVR1 gene encoding the BMP type I receptor activin-like kinase 2 (ALK2), characterized by the progressive heterotopic ossification (HO) of skeletal muscle and connective tissue leading to widespread joint immobilization, with significant morbidity and premature mortality. There are currently no effective treatments for FOP. The goal of this thesis is to develop and characterize highly selective BMP type I receptor inhibitors targeting ALK2 for the treatment of FOP.
Despite the high degree of structural homology between all the BMP and TGF-p type I receptors, I hypothesized that potent and selective inhibitors targeting a single BMP type I receptor, ALK2, could be developed based on a previously identified pyrazolo[1,5-a]pyrimidine core scaffold. I screened a library of pyrazolo[1,5-a]pyrimidine derivatives in a high throughout sensitive radiometric assay of BMP and TGF-P type I receptor kinase activities. I identified a derivative with a unique chemical moiety (5-quinoline) that demonstrated high selectivity for ALK2, but with lower potency than the parent molecule. We synthesized a new 5-quinoline derivative with increased potency and selectivity for ALK2 over the other BMP type I receptors and greatly improved selectivity against the TGF--P type I receptors. I used this highly selective compound to examine ALK2-mediated BMP signaling in vitro and demonstrated in vivo efficacy in two mouse models of HO.
In a complementary approach, we generated a library of novel BMP type I receptor inhibitors based on the 2-aminopyridine core scaffold. I developed a structure activity relationship to determine the key structural elements responsible for potency and selectivity. We
identified a several novel derivative compounds with improved potency and selectivity for ALK2 over the parent. We successfully used this set of derivatives to address a specific question in FOP biology, of whether ATP-competitive kinase inhibitors exert differential activity against wild-type or diverse FOP-causing ALK2 mutants.
Finally, in our SAR of pyrazolopyrimidine compounds, we identified a highly potent inhibitor of both BMP and TGF-$ type I receptor activity. I characterized the ability of this compound to inhibit ligand-induced BMP and TGF- signaling in a variety of cell culture models, as well as inhibit the activity of individual type I receptors. We then used this compound to examine the contribution of individual BMP and TGF-P receptors to signal transduction. We used the broad activity of this inhibitor to limit signaling of all endogenous BMP and TGF-P type I receptors in cells, while reconstituting the activity of specific type I receptors using engineered, inhibitor-resistant mutant receptor kinases which we developed by modifying gatekeeper residues critical for interactions with inhibitor. These mutant receptor kinases demonstrated preserved basal and ligand-mediated signaling functions which were unaffected by inhibitor. These results demonstrate proof-of-principle of a system for examining the function of individual receptors of this pathway in isolation.
The work presented in this thesis advances the development of novel BMP type I receptor kinase inhibitors of high selectivity and potency which could serve as important tools for the study of BMP signaling and as therapies for diseases of excessive BMP signaling such as FOP. Development of highly potent and selective inhibitors of ALK2 offers the hope of rational disease modifying therapy for the treatment of FOP.
Thesis Supervisor: Paul B. Yu
Acknowledgments
I dedicate this thesis to my grandfather, Umberto Panza, who has inspired me throughout my life. He has served as a standard to which I hold myself and hope to one day match.
This thesis would not have been possible without the love, commitment, and patience of my fiance Megan Baugh. I thank you most of all.
I would like to thank my parents Teresa and Sergio Mohedas, who from an early age instilled in me the importance of education and the power of hard work. I would not be where I am today without the unconditional support they have given me throughout my life. Thank you to my brother Ibrahim and sister Jimena for the fond memories and insults we've shared.
I thank my mentor and thesis advisor Dr. Paul Yu who throughout these years has been nothing but supportive of my research and goals. To my lab colleagues Kelli Armstrong, Dr. Jana Bagarova, Patrick McManus, Dr. Ivana Nikolic, Sam Paskin-Flerlage, Ashley Vonner, and Dr. Lai-Ming Yung thank you for putting up with me all of these years!
Thank you to my thesis committee members Dr. Collin Stultz, Dr. Thomas Michel, and Dr. Greg Cuny who listened carefully and provided crucial guidance. The work presented in this thesis is the result of multiple fruitful collaborations and I am forever grateful to Dr. Alex Bullock, Dr. Greg Cuny, and Dr. Joerg Ermann. I want to also thank the other scientists that contributed to this work; specifically Dr. Xuechao Xing and Dr. You Wang for synthesizing compounds and Dr. Caroline Sanvitale for solving crystal structures and assaying compounds.
I am also thankful to have had the opportunity to work with Dr. Arthur Lee and all of the great people at the Therapeutics for Rare and Neglected Diseases program at the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH). I hope my contributions to this program have in some small way helped advance the development of treatments for FOP.
Finally, I would like to thank all of my friends and the unique people that I've had the pleasure of knowing during my time at MIT. It has been quite a journey.
Table of Contents
Chapter 1 Introduction...17
Chapter 2 Background and M otivation... 20
2.1 The TGF-p Fam ily ... 20
2.1.1 H istorical perspective... 21
2.1.2 Canonical TGF-p and BM P signaling... 21
2.1.3 Role of TGF-P and BMP signaling in development and homeostasis ... 23
2.2 D iseases of TGF-3 and BM P Signaling ... 24
2.2.1 TGF-P and BM P signaling in disease ... 24
2.2.2 Fibrodysplasia ossificans progressiva... 26
2.3 K inase Inhibitors ... 28
2.3.1 K inase inhibitor developm ent and clinical use... 28
2.3.2 TGF-p type 1 receptor kinase inhibitors ... 30
2.3.3 BM P type 1 receptor kinase inhibitors ... 30
2.4 Sum m ary ... Error! Bookm ark not defined. Chapter 3 Characterization of dorsom orphin derivatives ... 33
3.1 Background and M otivation... 33
3.2 Experim ental M ethods ... 34
3.2.1 Traditional radiom etric kinase assay... 34
3.2.2 H igh throughput radiom etric kinase assay ... 36
3.3 Results and D iscussion...37
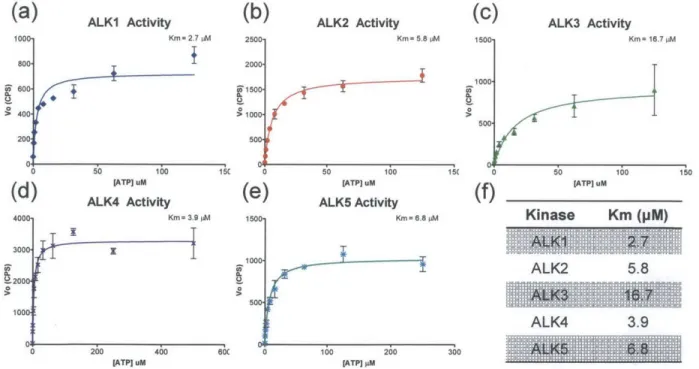
