HAL Id: tel-01140301
https://tel.archives-ouvertes.fr/tel-01140301
Submitted on 8 Apr 2015HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
d’interféron de type I par les cellules dendritiques
plasmacytoïdes
Liliane Khoryati
To cite this version:
Liliane Khoryati. Propriétés immuno-modulatrices des IgE dans le lupus érythémateux systémique : impact sur la sécrétion d’interféron de type I par les cellules dendritiques plasmacytoïdes. Biologie cellulaire. Université de Bordeaux, 2014. Français. �NNT : 2014BORD0159�. �tel-01140301�
THÈSE PRÉSENTÉE POUR OBTENIR LE GRADE DE
DOCTEUR DE
L’UNIVERSITÉ DE BORDEAUX
ÉCOLE DOCTORALE SCIENCES DE LA VIE ET DE LA SANTÉ (ED SVS) SPÉCIALITÉ MICROBIOLOGIE-IMMUNOLOGIE
Par Liliane KHORYATI
Propriétés immuno-modulatrices des IgE dans le
lupus érythémateux systémique :
impact sur la sécrétion d’interféron de type I par les
cellules dendritiques plasmacytoïdes
!
Sous la direction du Pr. Christophe RICHEZ
Soutenue le 07 octobre 2014
Membres du jury
M. le Professeur Patrick BERGER, PU-PH, Université de Bordeaux Président Mme le Professeur Pascale JEANNIN, PU-PH, Université d’Angers Rapporteur
M. le Professeur Frédéric BATTEUX, PU-PH, Université Paris 5 Rapporteur
M. le Docteur Nicolas CHARLES, CR, INSERM, Université Paris 7 Examinateur M. le Professeur Patrick BLANCO, PU-PH, Université de Bordeaux Invité
Monsieur le Professeur Patrick BERGER d’avoir présidé le jury de thèse
Madame et Monsieur les Professeurs Pascale JEANNIN et Frédéric BATTEUX d’avoir pris le temps de juger ce travail
Monsieur le Docteur Nicolas CHARLES de m’avoir fait l’honneur d’examiner cette thèse.
Je remercie également Monsieur le Professeur Jean-François Moreau et Madame le Docteur Julie Déchanet-Merville. Merci pour l’attention que vous avez accordée à mon projet de thèse. Merci pour votre disponibilité et le soutien que vous m’avez apporté. Je souhaite remercier Monsieur le Professeur Patrick Blanco de m’avoir accueillie dans son équipe. Merci pour votre confiance, votre support et vos conseils. Merci pour tous les moments que vous avez accordés à ce projet.
Je remercie particulièrement Monsieur le Professeur Richez !
Christophe, merci de m’avoir encadrée durant cette thèse, merci pour ton expérience que tu as partagée avec moi, merci pour le temps que tu m’as consacré, pour tes conseils, tes encouragements, ta gentillesse, ton sourire et ton amitié ! Merci !
Je remercie vivement toute l’équipe de l’EFS Aquitaine-Limousin qui s’occupait des poches d’hémochromatose et tous les patients sans qui ce projet n’aurait pu être réalisé !
Je remercie toutes les personnes du CIRID, Immuno et Cellules Souches, et particulièrement mon équipe ! Merci pour chaque souvenir que vous avez laissé en moi. Merci pour votre soutien, scientifique et amical. Merci à Isa, Cécile, Pierre, Marie-Elise, Esti et je pense aussi à Clem, Emmanuel, Irène, Anne, Béa et Jorge.
aide et ta disponibilité.
Maria ! Grand merci pour tout ce que tu m’as appris, depuis le master 1… Merci d’avoir partagé avec moi ton expérience d’enseignement et d’avoir été ma tutrice, et toujours dans la bonne humeur !
Je pense à mes collègues de bureau. Sophie, merci pour tout ce que tu m’as appris en biochimie, pour ta disponibilité, mais surtout pour ton soutien ! Sonia, merci de m’avoir appris la PCR et toujours avec le grand sourire ! Séverine, je n’oublierai jamais tes blagues, merci d’avoir été si proche, merci pour ton soutien !
Je remercie sincèrement Annie Muniesa et Richard Vinuesa. Merci d’être toujours présents pour gérer les commandes, les papiers administratifs et répondre à tous les besoins. Merci pour votre gentillesse et votre aide.
Une pensée particulière à Dada, Camillou, Rémi et Alice. Courage, bientôt la fin ! Merci pour tous les moments sympathiques que nous avons passés ensemble !
Aya, merci pour ton encouragement, ton aide, et ton amitié.
Sinisha, merci pour ton amitié, merci d’avoir tellement pris soin de moi ces derniers jours, merci pour tes encouragements, merci d’être toujours là…
et toi Jenny…my dearest Jenny ! Thank you for your lovely care, for all the moments you were by my side ! We shared everything during these three years, our happy moments as well as the saddest ones, our crazy labs evenings, our worries, our tears and our faith ! thank you for being the loveliest PhD sis !
A toute ma chère famille, mes cousins et tous mes amis : grand merci pour votre support et vos encouragements! Talia, Pierre, Georges, Georges et Sami : je pense fort à vous. C’est bientôt fini ! Courage !
Pour leur confiance en moi
Pour leur présence et soutien infaillibles Pour leur amour inconditionnel
A Maroun, A ma très chère, ma sœur Jihane,
Khoryati L, Shipley E, Contin-Bordes C, Douchet I, Truchetet ME, Lazaro E, Hourdille
A, Duffau P, Jacquemin C, Vacher P, Schaeverbeke T, Blanco P, Richez C. Plasmacytoid dendritic cell interferon-alpha production induced by TLR-7 and -9 ligands is downregulated in presence of IgE in SLE. En cours de soumission pour Journal of Autoimmunity
Gazal S, Sacre K, Allanore Y, Teruel M, Goodall AH; (The CARDIOGENICS consortium), Tohma S, Alfredsson L, Okada Y, Xie G, Constantin A, Balsa A, Kawasaki A, Nicaise P, Amos C, Rodriguez-Rodriguez L, Chioccia G, Boileau C, Zhang J, Vittecoq O, Barnetche T, Gonzalez Gay MA, Furukawa H, Cantagrel A, Le Loët X, Sumida T, Hurtado-Nedelec M, Richez C, Chollet-Martin S, Schaeverbeke T, Combe B,
Khoryati L, Coustet B, El-Benna J, Siminovitch K, Plenge R, Padyukov L, Martin J,
Tsuchiya N, Dieudé P. Identification of secretedphosphoprotein 1 gene as a new rheumatoid arthritis susceptibility gene. Ann. Rheum. Dis. (2014).
doi:10.1136/annrheumdis-2013-204581
Richez C*, Barnetche T*, Khoryati L*, Duffau P, Kostine M, Contin-Bordes C, Blanco P, Schaeverbeke T. Tocilizumab treatment decreases circulating myeloid dendritic cells and monocytes, 2 components of the myeloid lineage. J. Rheumatol. 39, 1192–7 (2012).