3.3.1 K m determ ination for A LK 1-5 ... 37
3.3.2 Screening of dorsom orphin deriviatives against ALK 1-5... 38
3.4 Conclusion ... 44
Chapter 4 Developm ent of potent and selective ALK2 inhibitors... 45
4.1 Background and M otivation... 45
4.2 Experim ental M ethods ... 46
4.2.1 Cell culture ... 46
4.2.2 Luciferase reporter assay... 46
4.2.3 Ligand induced SM A D phosphorylation ... 47
4.3 Results and D iscussion...48
4.3.2 LDN-212854 binding mode...55
4.3.3 Kinase profiling of LDN-193189 and LDN-212854...57
4.4 Concluson...5
4.4 Conclusion ... 62
Chapter 5 Study of BM P signaling using an ALK2 selective inhibitor... 64
5.1 Background and M otivation...64
5.2 Experimental M ethods...65
5.2.1 BM P-Induced ALP activity... 65
5.2.2 IL-6 induced hepcidin expression ... 65
5.2.3 caALK2 (Q207D) M ouse M odel of FOP... 66
5.3 Results and Discussion...66
5.3.1 LDN-212854 preferentially inhibits ALK2... 66
5.3.2 IL-6 induced hepcidin expression is predominantly mediated by ALK3...67
5.3.3 LDN -212854 inhibits ALK2Q207D-induced heterotopic ossification ... 69 5.3.4 LDN-212854 inhibits heterotopic ossification in Bmall-'- m ice ... 71
5 .4 C o n c lu sio n ... 7 6 Chapter 6 Development of 2-aminopyridine BM P kinase inhibitors ... 77
6.1 Background and M otivation... 77
6.2 Experimental M ethods ... 78
6.2.1 Therm al shift kinase assay ... 78
6.2.2 Cell viability assay ... 78
6.3 Results and Discussion...79
6.3.1 Structure activity relationship (SAR) of solvent exposed group... 79
6.3.2 Structure activity relationship (SAR) of hydrophobic pocket position ... 83
6.3.3 Structure activity relationship (SAR) of hinge binding position... 85
6.3.4 Structure activity relationship (SAR) of K02288 and LDN-193189 hybrid molecules...86
6.3.5 Kinome selectivity of LDN-212838 and LDN-214117 ... 88
6.3.6 FOP mutations, inhibitor binding affinity, and implications for therapeutics...96
6.3.7 Cytotoxicity of kinase inhibitors... 98
6.3.8 Structural basis of inhibitor binding... 99
6.4 Conclusion ... 102
Chapter 7 Development of a potent dual BM P/TGF-p inhibitor ... 104
7.1 Background and M otivation...104
7.2.1 Kinase assay ... 105
7.2.2 Luciferase reporter assay...105
7.2.3 Cell viability assay ... 106
7.3 Results and Discussion...107
7.3.1 Lead optimization of LDN-193189 to improve metabolic stability...107
7.3.2 SAR of LDN-193189 derivatives reveals 2-methylquinoline reduces selectivity ... 108
7.3.3 In vivo metabolism of TRND-343765 ... 109
7.3.4 Characterization of TRND-343765 as a potent dual BMP and TGF-P inhibitor ... 111
7.3.5 M utant type I receptors and TRND-343765 as in vitro probes of signaling ... 113
7 .4 C o n clu sio n ... 1 16 Chapter 8 Conclusions...117
8.1 Summary of the thesis...117
8.2 Suggested future work...118
8.2.1 Optimization of 5-quinoline position ... 118
8.2.2 Role of BM P signaling in heterotopic ossification in Bmall-.- ... 119
8.2.3 Improved selectivity for TRND-343765 ... 119
8.2.4 Resistant BM P and TGF-P type I receptors to study signaling ... 120 B ib lio grap hy ... 12 1
List of Figures
Figure 2.1. Phylogenetic tree of the TGF-p super family of signaling ligands, receptors, and intracellular transduction proteins (SM ADs). Adapted from [2]. ... 20 Figure 2.2. A schematic representation of the canonical BMP and TGF- signaling pathways
demonstrating ligand binding to tetrameric complexes of type I and type II receptors that phosphorylate R-SMADs, which translocate to the nucleus and effect specific transcriptional programs. ... 22
Figure 2.3. (a) A color coded grid with the percent sequence identity for each BMP and TGF-0 type I receptor was determined using the alignment tool (http://www.uniprot.org/blast/). (b) Superposition of ALKI and ALK2 demonstrating the high degree of similarity between these receptors despite their unique fu n ctio n s...2 3 Figure 2.4. (a) FOP-causing mutations in both the GS loop and kinase domain. (b) Soft tissue nodules
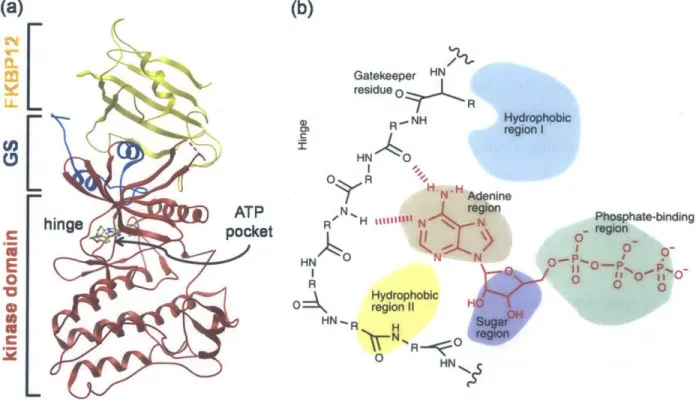
and HO lesions on the back of a 4-year-old child affected by FOP. (c) The same child at 8 years of age showing extensive HO of the back. Adapted from [73] and [74]. ... 27 Figure 2.5. (a) Structural overview of ALK2 demonstrating the bilobular structure of kinases including the kinase domain, hinge, GS domain, ATP binding pocket, and interacting regulatory protein FKBP 12.
(b) The highly conserved ATP binding region of kinases showing hydrogen bonding interactions between adenine and the hinge as well as hydrophobic pockets that are amenable to inhibitor binding. Adapted fro m [8 8 ]. ... 2 9 Figure 3.1. (a) Structure of the pyrazolo[1,5-a]pyrimidine core with R-groups at the 5-position of the pyrimidine (RI) and the 2-(R2) and 3-(R3) positions of the pyrazolo. (b) The structure of dorsomorphin with a piperidinyl ethoxy phenyl at R1, hydrogen at R2, and 4-pyridyl at R3. (c) The structure of
LDN-193189 with a phenylpiperazine at Ri, hydrogen at R2, and 4-quinolyl at R3. ... 33 Figure 3.2. Schematic representation of a traditional kinase assay with the kinase bound radiolabeled ATP (a) phosphorylating its protein substrate (b) which is then transferred to phosphocellulose paper (c) and finally placed in a vial with scintillation fluid (d) for light output measurement...35 Figure 3.3. (a)Traditional kinase assay setup including 96 scintillation vials, caps, and racks. (b) Equivalent high throughput radiometric kinase assay setup with 96-well plate. (c) Comparison metrics between traditional kinase assay and our optimized high throughput assay...36 Figure 3.4. Km determinations for (a) ALK1, (b) ALK2, (c) ALK3, (d) ALK4, and (e) ALK5. (f) Summary of Km values for BMP and TGF-p type I receptor kinases...37 Figure 3.5. Structure-activity relationship of 4-methoxyphenyl pyrazolo[1,5-a]pyrimidine derivatives. (a) In vitro kinase assay IC50 measurements against BMP and TGF-P type I receptors show only the
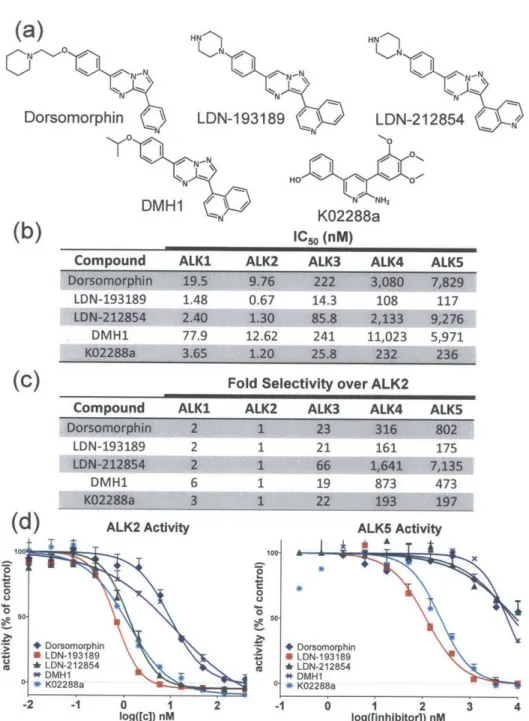
4-quinoline (LDN-193688) and 5-4-quinoline (LDN-193719) derivatives are active. (b) Structure of five pyrazolo[1,5-a]pyrimidine derivatives with varying quinoline attachment sites. (c) Fold selectivity of active inhibitors against ALK2 versus BMP and TGF-p type I receptors. Although slightly less potent against ALK2 than LDN-193688, the 5-quinoline derivative LDN-193719 has much greater selectivity for B M P vs. T G F-p type I receptors. ... 43
Figure 4.1. Synthetic scheme of LDN-212854. (a) AcOH, MeOH, 80 C, (61%). (b) Pd(PPh3)4, 2.0 M
Na2CO3, dioxane, 101
0
C, (82-95%). (c) NBS, DCM, (63%). (d) TFA, DCM, then sat. NaHCO3, (55%).
... 4 7 Figure 4.2. Potency and selectivity of BMP inhibitors based on in vitro kinase assay. a) Structure of previously described BMP inhibitors and LDN-212854. b) In vitro kinase assay measurements of IC5 0 for BMP and TGF-p type I receptors show LDN-193189 is the most potent inhibitor of ALK2, followed
closely by K02288a and LDN-212854. Both DMH1 and dorsomorphin exhibited 10-fold lower potency against BMP type I receptors. c) Fold selectivity of these inhibitors against ALK2 versus closely related BMP and TGF-p type I receptors. d) Inhibition of ALK2 (BMP) and ALK5 (TGF-p) kinase activity demonstrates LDN-212854 exhibits increased selectivity for ALK2 versus ALK5. Data shown are
representative of 2-3 independent experim ents... 50 Figure 4.3. Selectivity of known BMP inhibitors and novel inhibitor LDN-212854 based on in vitro kinase assay and cell-based BMP (BRe-Luc) and TGF-P (CAGA-Luc) transcriptional activity mediated by constitutively active ALK 1-5. (a) Graphical representation of the fold selectivity of these inhibitors in kinase assay against ALK2 versus closely related BMP and TGF-p type I receptors. (b) and (c) A graphical representation of the fold selectivity of inhibitors for caALK2 over closely related BMP and TGF-s type I receptors. LDN-212854 has 6-10 fold selectivity for caALK2 versus other BMP receptors (caALK I and caALK3) and much greater selectivity (>100 fold) over the TGF-p receptors (caALK4 and caALK5). Having much lower potency against caALK1, 2 and 3 in cells, both K02288a and DMH1 exhibit substantially lower selectivity. Data shown are calculated based on IC50 generated from at least 3 independent experiments, with data plotted as mean ± S.E.M ... 51
Figure 4.4. Potency Inhibition curves of BMP and TGF-3 transcriptional activity mediated by constitutively activated ALK 1-5 in a cell-based luciferase reporter assays. Representative inhibition curves for a) dorsomorphin, b) LDN-193189, c) LDN-212854, d) DMH1 and e) K02288a against constitutively active BMP (ALKI, 2 and 3) and TGF-P (ALK4 and 5) type I receptors. Dorsomorphin exhibits a similar selectivity profile to LDN-193189, but with approximately 10-fold decreased potency against all receptors. Despite showing potency similar to LDN-193189 in kinase assay, K02288a is less potent and selective in cells. DMH1 exhibits similar potency to dorsomorphin but with less selectivity. In contrast to dorsomorphin and LDN-193189, LDN-212854 demonstrates improved selectivity for ALK2 versus ALK4 and 5, as well as versus ALK 1 and ALK3. Data shown are representative of at least 3 independent experiments, with data plotted as mean ± S.E.M. (n=3 replicates per [c] point)...53 Figure 4.5. Potency and selectivity of BMP inhibitors based on on BMP (BRe-Luc) and TGF- (CAGA-Luc) transcriptional activity mediated by constitutively active ALK1-5. (a) In vitro cell-based assay IC50
measurements against constitutively-active BMP (caALK1, 2, and 3) and TGF-p (ALK4 and 5) type I receptors demonstrates LDN-212854 to be more selective for BMP versus TGF-p receptors while retaining low nanomolar potency against caALK2. (b-c) Inhibition of caALK1-5 by BMP inhibitors at various concentrations demonstrates that LDN-212854 preferentially inhibits caALK2 at concentrations near 100 nM, whereas other receptors are affected at higher concentrations. Data shown are calculated based on IC50 generated from at least 3 independent experiments, with data plotted as mean ± S.E.M...54
Figure 4.6. Comparison of potency and selectivity of LDN-193189 and LDN-212854 in modulating BMP and TGF-p ligand-mediated SMAD signaling. (a) Western blot analysis of BMP7 induced
LDN-193189 and LDN-212854. (b) Western blot analysis of TGF-31 induced phosphorylation of SMAD2 in wild-type PASMC revealed significant inhibition by LDN-193189 at concentrations greater than 1 gM,
and virtually no inhibition by LDN-212854 at concentrations up to 25 ptM. Data shown are representative of 2 independent experim ents. ... 55
Figure 4.7. (a) ATP pocket interactions of LDN- 193189 (magenta) co-crystallized with ALK2 (PDB
3Q4U). A single hydrogen bond to the hinge residue H286 is made by the central pyrazolo[1,5-a] pyrimidine. The pendant 4-quinoline moiety forms a water-mediated hydrogen bond to the aC-helix residue E248. Water is represented by a red sphere and labeled "Wat". Hydrogen bonds are shown as a dashed line. (b) Model for the binding of LDN-193189 (magenta) to ALK5. The inhibitor was located by the superposition of ALK2 (PDB 3Q4U) and ALK5 (SB-431542 complex; PDB 3TZM). ALK5 structures
show a conserved water position set further back in the ATP pocket between the aC-helix residues E245 and Y249. (c) Model for the binding of LDN-212854 (green) to ALK2. The pendant 5-quinoline group is predicted to form an alternative water-mediated hydrogen bond to the catalytic lysine (K23 5). The new water position was modeled from the ALK2-K02288 co-crystal structure (PDB 3MTF). (d) ATP pocket interactions of LDN-212854 (yellow and blue) in the ALK2 co-crystal structure. The pendant 5-quinoline moiety forms a water-mediated hydrogen bond with the catalytic lysine (K235) as predicted and also to the aC -helix glutam ate residue (E248). ... 56
Figure 4.8. Kinase inhibition profile of LDN-193189 and LDN-212854. Kinome dendrograms for (a)
LDN-193189 and (b) LDN-212854 showing both on-target hits from our kinase assay and the top off-target hits from a screen of 198 human kinases. (c) IC50 values for top off-target hits. RIPK2 was the most
potently inhibited off-target kinase followed by ABL 1 and PDGFR-p, while other kinases were inhibited at m uch higher concentrations. ... 57 Figure 5.1. LDN-212854 preferentially inhibits ALK2. (a) At low concentrations (34nM) LDN-212854 potently inhibits ALK2 (79%) whereas ALKI, ALK3, ALK4, and ALK5 remain largely active. In contrast, at 34nM LDN-193189 inhibits both ALK2 and ALK3 equally. a) Representative inhibition curves of caALK2 and b) caALK3 transcriptional activity (BRE-Luc) by LDN-193189 and LDN-212854 in C2C12 cells. Both compounds potently inhibit ALK2, whereas LDN-212854 is substantially weaker against ALK3. Data shown are representative of at least 3 independent experiments, with data plotted as m ean ± S.E.M . (n=3 replicates per [c] point)... 67 Figure 5.2. LDN-212854 provides useful selectivity as a probe of signaling mediated by ALK2 versus ALK3, and their respective ligands. (a) Alkaline phosphatase (ALP) activity induced in C2C 12 cells by BMP6, which signals primarily through ALK2, was inhibited with comparable potency by LDN- 193189 and LDN-212854. (b) ALP activity induced in C2C12 cells by BMP4, which signals primarily through ALK3, was more potently inhibited by LDN-193189 than LDN-212854. Data shown are representative of at least 3 independent experiments, with data plotted as mean ± S.E.M. (n=3 replicates per [c] point)...68 Figure 5.3. IL-6 induced hepcidin expression in HepG2 cells was potently inhibited by LDN- 193189 and less potently by LDN-212854, consistent with a primarily ALK3-dependent mechanism of IL-6 induced hepcidin expression. Data shown are representative of at least 2 independent experiments, with data plotted as mean ± S.E.M . (n=4 replicates per [c] point) ... 69 Figure 5.4. In vivo efficacy of LDN-212854 in a mouse model of fibrodysplasia ossificans progressiva (FOP). Mice expressing an inducible constitutively-active ALK2Q207D (CAG-Z-EGFP-caALK2) transgene were treated with (a) vehicle, (b) LDN-193189 or (c) LDN-212854 at 6 mg/kg IP BID.