Propriétés immuno-modulatrices des IgE dans le lupus érythémateux systémique: impact sur la sécrétion d’interféron de type I par les cellules
dendritiques plasmacytoïdes
Les cellules dendritiques plasmacytoïdes (pDCs) sont caractérisées par leur capacité unique de sécrétion massive d’interféron de type I (IFN-I) suite à la stimulation des Toll-like récepteurs (TLR) 7 et 9. Un rôle fondamental des pDCs a été démontré dans le lupus érythémateux systémique via la production d’IFN-I. Les pDC expriment le récepteur de forte affinité aux immunoglobulines de type E (IgE), FcεRI, impliqué dans la régulation négative de la sécrétion d’IFN-I. L’objectif de notre étude est d’explorer, dans le contexte lupique, les effets du traitement par les IgE sur les fonctions des pDC, particulièrement sur la production d’IFN-I. In vitro, le traitement des pDC par des IgE monoclonales permet la surexpression du FcεRI à leur surface et diminue le taux de transcrits des TLR7/9 et de l’IRF7. De plus, les pDC traitées par des IgE diminuent leur production d’IFN-I et l’expression de marqueurs de maturation, induites par leur stimulation par des ligands des TLR7/9 et des complexes immuns lupiques. En outre, ces pDC pré-traitées par des IgE induisent la différenciation de LT4 naïfs allogéniques en LT4 produisant de l’IL-10. In vivo, les patients lupiques en phase quiescente de la maladie présentent des taux plus élevés d’IgE totales comparés aux patients en phase active (indépendamment d’allergies et d’infestations parasitaires). Chez les patients lupiques, le taux d’IgE totales est inversement corrélé au taux d’anti-ADN et à l’activité de la maladie (SLEDAI). L’ensemble de nos résultats suggère un rôle protecteur des IgE dans le lupus à travers la modulation de la réponse inflammatoire des pDC.
Mots clés : Lupus érythémateux systémique, cellule dendritique plasmacytoïde,
Immunomodulatory properties of IgE in systemic lupus erythematosus : impact on type I interferon secretion by plasmacytoid dendritic cells
Plasmacytoid dendritic cells (pDCs) are characterized by their unique ability to produce large amounts of type I interferon (IFN-I) upon Toll-like receptors (TLR) 7 and 9 triggering. A fundamental role for pDCs has been shown in systemic lupus erythematosus (SLE) through IFN-I production. pDCs express the high affinity Fc receptor for immunoglobulin E (IgE), FcεRI, involved in the negative regulation of IFN-I secretion. The objective of our study is to investigate, in the context of SLE, the effects of IgE treatment on pDCs functions, especially on IFN-I production. In vitro, monoclonal IgE treatment of pDCs upregulate their surface expression of FcεRI and decrease transcripts levels of TLR7/9 and IRF7. IgE-treated pDCs decrease IFN-α secretion and downregulate maturation markers expression induced by TLR7/9 and immune complexes triggering. Moreover, the coculture of IgE pretreated pDCs with allogeneic naive LT4 promotes their differentiation into IL-10-secreting cells. In vivo, patients with quiescent SLE have higher IgE levels than patients with active disease (independently of allergy or parasitic infection). In SLE patients, IgE levels are inversely correlated to anti-DNA antibodies and disease activity (SLEDAI). All together, our data suggest a protective role for IgE in SLE through the modulation of the inflammatory response by pDC.
Keywords : Systemic lupus erythematosus, plasmacytoid dendritic cell, interferon type
INTRODUCTION ... 1
1 Le lupus érythémateux systémique (SLE): maladie auto-immune IFN-I-dépendante ... 2 1.1 Généralités ... 2 1.2 Clinique ... 2 1.3 Physiopathologie ... 5 1.3.1 La composante génétique ... 5 1.3.2 La composante environnementale ... 7 1.3.3 La composante immunitaire ... 9
2 Les cellules dendritiques plasmacytoïdes: dans l’immunité et l’auto-immunité via l’IFN-I ... 15
2.1 Découverte des pDC ... 15
2.1.1 La découverte de cellules plasmacytoïdes ... 15
2.1.2 La découverte de cellules productrices de taux importants d’IFN-I ... 15
2.1.3 La découverte des cellules dendritiques plasmacytoïdes ... 16
2.2 Ontogénie des pDC ... 17
2.2.1 Arguments en faveur d’une origine lymphoïde des pDC ... 17
2.2.2 Arguments en faveur d’une origine commune des cDC et des pDC ... 17
2.3 Cytokines et facteurs de transcription impliqués dans le développement des pDC ... 19
2.3.1 Les cytokines ... 19
2.3.2 Les facteurs de transcription ... 21
2.4 Caractérisation phénotypique des pDC ... 22
2.4.1 Profil d’expression des marqueurs de surface ... 22
2.4.2 Morphologie des pDC ... 23
2.5 Localisation et migration des pDC ... 24
2.5.1 Dans les conditions homéostatiques ... 24
2.6.1 Fonctions innées des pDC ... 27
2.6.2 Fonctions adaptatives des pDC ... 27
2.7 La pDC : principale cellule productrice d’IFN-I ... 30
2.7.1 Production d’IFN-I par la pDC ... 30
2.7.2 L’IFN-I des pDC dans l’auto-immunité : cas du SLE ... 42
2.7.3 Synthèse sur la production de l’IFN-I par les pDC ... 50
2.7.4 Régulation négative de la production d’IFN-I par les pDC ... 51
3 Les immunoglobulines E (IgE) : focus sur leur rôle dans l’auto-immunité ... 63
3.1 Introduction générale ... 63
3.2 Description de la molécule d’IgE ... 63
3.3 Production des IgE ... 64
3.4 Les récepteurs de l’IgE ... 67
3.4.1 Le récepteur de forte affinité ou FcεRI ... 67
3.4.2 Le récepteur de faible affinité ou FcεRII ... 68
3.5 Rôle physiologique et pathologique des IgE ... 70
3.5.1 Les IgE dans la défense anti-parasitaire contre les infestations par les helminthes ... 70
3.5.2 Les IgE dans les réactions d’hypersensiblité de type I ... 72
3.6 Le yin et le yang des IgE dans les maladies auto-immunes ... 76
3.6.1 L’aspect pathogène des IgE dans l’auto-immunité ... 76
3.6.2 L’aspect protecteur des IgE contre l’auto-immunité ... 81
RESULTATS ... 87 HYPOTHESE... 88 OBJECTIFS ... 89 RESULTATS I ... 90 ARTICLE I ... 90 RESULTATS II ... 95 CONCLUSION ET PERSPECTIVES ... 103 BIBLIOGRAPHIE ... 111
! ! !