Heterotopic ossification following injection of Ad.Cre was observed by X-ray (top panels) and staining for alizarin red (calcium) and alcian blue (glycosaminoglycans) (bottom panels). Heterotopic ossification following Ad.Cre injection was observed 100% of vehicle-treated mice, whereas ossification was
essentially absent in mice treated with LDN-193189 or LDN-212854. Transgene-mediated expression of GFP (middle panel) was observed at the site of Ad.Cre injection to confirm recombination and ALK2Q2 7 D expression. (d) Passive range-of-motion was progressively impaired in vehicle-treated mice starting on day 10, whereas mobility was almost entirely preserved in mice treated with 193189 and LDN-2 1LDN-2 8 5 4 ... . ... 7 0 Figure 5.5. LDN-212854 was significantly better tolerated than LDN-193189. (a) Weight change of mouse pups throughout the treatment period from P7 to P35. (b) LDN-193189 resulted in a 32%
reduction in the rate of growth, while LDN-212854 resulted in a significantly lower reduction of 14%...71 Figure 5.6. Voluntary wheel-running behavior in mice. Dark bars represent periods of wheel-running. Wild-type mice (a) and (b) Bmallknock-out mice were subjected to day-night cycles (day 0 - 21)
followed by complete darkness (day 22-70). Wild-type mice maintained circadian rhythms with a period of 23.6 hours, while Bmal-/- mice did not. Figure adapted from [126]. ... 71 Figure 5.7. Progressive arthropathy and joint ankylosing seen in Bmal' mice both in the intervertebral joints and at the Achilles anthesis. Adapted from [136]. ... 72
Figure 5.8. Relative expression of Bmallin the liver and the Achilles enthesis of Bmallf-Prx1-cre mice compared to BmaIf mice. Prx]-cre mice demonstrated disrupted and abnormal cycling of Bmall over 24 hours in the peripheral tissue of the Achilles enthesis, while maintaining normal circadian rhythm in the liv e r. ... 7 3 Figure 5.9. Heterotopic ossification (HO) at the Achilles enthesis. (a) Bmall1 Prx]-Cre mice develop
Achilles HO which is first visible by X-ray at 8 weeks of age. The HO continues to develop increasing in severity until 27 weeks of age. (b) Penetrance is of the Achilles enthesis HO is 100% but the severity of H O at 8 w eeks is highly variable. ... 74 Figure 5.10. Quantification of heterotopic ossification in Bmall1
Prx1-Cre mice. (a) A region of interest
(ROI) is selected just above the calcaneus and lateral to the tibia in the area of the Achilles tendon. Using ImageJ a threshold of 42 is set and the number of pixels above this threshold is counted. (b)
Quantification of HO in Bmall] PrxJ-Cre mice treated with vehicle, LDN-212854, or ALK3-Fc shows significant inhibition of H O by LDN -212854... 75 Figure 6.1. Superposition of the ALK2 and ALK5 co-crystal structures with K02288 and SB431542, respectively, showing selected interactions of ALK2 with K02288. The ATP pocket in many ALK5 co-crystal structures shows a more open conformation with a subtle movement of the N-lobe away from C-lobe. Such conformational differences, which change the shape, volume and dynamics of the ATP pocket, are likely to im pact inhibitor selectivity... 80 Figure 6.2. Potency and selectivity of K02288 derivatives based on thermal shift, biochemical kinase activity, and ligand induced transcriptional activity assays. (a) The 2-aminopyridine scaffold of K02288 (b) Modifications to the solvent exposed domain (R1) of K02288. (c) Thermal shift (ATm), biochemical
enzymatic inhibition (IC5 0) for ALK2 and ALK5 kinase proteins, and inhibition of cell-based BMP6 and
TGF-3 1-induced transcriptional activity (IC5o) by K02288 derivative compounds (nd = not determined).
Figure 6.3. A strong negative log-linear correlation is seen between thermal shift and biochemical IC5 0
for both (a) BMP (ALK2) and (b) TGF-p (ALK5) type 1 receptors. A strong negative log-linear
correlation is seen between thermal shift of BMP type I receptors and ligand induced cell-based IC50 for
both (c) BM P6 (ALK2) and (d) TGFbI (ALK5). ... 82
Figure 6.4. Potency and selectivity of compound 15 derivatives based on thermal shift, biochemical kinase activity, and ligand induced transcriptional activity assays. (a) The 2-aminopyridine scaffold of 15 (b) Modifications to the ATP-binding pocket hydrophobic domain (R2) of compound 15. (c) Thermal shift
(ATm), biochemical enzymatic inhibition (IC5o) for ALK2 and ALK5 kinase proteins, and inhibition of
cell-based BMP6 and TGF-1 -induced transcriptional activity (IC5o) by compound 15 derivatives (nd =
not determined). (d) Correlation of thermal shift and cell-based BMP/TGF-p inhibition assays...84 Figure 6.5. Potency and selectivity of K02288 derivatives based on thermal shift, biochemical kinase activity, and ligand induced transcriptional activity assays. a) The 2-aminopyridine scaffold of 15 b) Modifications to the primary amine kinase hinge binding domain (R3) of compound 15. c) Thermal shift (ATm), biochemical enzymatic inhibition (IC5o) for ALK2 and ALK5 kinase proteins, and inhibition of
cell-based BMP6 and TGF-p 1-induced transcriptional activity (IC5o) by compound 15 derivatives (nd =
not determined). d) Correlation of thermal shift and cell-based BMP/TGF- inhibition assays. ... 86 Figure 6.6. Potency and selectivity of K02288 derivatives based on thermal shift, biochemical kinase activity, and ligand induced transcriptional activity assays. (a) Structure of hybrid derivatives. (b) Thermal shift (ATm), biochemical enzymatic inhibition (IC50) for ALK2 and ALK5 kinase proteins, and
inhibition of cell-based BMP6 and TGF-3 1-induced transcriptional activity (IC5o) by hybrid molecules
(nd = not determined). (c) Correlation of thermal shift and cell-based BMP/TGF-p inhibition assays...88 Figure 6.7. Kinome dendrogram plot for (a) LDN-212838 (15) and (b) LDN-214117 (10) showing an
improved selectivity profile for LDN-214117, albeit with reduced potency for BMP type I receptor k in a se s...8 9 Figure 6.8. FOP causing ALK2 mutations do not affect inhibitor binding. a) Strong correlation of
thermal shift data for ATP competitive kinase inhibitors binding to wild-type ALK2 versus known FOP causing GS domain mutations of ALK2 and b) known FOP causing kinase domain mutations suggests the potency of ATP competitive inhibitors are not affected by these disease causing mutations. m = slope, R2
= correlation coefficient...97 Figure 6.9. Cell viability. HepG2 cells were exposed to 1, 10, and 100 tM of compounds for 4 or 24 hours. The average cell viability of three experiments is shown with green indicating >75%, orange indicating 25-75% , and red <25% ... 99 Figure 6.10. Plots of cell-based BMP (a) and TGF-p (b) IC50 versus cell viability show no correlation
betw een potency and toxicity ... 100 Figure 6.11. Binding mode of 26. (a) The inhibitor (yellow) forms a single hydrogen bond to the hinge amide of H286 as well as a water-mediated bond to the catalytic lysine K235. (b) Plot of the interactions of the inhibitor (purple) in the binding pocket of ALK2. The plot was generated by LigPlot+.[154]...101 Figure 6.12. Docking model for 10. Docking was performed using the ICM-Pro software package (Molsoft) and the ALK2-26 structure as a template. Compound 10 (cyan) is predicted to bind similarly to the parent molecule K02288 (PDB 3MTF) as well as the close derivative 26 (PDB 4BGG). The hinge
binding orientation of this 2-aminopyridine series differs compared to the pyrazolo[1,5-a]pyrimidine scaffold of LDN-193189 (dark blue thin sticks; PDB 3Q4U). ... 101 Figure 7.1. (a) In vitro studies of metabolism for LDN-193189 revealed major metabolism via oxidation of the quinoline (NIH-Q55) mediated by aldehyde oxidase. Analine formation at the phenylpiperazine position was also observed. (b) In vivo studies confirmed the in vitro findings and found that
LDN-193189 is quickly metabolized by the liver into a low potency metabolite, NIH-Q55. Furthermore, the metabolite accumulates in tissues (e.g. muscles) at concentrations far exceeding those of LDN-193189.