Figure 1. Rôle central de l’IFN-I dans la dérégulation du système immunitaire dans le
SLE ... 14
Figure 2. Ontogénie des pDC ... 19
Figure 3. Morphologie des pDC humaines ... 23
Figure 4. Localisation des pDC dans les ganglions lymphatiques humains ... 24
Figure 5. Structure des TLR comparée à celle de l'IL-1R ... 35
Figure 6. Squelette de l'ADN naturel et de l'ODN ... 37
Figure 7. Voies de Signalisation des TLR7-9 dans les pDC ... 40
Figure 8. Production d’IFN-I par les pDC dans le SLE ... 48
Figure 9. Voie de signalisation ITAM dans les pDC ... 61
Figure 10. Structure de l'IgE comparée à l'IgG ... 65
Figure 11. Structure du FcεRI ... 68
Figure 12. Structure du CD23 membranaire ... 69
Figure 13. Fonctions effectrices des IgE dans la réponse anti-parasitaire ... 72
Figure 14. Fonctions effectrices des IgE dans les réactions d'hypersensibilité 1 ... 75
Figure 15. Effet paradoxal des IgG et des IgE dans le SLE ... 109
!
LISTE DES TABLEAUX
Tableau 1. Critères de l'ACR pour la classification du SLE ... 4Tableau 2. Principaux gènes associés à un risque de développer un SLE en fonction des mécanismes/voies physiologiques ou immunologiques impliquées dans la pathogenèse de la maladie. ... 6
Tableau 3. Modèles animaux d’infestations conférant une protection contre le développement de maladies auto-immunes ... 85
A
ADAM10 A disintegrin and metalloproteinase 10
ADN Acide désoxyribonucléique
ANA Antinuclear antibody
Anti-ds DNA Anti-double stranded DNA
Anti-Sm Anti-Smith
Anti-SSA/B Anti-Sjögren’s syndrome A/B
APC Antigen presenting cell
B
BCAP B cell adapter for phosphoinositide 3-kinase
BCR B cell receptor
BDCA Blood dendritic cell antigen
bHLH Basic helix-loop-helix
BLNK B cell linker
BST2 Bone marrow stromal antigen 2
BTK Bruton’s tyrosine kinase
C
CCL CC chemokine ligand
CCR CC chemokine receptor
CD Cluster of differentiation
CD40-L CD40 ligand
cDC Conventional dendritic cell
CDP Common dendritic cell progenitors
ChemR23 Chemerin receptor 23
CIC Circulating immune complexes
CLP Common lymphoid progenitors
CLR C-type (calcium dependent) lectin receptors
CMH Complexe majeur d’histocompatibilité
CMP Common myeloid progenitors
CMV Cytomegalovirus
CRAMP Cathelicidin-related antimicrobial peptide
CRP C-reactive protein
CSH Cellule souche hématopoïétique
DAG Diacylglycerol
DAMPs Damage-associated molecular pattern
DCIR Dendritic cell immunoreceptor
DNA Deoxyribonucleic acid
DNase Deoxyribonuclease
E
EBV Epstein-Barr virus
Eph-B1 Ephrin type-B receptor 1
F
FLT3 FMS-like tyrosine kinase 3
FLT3L FLT3-ligand
H
HEV High endothelial venules
HIV Human immunodeficiency virus
HLA Human leukocyte antigen
HMGB1 High-mobility group box 1
HNP Human neutrophil peptide 1
HPC Hematopoietic progenitor cell
HSP Heat shock proteins
HSV Herpes simplex virus
hToll Human Toll
I
ICAM Intercellular adhesion molecule 1
Id Inhibitor of DNA binding
IFN Interferon
IFNAR IFN-alpha/beta receptor
Ig Immunoglobulin
IL Interleukin
ILT7 Immunoglobulin like transcript
IP3 Inositol triphosphate
IPC Interferon producing cell
IRAK Interleukin-1 receptor-associated kinase
IRF Interferon regulatory factor
ISRE Interferon-stimulated response element
K
KO Knockout
L
LB Lymphocyte B
LGP2 Laboratory of genetics and physiology 2
LPS Lipopolysaccharide
LT Lymphocyte T
LT4 Lymphocyte T auxilliaire CD4+
LT8 Lymphocyte T cytotoxique CD8+
M
M-CSF Macrophage-colony stimulating factor
M-CSFR M-CSF receptor
MALT Mucosa-associated lymphoid tissue
MAPK Mitogen-activated protein kinase
MCMV Murine CMV
MDA5 Melanoma differentiation-associated protein 5
mDC Myeloid dendritic cell
MDP Macrophage-dendritic cell progenitors
MyD88 Myeloid differentiation factor 88
N
NF-κB Nuclear factor-κB
NK Natural killer
NKp44 Natural killer cell p44-related protein
NLR Nod-like receptors
O
ODN Oligodeoxynucleotides
P
PAMP Pathogen-associated molecular pattern
PD Phospho-diester
pDC Plasmacytoid dendritic cell
PI3K Phosphoinositide 3-kinase
PRR Pattern recognition receptor
PS Phospho-thioate
PSGL-1 P-selectin glycoprotein ligand 1
PTK Protein tyrosine kinase
R
RIG-I Retinoic acid-inducible gene 1
RLR RIG-I-like receptors
RNA Ribonucleic acid
RNase Ribonuclease
RNP Ribonucleoprotein
S
SH2 Src homology 2
Si RNA Small interfering RNA
SLE Systemic lupus erythematosus
ssRNA Single-stranded RNA
T
TAK-1 TGF-β activated kinase 1
TCR T cell receptor
TGF-β Transforming growth factor beta
Th T helper
TLR Toll-like receptor
TNF-a Tumor necrosis factor alpha
TPO Thrombopoietin
TRAF6 TNF receptor-associated factor 6
V
VEGFA Vascular endothelial growth factor A
VSV Vesicular stomatitis virus
W
!
La fonction cardinale du système immunitaire, par ses deux composantes innée et adaptative, est la protection de l’organisme contre toute agression extérieure afin de préserver son intégrité. Toutefois, un dérèglement de la réponse immune entraîne le développement de pathologies immunitaires, telles que les immunodéficiences et les maladies auto-immunes.
Dans notre équipe de recherche, l’équipe du Pr. Blanco au laboratoire CIRID CNRS UMR 5164, nous nous intéressons à l’étude des mécanismes impliqués dans la survenue de maladies auto-immunes, et plus particulièrement, le lupus érythémateux systémique. Nos thématiques de recherche se focalisent sur le dysfonctionnement des cellules dendritiques et des lymphocytes T dans le contexte d’auto-immunité.
Le lupus érythémateux systémique (SLE) est une maladie auto-immune dont l’étiologie reste mal connue à ce jour. Une caractéristique majeure du dérèglement immunitaire au cours du SLE est la production excessive et non-contrôlée d’IFN de type I (IFN-I) jouant un rôle central dans l’entretien de l’activation des cellules de l’immunité innée et adaptative. Une sous-population de cellules dendritiques, les cellules dendritiques plasmacytoïdes (pDC), a été identifiée comme étant la source majeure d’IFN-I dans le SLE. De nombreuses études se sont intéressées à l’activation des pDC aboutissant à la sécrétion massive d’IFN-I. Toutefois les mécanismes régulateurs de la production d’IFN-I par les pDC sont mal explorés dans le SLE.
En effet, dans les conditions physiologiques, les pDC sont dotées de récepteurs de surface ayant pour rôle de contrôler leur réponse inflammatoire en régulant négativement leur production d’IFN-I. Mon projet de recherche s’intéresse à l’étude de la modulation de la sécrétion d’IFN-I par les pDC via l’un de ses récepteurs, le FcεRI, récepteur de forte affinité aux IgE.
Dans un premier temps, j’ai mis au point un modèle, in vitro, de culture des pDC en présence d’IgE permettant une sur-expression du FcεRI à la surface de ces cellules. Après avoir sélectionné les conditions optimales de culture, j’ai exploré l’effet direct du
mécanismes moléculaires mis en jeu ont été étudiés.
Afin de mieux situer le contexte et les objectifs de mes travaux de thèse, l’introduction de ce manuscrit exposera :
- Une présentation générale des principaux traits de la physiopathologie du SLE en s’intéressant plus particulièrement au rôle de l’IFN-I dans la dérégulation de la réponse immunitaire lupique ;
- Une description des pDC et de ses fonctions dans l’immunité et l’auto-immunité ; - Une description des IgE et de leurs fonctions physiologiques et pathogènes. Puis, les hypothèses de travail, les objectifs et les résultats obtenus seront présentés et discutés.
1 Le
lupus
érythémateux
systémique
(SLE):
maladie auto-immune IFN-I-dépendante
! !
1.1 Généralités
!Le lupus érythémateux systémique (SLE) est une maladie auto-immune systémique et chronique, d’origine multifactorielle. Cette pathologie touche préférentiellement la femme jeune (9 femmes pour 1 homme) en âge de procréer (15-40 ans)1. La prévalence de la maladie varie d’un pays à l’autre, allant de 15 à 50 cas pour 100 000 habitants.
Le SLE évolue par des poussées entrecoupées de phases de rémissions. Les manifestations cliniques sont très polymorphes et le pronostic dépend de la nature des lésions viscérales, particulièrement, les atteintes rénales et neurologiques2.
1.2 Clinique
!Les manifestations polymorphes du SLE peuvent aller de simples atteintes cutanéo-articulaires à des atteintes viscérales (rénales, cardiaques, pulmonaires et neurologiques) pouvant engager le pronostic vital. Ainsi, deux types de tableaux cliniques sont distingués3:
- Formes bénignes et fréquentes cutanéo-articulaires
Les lésions cutanées, pouvant précéder les signes systémiques de plusieurs années, sont souvent localisées dans les zones photo-exposées. L’aspect typique du SLE se caractérise par un érythème du visage d’aspect maculeux ou maculo-papuleux avec une topographie en Vespertilio ou loup. D’autres types de lésions sont également observés : livedo, urticaire, purpura vasculaire, alopécie, lésions muqueuses érosives.
Les atteintes articulaires se manifestent par une polyarthralgie ou polyarthrite non destructrice aiguë, bilatérale et symétrique avec atteinte préférentielle des articulations interphalangiennes proximales, métacarpophalangiennes, du carpe et des genoux.
- Formes plus rares et sévères avec atteintes viscérales
L’atteinte rénale représente un facteur majeur du pronostic et se révèle le plus souvent par un syndrome néphrotique pur ou impur.
L’atteinte cardiaque se caractérise le plus souvent par une péricardite. Les myocardites et les endocardites lupiques sont plus rares.
L’atteinte pleuro-pulmonaire se caractérise par une pleurésie lymphocytaire, dans la majorité des cas.
Les atteintes vasculaires se manifestent par un phénomène de Raynaud, des thromboses artérielles ou veineuses et une vascularite. En outre, les patients atteints de SLE présentent une athérosclérose accélérée.
Enfin, des atteintes neurologiques ou psychiatriques peuvent également être observées.
Le diagnostic du SLE repose sur un examen clinique approfondi et un bilan biologique adapté. Sa classification est basée sur les onze critères de l’ACR (American College of Rheumatology) révisés en 19974,5 (tableau 1). Pour classer une maladie comme lupique, la présence simultanée ou successive de quatre critères, parmi les onze, est nécessaire.
Les critères publiés par le groupe SLICC (Systemic Lupus International Collaborating Clinics) en 2012, sont complémentaires des critères de l’ACR et permettent également la classification de la maladie lupique6.
1. Erythème facial en aile de papillon 2. Lupus discoïde
3. Photosensibilité
4. Ulcération buccale ou nasopharyngée
5. Arthrite non déformante, atteinte d’une ou plusieurs articulations périphériques avec douleurs à la mobilisation, sensibilité, épanchement ou gonflement des parties molles sans déformation
6. Pleurésie ou péricardite (sérite)
7. Atteinte rénale : protéinurie persistante > 0,5 g/24 heures ou cylindres urinaires
8. Atteinte neurologique : psychose ou convulsion (en l’absence de cause médicamenteuse et/ou métabolique)
9. Atteinte
hématologique
Leucopénie < 4 000/mm3 à deux reprises
Thrombopénie < 100 000/mm3 (en l’absence de drogue cytopéniante)
Lymphopénie < 1 500/mm3 à deux reprises Anémie hémolytique
10. Anomalies immunologiques
Présence d’anti-ADN à un taux significatif Présence d’anti Sm
Présence d’anticorps anti-phospholipides : soit sérologie syphilitique dissociée positive à 2 déterminations à 6 mois d’écart, soit anticoagulant circulant de type lupique ou titre anormal d’anticorps anti-cardiolipine et IgG et/ou IgM
11. Taux anormal d’anticorps anti-noyaux identifié par immunofluorescence ou une autre technique, persistant et en l’absence de médicaments inducteurs
1.3 Physiopathologie
!L’étiologie du SLE reste imparfaitement connue. Cependant, comme toute maladie auto-immune, sa physiopathologie est complexe et multifactorielle. Un terrain de prédisposition génétique, des facteurs environnementaux et des dysfonctionnements du système immunitaire semblent être impliqués et intriqués.