... 1 0 8 Figure 7.2. (a) Methylation at the 2-position of the quinoline in LDN-193189 and derivatives was used as a strategy to block aldehyde oxidase metabolism at this position. (b) In each case there was a dramatic decrease in B M P versus TG F-p selectivity...109 Figure 7.3. Significant improvements to in vivo metabolic stability seen with 343765. (a)
TRND-343765 had improved half-life, AUC, and oral bioavailability (F) as compared with LDN-193189. (b)
TRND-343765 demonstrated superior pharmacokinetics to LDN-193189 with high plasma concentrations at 7 hours after administration, while LDN-193189 was almost completely cleared. ... 110 Figure 7.4. Characterization of the inhibitory profile of TRND-343765 compared with LDN-193189. (a) Kinase assay inhibition curves for ALKI-6 showing that at 200 nM TRND-343765 completely inhibits all BMP and TGF-p type I receptor kinases, while LDN-193189 only inhibits BMP type I receptor kinases. (b) IC5o and 1C9o values for LDN-193189 and TRND-343765 showing potent dual BMP and
TGF-inhibition. (c) Cell-based ligand induced transcriptional activity inhibition curves. ... 111 Figure 7.5. Cell-based inhibition of BMP and TGF-3 transcriptional activity. (a-c) LDN-193189 and TRND-343765 both potently inhibited all BMP ligand and constitutively active type I receptor
transcriptional activity (d-f) Only TRND-343765 potently inhibited all TGF-p ligand and constitutively active type I receptor transcriptional activity...112 Figure 7.6. (a) Abstract representation TRND-343765 used in conjunction with inhibitor resistant type I receptors. (b) Deep hydrophobic pocket of ALK2 showing threonine gatekeeper residue that was mutated to isoleucine to generate inhibitor resistant mutants. (c) Baseline and ligand-induced signaling of wild type ALK2 and caALK2 is inhibited by TRND-343765 while T2831 mutants show inhibitor resistance in b o th c a se s...1 15
List of Tables
Table 2.1. Members of the TGF-p and BMP signaling family including receptors, ligands, co-receptors, and endogenou s inhibitors...24 Table 3.1. Dorsomorphin derivatives and their respective IC5 0 (nM) values for BMP4 induced
pSMAD1/5/8 in cell western and biochemical kinase assay for ALK1-5. * n.a. = no activity...42 Table 4.1. Kinome profiling for LDN-193189 and LDN-212854 at 100 nM and 1IM ranked by kinases with the greatest inhibition of enzym atic activity. ... 62 Table 6.1. Comparison of compounds 15, 26, and 10 across multiple assays including thermal shift kinase assay, ligand induced transcriptional assay, and constitutively active ALK 1-5 transcriptional activity demonstrates increased selectivity for ALK2 for compound 10 albeit with a reduction in potency. ... 87 Table 6.2. Kinome profiling for LDN-212838 and LDN-214117 at 100 nM and I pM for >200 kinases representing a wide sampling of the human kinome ranked by kinases with the greatest inhibition of enzy m atic activ ity . ... 9 5 Table 6.3. Km values for wild type ALK2 and various constitutively active mutant versions of ALK2 seen in F O P ... 9 6
Chapter 1 Introduction
Bone morphogenetic proteins (BMP), which belong to the transforming growth factor beta (TGF-p) family of over 30 structurally diverse ligand molecules, are critical for development and homeostasis. BMPs, like TGF-p ligands, signal through binding to signaling complexes of type I and type II receptors, whose intracellular kinase domains phosphorylate and activate SMAD effector proteins. Activated SMADs serve as broadly-acting transcriptional regulators, modulating a wide variety of cellular mechanisms including differentiation, proliferation, and migration. Novel reagents such as selective inhibitors of the type I receptor kinases, have become critical tools for studying this complex signaling pathway.
BMP and TGF-p signaling have been implicated in a wide variety of diseases, including fibrosis, cancer, and anemia. In particular, an activating mutation of the BMP type I receptor kinase, activin-like kinase 2 (ALK2), is the known cause of an extremely rare genetic disorder known as fibrodysplasia ossificans progressiva (FOP). FOP-causing mutations in ALK2 lead to increased BMP signaling and the development of heterotopic ossification (HO) lesions in skeletal muscle and connective tissue. This pathological bone formation begins in the first decade of life, progresses inexorably, and causes disability through ankylosis or fusion of joints. FOP is a life-shortening and highly morbid disease with no effective treatments.
This thesis was motivated by the need for novel, highly refined inhibitors of the BMP type I receptor kinases. These compounds could serve as important scientific tools for the study of BMP signaling in vitro and in vivo. Additionally, the development of highly selective ALK2 inhibitors would offer a rational therapeutic approach to treating FOP. This thesis builds on previous work in which a low potency compound was modified to yield a potent, albeit, less selective BMP type I receptor kinase inhibitor. This thesis describes the work done to further expand the understanding of the chemical structural features that modulate BMP type I receptor kinase potency and selectivity. The thesis is divided into several chapters as outlined below.
Chapter 2 provides a detailed overview of the TGF-p and BMP signaling pathways including a historical perspective on their discovery, the mechanisms by which a variety of ligands signal and exert their pleiotropic effects, the role of these pathways in development and tissue homeostasis, and finally the role of these pathways in a variety of diseases. We focus on
the example of FOP and how this work could contribute to the development of therapy for this devastating disease. We close the chapter by describing the field of kinase inhibitors, their development as therapeutics, and the work to date on TGF-p and BMP kinase inhibitors.
In Chapter 3 we describe the characterization of a previously synthesized but incompletely characterized library of dorsomorphin derivatives. Dorsomorphin is a low potency BMP type I receptor kinase inhibitor discovered previously by our laboratory. By analyzing this derivative library using a highly sensitive radio-kinase assay for the BMP and TGF-P type I receptors ALKi, ALK2, ALK3, ALK4, and ALK5 we were able to discover a unique structural insight that greatly improved selectivity for BMP versus TGF- signaling.
In Chapter 4 we describe the synthesis and characterization of a novel potent and selective inhibitor of ALK2 based on the structural findings in Chapter 3. We compare this novel derivative with all of the previously described BMP type I receptor kinase inhibitors in biochemical and cell-based assays of BMP signaling. We demonstrate this novel inhibitor provides superior potency and selectivity as compared to other inhibitors. We also identified key off-targets across the human kinome. Finally, we describe an inhibitor-ALK2 co-crystal structure that highlights aspects of the binding mode that impart a high level of selectivity in this compound series.
In Chapter 5 we build on the characterization of the compound developed in Chapter 4 and show its utility as a scientific tool for the study of BMP signaling both in vitro and in vivo. In particular, we demonstrate this compound has sufficient selectivity for ALK2 versus other BMP type I receptors, such as ALK3, and that it can be used to ascertain the relative contribution of these type I receptors in signaling phenomena. We also use this compound in a mouse model of FOP and demonstrate complete prevention of pathological bone formation as well as improved tolerability as compared to a previously developed less selective inhibitor. Finally we use this compound to demonstrate that BMP signaling plays a role in the development of heterotopic ossification in a novel model of arthropathy suggesting a potential for using BMP type I receptor kinase inhibitors in other diseases such as ankylosing spondylitis and trauma induced heterotopic
ossification.
Chapter 6 describes the development of a unique set of kinase inhibitors based on a 2-aminopyridine scaffold discovered by our collaborator. We developed a large set of derivatives and explored the structure-activity relationships that modulate potency and selectivity. We were
able to discover a potent and selective BMP type I receptor kinase inhibitor that demonstrated very low cytotoxicity and improved kinome-wide selectivity. Finally, we used this derivative library to answer a particular question in the field of FOP concerning the ability of BMP type I receptor kinase inhibitors developed against wild type ALK2 protein to inhibit the many different FOP-causing ALK2 mutants.
In Chapter 7 we describe a novel compound that was discovered as part of our collaboration with the NIH that is a potent inhibitor of both BMP and TGF-$ type I receptor kinase inhibitors. In addition this compound demonstrated superior pharmacokinetic properties as well as significantly less cytotoxicity. We fully characterized the ability of this compound to inhibit both BMP and TGF- signaling in cells in response to a wide-variety of ligands. Finally, we demonstrate a proof of concept that we can use this dual BMP and TGF-p inhibitor as a way of studying this complex signaling pathway. By engineering BMP and TGF-p type I receptors with mutations at the conserved gatekeeper residues we can create mutant receptors that are resistant to the inhibitor. We can then apply the inhibitor to cells effectively shutting down all BMP and TGF-p type I receptor kinase activity and then selectively express resistant mutant kinases to assess the function of that receptor or combinations of resistant mutant receptors for particular biological processes.
Chapter 8 provides a summary of the main conclusions of the thesis and their implications. I also discuss future work based on the results presented.
Chapter 2 Background and Motivation
The purpose of this chapter is to provide a high level background and context for the work described in this thesis. We begin with an overview of the BMP and TGF-3 signaling pathways and their role in development, homeostasis, and disease. We pay particular attention to fibrodysplasia ossificans progressiva, a very rare disease of BMP signaling. We then discuss kinase inhibitor development and clinical use. Finally we look specifically at the development of both TGF-3 and BMP kinase inhibitors and present the motivation for this thesis work
2.1 The TGF-P Family
The transforming growth factor beta (TGF-P) superfamily (Figure 2.1) of signaling ligands, transmembrane receptors, and intracellular transduction proteins is essential for embryonic development and the homeostatic regulation of a multitude of cellular processes, including cell proliferation, differentiation, migration, and apoptosis[1, 2]. The TGF- superfamily also regulates many processes of disease pathophysiology and is implicated in cancer, fibrosis, atherosclerosis as well as a multitude of hereditary disorders including familial pulmonary arterial hypertension (PAH), Hereditary hemorrhagic telangiectasia (HHT), juvenile polyposis syndrome (JPS), and fibrodysplasia ossificans progressiva (FOP) [3, 4].
UgaNdS__ Receptors
BMP2 ACVRXc (ALK7M lypt I mmcpw r
DPP (Decapentaplegic) ( GBAB (BA ) TKV (Thickveins) GD2AVRLI0I BMP5 SX(aophone) r MPAType Uecps BMP8B TGFRW (rGFORwI
GBB (Glass bottom boat) AMHR2 (MISRJ)
SCW (Screw)
Tful? thinking)
GD n
ACVR2 ACM QAcvR2(AcC1u)
BMP3 ) .. GDF PUT (Punt) - MsTN ynG SMADs Myoglianin SMADI RMADs =7TU1 2 SMAO5 SMAD9 (SMAD8)
T (Maverick) MAD (Mothers against dpp)
GDF 5 SMAD2
AMH 3 SMAD3 SMOX (Smad on X)
Alp23B (Dawdle) INHB (activin ) NHBE (acfdW-Activin I4MADs SMAD6 BIMP1S ShAD7 GDF9DAD (Daughters against dpp)
Figure 2.1. Phylogenetic tree of the TGF-p super family of signaling ligands, receptors, and intracellular transduction proteins (SMADs). Adapted from [2].
2.1.1 Historical perspective
TGF-Ps were first described in 1978 by Joseph de Larco and George Todaro at the National Cancer Institute [5]. They describe the secretion by sarcoma virus-transformed mouse fibroblasts of polypeptide growth factors with the ability impart transformed-like properties, such as cell proliferation and anchorage independent growth onto normal fibroblasts. Further work in the early 1980s led to the purification of TGF-Ps from many non-neoplastic tissues, including human placenta and their characterization as 25-kD dimers active at 2-3 pM and critical to wound healing [6, 7]. As work in the TGF-$ progressed throughout the 1980s, the complexity of this signaling pathway was just beginning to be uncovered with the discovery of high affinity type I, type II, and type III receptors as well as the seemingly paradoxical effects of TGF-s [8, 9]. For example, in normal epithelial cells TGF-Ps inhibit growth and suppress tumor formation while in cancer cells they can promote tumor progression and metastasis [10, 11]. Despite the identification of many TGF- signaling family members (Figure 2.1) and almost 60,000 publications to date, there remains a significant need to further understand the intricacies of this complex signaling family in development, homoeostasis, and particularly in disease.