1.3.1 La composante génétique
!
L’une des premières évidences en faveur d’une prédisposition génétique au développement du SLE est la forte agrégation familiale observée. En effet, 10 à 12% des patients ont un apparenté du premier degré atteint7. Les différences entre les taux
de concordance chez les jumeaux monozygotes et dizygotes confirment également l’implication des gènes dans la physiopathologie du SLE. Lorsqu’un jumeau est malade, la probabilité que le second soit atteint est de 24 à 69% chez les jumeaux monozygotes alors qu’elle n’est que de 2 à 9% chez les jumeaux dizygotes8,9. Enfin, l’héritabilité de la maladie est estimée à 66%, ce qui reflète le poids de la composante génétique dans le SLE10.
Outre ces données, de nombreux variants génétiques prédisposent au développement du SLE11. Les principaux gènes associés à la maladie peuvent être regroupés en fonction des mécanismes et des voies, physiologiques ou immunologiques, potentiellement impliqués dans la pathogénèse du SLE12 (tableau 2).
Mécanisme/voie impliqué(e) Gène
Dégradation de l’ADN, apoptose, clairance des débris cellulaires
FCGR2B, ACP5, TREX1, DNASE1, DNASE1L3, ATG5
Signalisation TLR et IFN-I
TLR7, IRF5, IRF7/PHRF1, IRF8, IRAK1, IFIH1, TYK2, PRDM1, STAT4, TREX1, ACP5
Signalisation NF-κB IRAK1, TNFAIP3, TNIP1, UBE2L3,
SLC15A4, PRKCB Clairance et phagocytose des complexes
immuns
C1Q, C1R/C1S, C2, C4A/B, FCGR2A/B, FCGR3A/B
Signalisation et fonctions des
lymphocytes B
FCGR2B, BLK, LYN, BANK1, PRDM1, ETS1, IKZF1, AFF1, RASGRP3, IL10, IL21, NCF2, PRKCB, HLA-DR2 & DR3, MSH5, IRF8
Signalisation et fonctions des
lymphocytes T
PTPN22, TNFSF4, CD44, ETS1, IL10, IL21, TYK2, STAT4, PRDM1, AFF1, IKZF1, HLA-DR2 & DR3
Signalisation et fonctions des neutrophiles et des monocytes
ITGAM, ICAMs, FCGR2B, FCGR3A/B, IL10, IRF8
Tableau 2. Principaux gènes associés à un risque de développer un SLE en fonction des mécanismes/voies physiologiques ou immunologiques impliquées dans la pathogenèse de la maladie.
[TLR, Toll-like receptor; IFN, interferon; NF-κB, nuclear factor kappa B; FCGR, Fc fragment of IgG receptor; ACP5, acid phosphatase 5 tartrate resistant; TREX1, three prime repair exonuclease 1; DNase, deoxyribonuclease; ATG5, autophagy related 5; IRF, IFN regulatory factor; PHRF1, PHD and ring finger domains 1; IRAK, interleukin-1 receptor-associated kinase; IFIH1, interferon induced with helicase C domain 1; TYK2, tyrosine kinase 2; PRDM1, PR domain containing 1 with ZNF domain; STAT, signal transducer and activator of transcription; TNFAIP3, tumor necrosis factor (TNF) alpha-induced protein 3; TNIP1, TNFAIP3 interacting protein 1; UBE2L3, ubiquitin-conjugating enzyme E2L 3; SLC15A4, solute carrier family 15 member 4; PRKCB, protein kinase C beta; BLK, B lymphoid tyrosine kinase; LYN, Lck/Yes-related novel protein tyrosine kinase; BANK1, B-cell scaffold protein with ankyrin repeats 1; ETS1, protein C-ets-1; IKZF1, IKAROS family zinc finger 1; AFF1, AF4/FMR2 family member 1;
RASGRP3, RAS guanyl releasing protein 3; IL, interleukin; NCF2, neutrophil cytosolic factor 2; HLA-DR, human
adhesion molecule]
1.3.2 La composante environnementale
!
Les disparités majeures observées au sein de populations génétiquement liées plaident pour une implication des facteurs environnementaux dans la pathogénèse du SLE13–16.
Certains de ces facteurs sont exogènes (agents infectieux, médicaments, rayonnement ultraviolet) et d’autres sont endogènes (facteurs hormonaux).
1.3.2.1 Les facteurs exogènes
!
- Les agents infectieux
Parmi les agents viraux, ceux qui sont le plus fréquemment impliqués dans la genèse ou l’exacerbation de poussées de SLE sont le parvovirus B19, le cytomégalovirus (CMV) et l’Epstein-Barr virus (EBV)17–20.
Les infections bactériennes peuvent également contribuer à l’aggravation de la maladie, notamment les infections par le pneumocoque, Escherichia coli et
Chlamydia trachomatis21.
Outre leur rôle pathogène direct, ces agents infectieux contribuent à l’aggravation du SLE par différents mécanismes. Ils sont par exemple susceptibles de générer et d’entretenir une réponse auto-immune aberrante via un mimétisme moléculaire (réaction croisée à partir d’antigènes viraux ou bactériens) ou via des dégâts tissulaires générateurs d’auto-antigènes21,22.
- Les médicaments
La prise de certains médicaments peut induire de novo des manifestations cliniques et biologiques du SLE23,24, en favorisant une réponse auto-immune exacerbée.
Parmi les différents mécanismes évoqués, on retiendra: une induction d’une hypométhylation de l’ADN à l’origine d’une dérégulation de l’expression des gènes25,26, une augmentation de l’apoptose et une altération de la clairance des
corps apoptotiques27–29. Ceci aboutit au relargage et l’accumulation d’auto-antigènes.
Les vaccins pourraient également être des facteurs déclencheurs de poussées chez les patients SLE. La présence de produits bactériens ou viraux, de solvants ou d’adjuvants pourrait en effet favoriser une réaction auto-immune inappropriée. Toutefois, aucune étude n’a démontré une relation de causalité entre une vaccination récente et la survenue d’une poussée lupique. Ainsi, le risque d’aggraver le SLE par les vaccins reste, à ce jour, théoriquement non confirmé. - Les rayonnements ultraviolets
La photo-exposition solaire est un facteur de risque important dans le SLE. Elle favorise surtout le développement des lésions cutanées30. A titre d’exemple, l’exposition des kératinocytes aux UV induit leur apoptose et stimule l’expression de cytokines pro-inflammatoires à l’origine d’une activation immunitaire30,31.
Outre les facteurs cités, le tabac32, le stress33 et divers agents34,35 (pesticides, solvants, teintures…) sont considérés comme des facteurs de risque du SLE.
1.3.2.2 Les facteurs endogènes
!
- Les facteurs hormonaux
Le SLE touche principalement les femmes (85 à 90%), avec une prévalence maximale chez les femmes en âge de procréer. De plus, une rythmicité des poussées temporellement associée à la période prémenstruelle est observée. Ceci suggère un rôle potentiel des facteurs hormonaux, particulièrement des œstrogènes dans le SLE.
a. Les Oestrogènes
Les œstrogènes ont un effet immuno-modulateur établi via leurs récepteurs exprimés à la surface de la plupart des cellules immunitaires. Des données récentes leur attribuent un rôle dans la survie des LB auto-réactifs et la sécrétion d’IFN-α par les cellules dendritiques plasmacytoïdes36,37. L’utilisation d’œstroprogestatifs chez la patiente lupique était déconseillée pendant des années, par crainte d’une exacerbation de la maladie. Deux essais cliniques randomisés n’ont pas validé la
de poussées plus faibles dans le bras progestatifs seuls38,39. Ces résultats sont confirmés par d’autres études observationnelles40,41. Même si l’utilisation d’une contraception œstroprogestative ne semble pas influencer directement l’activité du SLE chez les patientes, un problème de risque thrombotique se pose, surtout en présence d’anticorps anti-phospholipides39,42–44.
b. La progestérone
La grossesse est reconnue comme étant un facteur de risque de poussée lupique45. Malgré les constatations épidémiologiques, le rôle délétère de la progestérone sur la maladie n’a pas été prouvé39. De plus, la progestérone pourrait avoir un rôle protecteur dans le SLE en limitant la sécrétion d’IFN-α par les cellules dendritiques plasmacytoïdes46.
1.3.3 La composante immunitaire
!
Dans le SLE, l’interaction entre un terrain de prédisposition génétique et les facteurs environnementaux cités est responsable d’une activation aberrante et exacerbée des cellules du système immunitaire inné et adaptatif.
La caractéristique majeure de la réponse immune dérégulée dans le SLE est la perte de tolérance vis-à-vis des antigènes nucléaires du soi, aboutissant à la production d’auto-anticorps dits ANA (Anti-nuclear antibody). Parmi ces ANA, on distingue les anti-dsDNA (double stranded DNA), et les anticorps antigènes nucléaires solubles : anti-Sm (anti-anti-Smith), anti-RNP (anti-ribonucleoprotein), anti-SSA/Ro (anti-Sjögren syndrom A), Anti-SSB/La (anti-Sjögren syndrom B). Une proportion non négligeable des patients développent aussi des anticorps anti-phospholipides47. Ces auto-anticorps forment avec les auto-antigènes nucléaires des complexes immuns circulants (CIC) qui jouent un rôle important dans le maintien de l’inflammation et la formation des lésions tissulaires. En outre, un rôle important a été décrit pour l’interféron de type I (IFN-I), principalement l’IFN-α dans la pathogenèse du SLE.
1.3.3.1 Description de la famille des IFN-I
!
Les IFN-I appartiennent à la famille des cytokines à hélice alpha de classe II, qui regroupe également les IFN de type II (IFN-γ), les IFN de type III (IFN-λ), l’interleukine 10 (IL-10) et ses homologues (IL-19, IL-20, IL-22, IL-24, IL-26)48–50.
Les IFN-I représentent une famille de cytokines constituée de plusieurs membres : les sous-types d’IFN-α, IFN-β, IFN-ε, IFN-κ, IFN-Ω, IFN-δ, IFN-τ. Les IFN-δ et IFN-τ ne sont pas trouvés chez l’homme. L’IFN-κ est exprimé par les kératinocytes. Du point de vue immunologique, les IFN-α et l’IFN-β sont les types les plus importants, dû à leur diverses fonctions immunitaires (implication dans le développement des lignées de cellules immunitaires ainsi que dans l’immunité innée et adaptative cellulaire et humorale)51. L’IFN-α est codé par une vingtaine de gènes dont 13 uniquement codent pour des polypeptides fonctionnels. Quand à l’IFN-β, il n’a pas de sous-types.
L’IFN-α et l’IFN-β partagent le même récepteur (interferon alpha/beta receptor IFNAR) exprimé de manière ubiquitaire. L’IFNAR est hétérodimérique formé de deux sous-unité IFNAR1 et IFNAR2. L’IFNAR2 est responsable de la liaison au ligand alors que les deux sous-unités sont nécessaires pour la transduction du signal52,53.
L’IFN-α et l’IFN-β interviennent à différents niveaux de la réponse immune. Ils participent notamment à la défense anti-virale au cours de laquelle ils inhibent l’expansion de l’infection à travers plusieurs mécanismes, tels que :
- L’inhibition de la réplication virale par la dégradation des ARN54 ou la diminution de la synthèse des protéines55 ;
- L’induction de l’apoptose des cellules infectées56.
Outre leur rôle physiologique, ces cytokines jouent un rôle pathogène dans plusieurs maladies auto-immunes dont le SLE57.
!
La première association entre le SLE et l’IFN, a été décrite en 1979 suite à la découverte de taux élevés d’IFN, suggéré IFN-γ, dans le sérum de patients SLE58. Plus tard, en 1982, cet IFN a été caractérisé comme étant l’IFN-α59. Depuis, plusieurs études ont montré que les taux sériques d’IFN-α sont corrélés à l’activité de la maladie ainsi qu’à ses différentes manifestations cliniques, et à l’activation du système immunitaire60. Des taux élevés de protéines induites par l’IFN-α, comme la 2’-5’ oligoadenylate synthetase61 et la MxA62, ont également été trouvés chez la plupart des patients SLE, prouvant que l’IFN-α produit est bioactif. D’ailleurs, de nombreuses études d’expression génétique ont mis en évidence, par la technique des gene-microarray, une signature IFN-I chez la majorité des patients SLE, associée à un tableau clinique plus sévère avec le développement de néphrite et de manifestations hématologiques63–66. Dans le SLE pédiatrique, la signature IFN a été également reportée dans les stades précoces de la maladie63. Ceci suggère que l’activation du système d’IFN-I serait particulièrement importante dans la pathogenèse du SLE. Enfin, une augmentation du taux sérique de chémokines induites par l’IFN-I est associée à un statut plus actif de la maladie67.
1.3.3.3 Source de l’IFN-I dans le SLE
!
La principale source d’IFN-I dans le SLE est la sous-population de cellules dendritiques nommées cellules dendritiques plasmacytoïdes (pDC). Les pDC jouent un rôle central dans la réponse inflammatoire du SLE, principalement via leur production excessive et continue d’IFN-I après activation par les CIC68. Les mécanismes aboutissant à cette sécrétion aberrante d’IFN-I par les pDC seront détaillés plus tard.