2.1.2 Canonical TGF-3 and BMP signaling
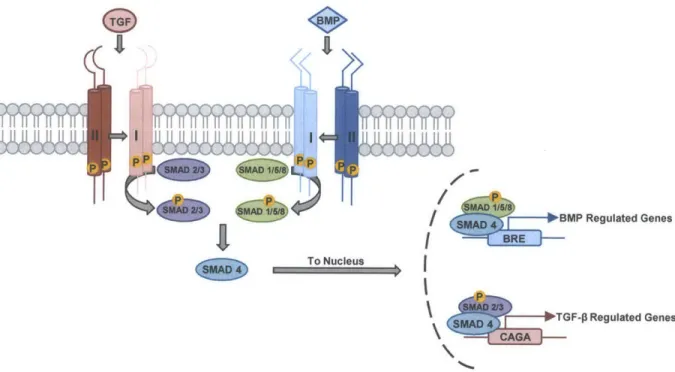
Bone morphogenetic proteins (BMPs) are members of the transforming growth factor-beta (TGF-) signaling family, which includes over 30 different ligands including TGF-Ps, growth and differentiation factors (GDFs), and Activins [2, 12, 13]. BMP signaling is essential for numerous processes including cell fate determination, embryonic patterning, and iron homeostasis [13, 14]. The BMP signaling cascade parallels that of TGF-$ signaling (Figure 2.2). BMP ligand dimers facilitate the assembly of tetrameric receptor complexes consisting of two constitutively-active type II receptor kinases (BMPRII, ACTRIIA, or ACTRIIB), which transphosphorylate and activate two type I receptor kinases (ALKI, ALK2, ALK3, or ALK6)
[15]. Activated type I receptors phosphorylate effector proteins (SMAD1/5/8) that complex with
SMAD4, translocate to the nucleus, and activate BMP responsive genes, such as the inhibitor of differentiation (Id) gene family. Activin and TGF-$ ligands similarly recruit TGF-s or Activin type II receptors (TGFpR2, ACTRIIA, or ACTRIIB) with a set of type I receptors (ALK4, ALK5, or ALK7) to activate SMADs 2 and 3, which translocate with SMAD4 to the nucleus to regulate distinct transcriptional programs.
T )1
I I
1) AD1I L5 BMP Regulated Genes
C W! D4To Nucleus
SNA 4 GF-p Regulated Genes
Figure 2.2. A schematic representation of the canonical BMP and TGF-P signaling pathways demonstrating ligand binding to tetrameric complexes of type I and type II receptors that phosphorylate R-SMADs, which translocate to the nucleus and effect specific transcriptional programs.
Biological specificity in BMP and TGF-P signaling is conferred in part by preferential binding of ligands with specific combinations of type I and type II receptors, although there is considerable functional redundancy among ligands and receptors [16]. Functional and anatomic specificity of BMP signaling is also regulated by the spatio-temporal expression of ligands and their cognate receptors, as well as the expression of endogenous BMP antagonists such as noggin
(Table 2.1) [2, 16]. The diversity of upstream ligand and receptors signals, and their pleiotropic
downstream effects raises questions of how specificity is recognized and translated into biological outcome in this pathway [17]. The various permutations of type 1 and type 2 tetrameric receptor complexes allows for context dependent and selective responses to the various permutations of BMP and TGF-P ligand dimers. However, there is considerable functional redundancy in this pathway at the level of ligands and receptors, posing some challenges for understanding the specific roles of its individual components. Despite serving diverse biological functions, there is a tremendous degree of structural homology between the receptors of the BMP and TGF-P signaling pathways (Figure 2.3). Kinase domain sequence identity is particularly high for ALK1 and ALK2 (79%), ALK3 and ALK6 (86%), and ALK4 and ALK5 (90%) [18].
(a)
Kinase Domain Sequence Identity
(b)
ACVRLi (ALKi) ALK1 ALK2 ALK3 ALK4 ALK5 ALK6 ALK7 ACVRi (ALK2)
ALK2 64.95 66.32 64.26 Hinge ALK3 "4.91 W54 63.86 AMK 67.37 ' AMK 68.04 ALK6 65.26 ALK7
Figure 2.3. (a) A color coded grid with the percent sequence identity for each BMP and TGF-P type I receptor was determined using the alignment tool (http://www.uniprot.org/blast/). (b) Superposition of ALK 1 and ALK2 demonstrating the high degree of similarity between these receptors despite their unique
functions.
2.1.3 Role of TGF-6 and BMP signaling in development and homeostasis
During embryogenesis BMP, nodal and activin signaling play major roles in tissue patterning and axis determination [19]. Concentration gradients of ligands (e.g. BMP4 and BMP7) and endogenous antagonists (e.g. noggin) lead to specific downstream signaling in the relevant cells that determines the ventral to dorsal axis [20, 21]. Additionally, nodal signaling through type I receptors ALK4 and ALK7 plays a role in the formation of the three germ layers: endoderm, mesoderm, and ectoderm [22, 23]. In addition to embryonal patterning members of
the TGF-P superfamily of signaling ligands are involved in the development of many organs. For example, TGF-p ligands are known to induce the epithelial-mesenchymal transition (EMT) of endocardial cells leading to invasion of the heart cushion and eventually heart valve formation [24]. BMPs and GDFs are also known to play critical roles in limb and digit formation [25, 26]. Long after development, the TGF-P superfamily continues to be important for the maintenance of tissue homeostasis. For example, signaling by BMP9/10 through ALKI and co-receptor endoglin serves as an important endothelial quiescence factor in the maintenance of the vasculature [27, 28]. TGF-p signaling has been shown to be important in wound healing and maintenance of extracellular matrix (ECM) [29, 30]. As can be appreciated from this brief review, signaling from the TGF-p superfamily of ligands is absolutely essential for normal
development and tissue homeostasis. Many diseases have been identified that involve disruptions in TGF-3 signaling and are described in 2.2.1.
TGF-P/Nodal Family BMP Family
Type I Receptors TGFPRII ActRila, ActRilb, BMPRII
Ligands TGFP1-3, Activins, Nodal BMPs
Table 2.1. Members of the TGF-p and BMP signaling family including receptors, ligands, co-receptors, and endogenous inhibitors.
2.2 Diseases of TGF-p and BMP Signaling
TGF-3 and BMP signaling have been implicated in many common diseases such as atherosclerosis to extremely rare conditions such as fibrodysplasia ossificans progressiva. In many cases TGF-P and BMP signals mediate certain aspects of disease pathophysiology but are not in and of themselves disease causing. In other cases, however, gain or loss of function in TGF-p and BMP signaling is directly responsible for the disease phenotype. In this section we provide a brief overview of the role of TGF-P and BMP signaling in disease with a particular focus on a rare genetic disorder caused by excessive BMP signaling.
2.2.1 TGF-3 and BMP signaling in disease
It has become increasingly appreciated that disordered BMP signaling contributes to developmental and postnatal disease [31, 32]. There are at least six known heritable disorders caused by germline mutations in members of the TGF- superfamily including heritable forms of pulmonary arterial hypertension (PAH), hereditary haemorrhagic telangiectasia (HHT1/2), juvenile polyposis syndrome (JPS), Loeys-Dietz syndrome, and fibrodysplasia ossificans progressiva (FOP). Familial PAH, caused by heterozygous loss-of-function mutations in the BMP type II receptor BMPR2 transmitted in an autosomal dominant fashion with incomplete penetrance (-20%), is characterized by pathological remodeling of the small arterioles (hypertrophy of medial smooth muscle and intimal thickening) in the lung leading to increased
resistance, elevated pressures (>25 mmHg), eventual right heart failure, and high mortality [27,
33-35]. HHT1 and HHT2, caused by loss-of-function mutations in the TGF-$/BMP co-receptor
endoglin and the BMP type I receptor ALKI, are characterized by dilated blood vessels at the surface of the skin and mucous membranes known as telangiectasias that often cause epistaxis and gastrointestinal bleeding and vascular abnormalities in other organ systems such as the lung and brain, which can lead to significant morbidity and mortality [3, 36]. Up to 50% of JPS cases are caused by loss-of-function mutations in the common TGF-$/BMP signaling mediator SMAD4 or the BMP type I receptor ALK3 [37, 38]. JPS is characterized by the development of hamartomatous gastrointestinal polyps and a up to 50% lifetime risk of developing cancer [13]. Loeys-Dietz syndrome, which shares many phenotypic similarities with Marfan syndrome caused by deficiencies in extracellular matrix protein fibrillin-1, is caused by loss-of-function mutations in TGF- type I and type II receptors ALK5 and TGFBR2, respectively, and is characterized by craniofacial abnormalities such as cleft palate, hypertelorism as well as aneurysms of the aortic root and other vessels [39, 40]. FOP, caused by gain-of-function mutations in BMP type I receptor ALK2, is a rare and debilitating disorder characterized by the development of progressive heterotopic ossification of the skeletal muscle and connective tissue leading to joint immobility, pain, and premature death [41, 42]. FOP is discussed in more detail in section 2.2.2. As can be appreciated from this brief review of the currently known monogenic diseases of TGF-p and BMP signaling, these pathways are critically important for homeostasis and maintenance of many tissues and their disruption can incur a multitude of deleterious and diverse effects. In addition to monogenic diseases, the TGF-P and BMP signaling pathways have been show to contribute to the complex pathogenesis of many diseases including fibrosis, atherosclerosis, cancer and anemia of inflammation [31, 43-45] [46].