Chez les patients SLE, les pDC sont retrouvées en nombre élevé dans les ganglions lymphatiques69, la peau70,71 et le tissu rénal72. Par contre, leur fréquence dans le sang périphérique est diminuée73–75. Ceci suggère un recrutement des pDC circulantes dans les tissus, suite à leur activation.
D’autres types cellulaires, comme les monocytes76 et les granulocytes77, peuvent aussi produire de l’IFN-I mais en quantités nettement plus faibles que les pDC.
1.3.3.4 Conséquences de la production d’IFN-I sur les cellules du système immunitaire
!
L’IFN-I produit en excès dans le SLE occupe une place centrale dans le développement de la maladie à travers l’activation du système immunitaire inné et adaptatif (figure 1). Les effets de l’IFN-I sur les différents types de cellules du système immunitaire sont résumés ci-dessous :
Sur les monocytes78
- Induction de la différenciation en cellules dendritiques myéloïdes (mDC) présentatrices d’antigènes
Sur les mDC78–81
- Augmentation de l’expression des molécules du CMH (complexe majeur d’histocompatibilité) de classe II (CMH-II), des molécules de co-stimulation CD80 et CD86 et la molécule de maturation CD83. Ceci permet une activation des fonctions de présentation des auto-antigènes aux lymphocytes T CD4+ (LT4) auto-réactifs
- Augmentation de la survie
- Promotion de la différenciation des lymphocytes T CD8+ (LT8) cytotoxiques effecteurs
- Production des cytokines BAFF (B-cell activating factor) et APRIL (a proliferation inducing ligand) favorisant la survie des lymphocytes B (LB), leur différenciation en plasmocytes et le switch isotypique des IgM vers les IgG Sur les neutrophiles82,83
- Activation des neutrophiles favorisant la production d’auto-antigènes (cette activation sera détaillée plus tard, dans le contexte de production d’IFN-I par les pDC dans le SLE)
provoquant ainsi une élévation du taux d’apoptose et le relargage d’auto-antigènes.
Sur les LB79,85
- Augmentation de la survie et de la différentiation des LB auto-réactifs favorisant la production d’auto-anticorps
Sur les LT486
- Orientation de la réponse immune vers la voie Th1 (T helper 1)
- Stimulation de la génération de LTfh (LT-follicular helper) impliqués dans l’activation des LB et la production d’anticorps.
Sur les lymphocytes T régulateurs (LTreg)87
- Altération des fonctions immuno-suppressives des LTreg favorisant une dérégulation de la réponse inflammatoire et le développement des réponses auto-réactives.
Sur les LT881,84
- Augmentation de la cytotoxicité via l’expression de la perforine et du granzyme B favorisant le relargage de nucléosomes solubles et d’auto-antigènes.
!
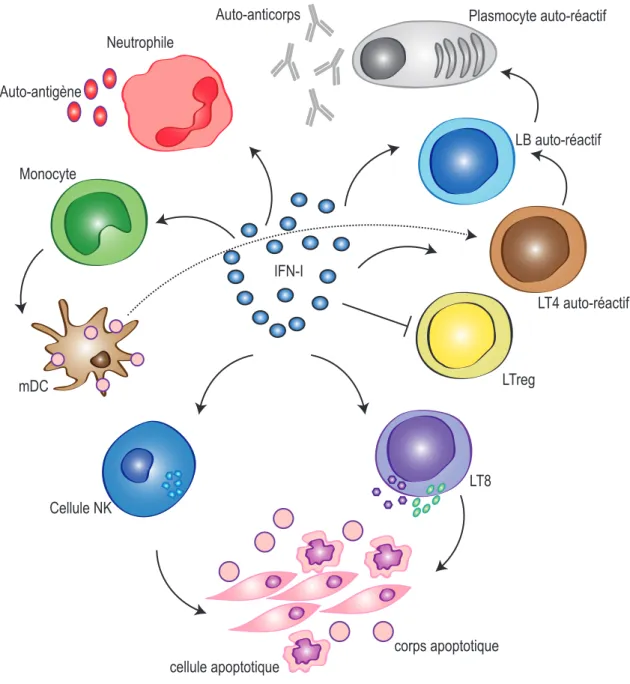
Figure 1. Rôle central de l’IFN-I dans la dérégulation du système immunitaire dans le SLE
L’IFN-I initie et maintient une réponse auto-immune, inflammatoire, exacerbée et dérégulée dans le SLE. Il active les fonctions cytotoxiques des cellules NK et des LT8 favorisant ainsi l’apoptose des cellules cibles et le relargage de corps apoptotiques contenant des auto-antigènes. L’IFN-I stimule également la différenciation des monocytes en cellules dendritiques myéloïdes (mDC) présentatrices d’antigènes. Les mDC captent les corps apoptotiques et les présentent aux LT4 auto-réactifs qui à leur tour activent la différenciation des LB réactifs en plasmocytes produisant des auto-anticorps. De plus, l’IFN-I inhibe les fonctions suppressives des LTreg et active le relargage d’auto-antigènes par les neutrophiles.
Neutrophile Monocyte Plasmocyte auto-réactif IFN-I mDC LB auto-réactif LT4 auto-réactif LTreg LT8 Cellule NK corps apoptotique cellule apoptotique Auto-anticorps Auto-antigène
2 Les
cellules
dendritiques
plasmacytoïdes:
dans l’immunité et l’auto-immunité via l’IFN-I
! !
2.1 Découverte des pDC
2.1.1 La découverte de cellules plasmacytoïdes
!
En 1958, des études pionnières menées par Lennert et Remmele ont mis en évidence un type cellulaire morphologiquement similaire au plasmocyte et abondant dans les zones paracorticales riches en LT des ganglions lymphatiques humains88. Ces cellules,
n’exprimant pas les marqueurs des LB ni ceux des plasmocytes, ont eu, pendant plusieurs années, différentes appellations par les pathologistes de l’époque. Elles ont été nommées : « plasmocytes associés aux cellules T »89, « cellules T plasmacytoïdes » trouvées dans des contextes inflammatoires90 et néoplasiques91 et « monocytes plasmacytoïdes » pour leur expression de certains marqueurs de myélomonocytes92.
2.1.2 La découverte de cellules productrices de taux importants
d’IFN-I
!
Dans le même temps, en 1978, Trinchieri et al. ont montré que des leucocytes humains du sang périphérique, activés par des virus, produisent une forte quantité d’IFN-I responsable d’une activité anti-virale. Par des techniques de fractionnement des leucocytes, ils ont observé que seule une petite fraction de ces cellules était responsable de la production d’IFN-I93. Dans les années suivantes, plusieurs études ont été menées pour caractériser ces cellules nommées « IFN-α producing cells » (IPC) ou cellules naturelles productrices de l’IFN94–97. Il a été montré que ces cellules
n’expriment pas les marqueurs des LT, LB, cellules NK, et monocytes. Elles expriment en revanche à leur surface la molécule HLA-DR du complexe majeur d’histocompatibilité de classe II98 mais sont complètement distinctes des cellules dendritiques conventionnelles trouvées dans le sang périphérique.