The role of TGF-P signaling as a tumor suppressor had long been suspected due to its ability to inhibit cellular proliferation [47]. Two papers published in 1995 reported the first direct evidence of TGF-P tumor suppression activity in both human disease, where mutations in TGFpR2 were identified in 8 colon cancer cell lines, and in a mouse model of breast cancer, where in vivo overexpression of TGFp1 significantly reduced tumor development [48, 49]. Since those early findings, a multitude of components of the TGF-P and BMP signaling pathway have been implicated in cancer development and progression including SMAD4, ALK3, and ALK2 [50, 51]. Despite serving a tumor suppression function, significant evidence has
demonstrated that after carcinogenesis TGF- signaling promotes proliferation and metastasis through effects on tumor microenvironment, cancer stem cells and epithelial to mesenchymal transition (EMT) [11, 52, 53]. Recently, BMP signaling through ALKI has been shown to be
pro-angiogenic and could serve as a therapeutic target against tumor vascularization and progression [54, 55]. Fibrosis is the accumulation of extracellular matrix connective tissue as a result of injury repair mechanisms. When it occurs inappropriately or in excess, fibrosis leads to a loss of normal function in affected tissues. Excess TGF- signaling has been demonstrated in multiple models of fibrosis including pulmonary, hepatic, renal, cardiac, and scleroderma [44]. The TGF-s and BMP signaling pathways play critical roles in all stages of cancer and may potentially serve as therapeutic targets.
Anemia of inflammation (AI) is typically a mild to moderate normocytic anemia associated with chronic diseases including neoplastic disorders, rheumatoid arthritis, and multiple myeloma [56, 57]. It is thought that inflammatory signals such as IL-6 stimulate the synthesis of hepcidin in the liver to suppress iron absorption and bioavailability, leading to impaired erythropoiesis [58, 59]. Recently the role of hepatic BMP signaling has been implicated in the regulation of hepcidin synthesis in response to iron loading, IL-6 signaling, and other types of inflammation [14, 60-63]. Use of BMP type I receptor kinase inhibitor
LDN-1931879 was shown to normalize iron levels and blood counts associated with chronic inflammation [64]. These results suggest that therapeutic intervention with kinase inhibitors can be an effective means of treating diseases where the pathogenesis results from excess or potentially disregulated BMP signaling.
2.2.2 Fibrodysplasia ossificans progressiva
One of the most striking examples of BMP signaling-related disease is seen in fibrodysplasia ossificans progressiva (FOP), an extremely rare and disabling genetic disease with an estimated prevalence of 1 in 2 million affecting an estimated 3,000 people worldwide and with only 800 diagnosed patients [42]. In familial cases, FOP is inherited as an autosomal dominant disorder. Individuals with the classical form of FOP are nearly normal at birth except for cervical and hallux valgus joint deformities. However, during the first decade of life they develop progressive formation of endochondral bone in muscles, fascia, and ligaments leading to severe immobility, pain, and premature mortality a consequence of restrictive lung disease due to
thoracic involvement [41, 65]. A highly recurrent gain-of-function mutation in the glycine-serine (GS) rich domain of the BMP type-I receptor ALK2 (c.617G>A; p.R206H) accounts for more than 98% of cases of classic FOP [66]. Several other FOP-causing gain-of-function mutations in both the GS and kinase domains of ALK2 have also been described in non-classic or variant forms of FOP (Figure 2.4a) [67-70]. The arginine to histidine mutation within the GS activation domain of ALK2 is non-conservative and abrogates the normal requirement for phosphorylation for activation of the kinase. Structural modeling of the mutant and wild-type receptor predicts that the R206H mutation disrupts alpha helical structure of the GS domain and prevents intramolecular salt-bridging, exposing the active site and rendering the enzyme constitutively-active [71, 72]. It is thought that enhanced induced, and potentially ligand-independent BMP signaling potentiate osteogenic differentiation in mesenchyme-derived progenitors in these individuals. Constitutively-active mutant kinases, such as BCR-ABL have been successfully targeted by small molecule inhibitors such as imatinib". ALK2R206
H, with a
constitutively-active intracellular kinase domain, is unlikely to be affected by endogenous antagonists of BMP signaling such as chordin or noggin, which sequester BMP ligands, or by receptor- or ligand-neutralizing antibodies. ALK2 R26H thus represents an ideal therapeutic target for a highly selective small molecule kinase inhibitor in the treatment of FOP.
(a) (b) (
anQ207E ~ Pi47jJ96ensL
G328E
375P
Figure 2.4. (a) FOP-causing mutations in both the GS loop and kinase domain. (b) Soft tissue nodules and HO lesions on the back of a 4-year-old child affected by FOP. (c) The same child at 8 years of age showing extensive HO of the back. Adapted from [73] and [74].
2.3 Kinase Inhibitors
In the last decade protein kinases have become an increasingly important class of drug targets, accounting for 20% of preclinical research targets for pharmaceutical and biotechnology companies. Strategies to block receptor kinase signaling include monoclonal antibodies, ligand traps, and small molecule kinases inhibitors. The latter are particularly effective when targeting constitutively-active kinases that function independently of activation or ligand-mediated signaling, as is the case for many mutant kinases found in cancer. In fact, currently there are 22 FDA-approved small molecule kinase inhibitors, generating $30B in worldwide sales, 21 of which have been approved for cancer indications, the majority targeting a few oncogenic kinase such as BCR-AbL, VEGFR, EGFR, and RAF [75-77]. Just recently tofacitinib, which selectively targets JAK3, was the first kinase inhibitor approved for a non-cancer indication, rheumatoid arthritis. This development provides a proof of concept that kinase inhibitors can be successfully developed to treat non-cancer diseases. While certainly not all kinases will be druggable, with only a small fraction (4%) of the human kinome currently targeted by FDA-approved drugs there is ample opportunity for further development of novel kinase inhibitor therapeutics.
2.3.1 Kinase inhibitor development and clinical use
The human kinome consists of 518 protein kinases that share a bilobular structure and a highly conserved ATP binding pocket (Figure 2.5a) [77]. The development of highly selective kinase inhibitors is challenging, particularly given the conserved nature of the ATP binding site, but is critical for reducing the potentially dose-limiting effects of off-target inhibition [78]. The adenine ring of ATP forms hydrogen bonds at the kinase hinge that connects the amino and carboxy-terminal domains of the kinase [79]. There are four classes of small molecule kinase inhibitors: type I, type II, type III, and covalent. Most protein kinases have a conserved activation loop which regulates kinase activity, and is marked by a conserved DFG (corresponding to its one-letter amino acid sequence) motif at the start of the loop. Conformations of the activation loop include those that are catalytically active, and those that are inactive in which the activation loop blocks the substrate binding site, a.k.a., the "DFG-out" conformation. The vast majority of current kinase inhibitors mimic ATP by forming one to three hydrogen bonds to the hinge and typically also interact with adjacent hydrophobic regions for