2.1.3 La découverte des cellules dendritiques plasmacytoïdes
!
En 1994, O’Doherty et al. ont identifié dans le sang périphérique deux populations de cellules dendritiques, l’une mature HLA-DR+CD11c+ et l’autre immature HLA-DR+CD11c- 99. En 1997, Grouard et al. ont enfin isolé, à partir d’amygdales humaines, ces « cellules T plasmacytoïdes » jusqu’ici observées par les pathologistes dans les tissus lymphoïdes secondaires sans aucun rôle fonctionnel encore décrit. Ils ont montré que ces cellules sont identiques aux cellules dendritiques immatures décrites dans le sang périphérique. Elles nécessitent de l’IL-3 pour survivre in vitro et, en présence d’IL-3 et de CD40 ligand (CD40L), sont capables de se différencier en cellules dendritiques stimulant une forte prolifération de cellules T naïves allogéniques CD4+CD45RA+. C’est pour cette raison que ces cellules plasmacytoïdes ont été considérées comme des précurseurs de cellules dendritiques100.
Finalement, en 1999, les groupes de Liu et Colonna ont montré que les cellules dendritiques HLA-DR+CD11c- du sang périphérique et les cellules plasmacytoïdes
trouvées dans les organes lymphoïdes secondaires correspondent aux cellules professionnelles productrices de l’IFN-α (IPC) en réponse aux infections virales101,102. C’est ainsi que ces cellules conciliant les caractéristiques des cellules plasmacytoïdes et celles des IPC ont été classées comme une sous-population de cellules dendritiques, identifiée sous le nom de « cellules dendritiques plasmacytoïdes » (pDC).
2.2 Ontogénie des pDC
!Depuis la caractérisation des pDC en 1999, plusieurs études ont tenté de comprendre le développement de ces cellules. Différentes hypothèses ont été émises aboutissant à des interprétations divergentes et controversées.
Les pDC se développent dans la moelle osseuse à partir de progéniteurs dérivant des cellules souches hématopoïétiques (CSH).
2.2.1 Arguments en faveur d’une origine lymphoïde des pDC
!
Les premiers travaux ont suggéré une origine lymphoïde des pDC. En effet, il a été montré que la surexpression de Id2 ou Id3, protéines inhibitrices de la liaison à l’ADN, bloque le développement des LB, LT et des pDC mais n’affecte pas la sous-population de cellules dendritiques conventionnelles (cDC)103. De plus, il a été mis en évidence la présence de marqueurs associés à la lignée lymphoïde dans les pDC et pas dans les cDC. Parmi ces marqueurs, un réarrangement D-J des gènes de la chaîne lourde des immunoglobulines et des transcrits codant pour la chaîne epsilon du CD3 (CD3ε), la chaîne pre-Tα (pre-T cell receptor α), le récepteur ILT7 (immunoglobulin like transcript 7), le granzyme B, le facteur de transcription Spi-B et le récepteur tyrosine kinase Eph-B1 ont été trouvés104,105.
Cependant, ces observations ont été suivies par d’autres recherches qui ont amené à discuter cette origine lymphoïde unique.
2.2.2 Arguments en faveur d’une origine commune des cDC et des
pDC
!
Il a été démontré que la différenciation des pDC est abolie chez les souris déficientes pour le facteur de transcription IRF8 qui est essentiel au développement de la lignée myéloïde106. Une approche inverse consistant en une déplétion des progéniteurs lymphoïdes communs par un traitement à l’œstrogène n’a montré aucun effet sur le
développement des pDC, in vivo107. En outre, il a été montré que les deux
sous-populations de cellules dendritiques, pDC et cDC, peuvent se différencier in vitro et in vivo à partir de progéniteurs lymphoïdes communs (CLP) ou de progéniteurs myéloïdes communs (CMP), positifs pour le récepteur tyrosine kinase Flt3108–110. En se basant sur ces résultats, une nouvelle hypothèse a ainsi émergé, suggérant l’existence d’une voie de différenciation commune et restreinte aux cellules dendritiques.
En 2007, deux équipes différentes de recherche ont identifié un progéniteur commun pour les pDC et les cDC nommé progéniteur de cellules dendritiques commun (CDP). La première étude a identifié, dans la moelle osseuse murine, la présence de cellules progénitrices Lin-c-kitintFlt3+M-CSFR+ caractérisées par une importante activité proliférative et un profil d’expression génétique distinct. Ces cellules avaient le pouvoir de générer uniquement les sous-types de cellules dendritiques (pDCs et cDC), in vitro et in vivo111. La deuxième étude a montré que la culture de moelle osseuse murine en présence de Flt3-ligand, in vitro, génère deux précurseurs successifs spécifiques des cellules dendritiques : les pro-DC se divisant activement pour donner les pre-DC qui sont ensuite capables de se différencier pour donner l’ensemble des sous-populations de cellules dendritiques. Des équivalents in vivo des pro-DC ont également été isolés112.
Ces deux travaux ont donc mis en évidence, pour la première fois, une voie de différenciation unique, commune et spécifique aux cellules dendritiques à partir de progéniteur commun (CDP ou pro-DC). Dans des modèles murins, il a été montré que le CDP provient du progéniteur commun des macrophages et des cellules dendritiques (MDP) se différenciant lui-même à partir des CMP113,114.
Enfin, l’ensemble de toutes ces observations souligne un développement flexible caractéristique des pDC. Ces cellules, représentant une lignée hématopoïétique unique, semblent provenir de progéniteurs à différent potentiel de différenciation dérivant des CSH (Figure 2). Toutefois, un progéniteur ayant le potentiel unique de s’engager exclusivement dans la voie de différenciation en pDC reste à identifier115.
!
Figure 2. Ontogénie des pDC
Dans la moelle osseuse, à partir de cellules souches hématopoïétiques (HSC) se différencient des progéniteurs MPP (multi-potent progenitor) dont dérivent les progéniteurs lymphoïdes et myéloïdes communs (CLP et CMP, respectivement). Les CMP se différencient en MDP (macrophage-dendric cell progenitor) puis en CDP (common DC progenitor). Les CDP se différencient en pDC ou en cDC. Les CLP peuvent également se différencier en pDC.
2.3 Cytokines et facteurs de transcription impliqués dans le
développement des pDC
!2.3.1 Les cytokines
! 2.3.1.1 Flt3L !La cytokine clé dans le développement des pDC est le FMS-like tyrosine kinase 3 ligand (Flt3L) ayant pour récepteur le Flt3 (CD135). Plusieurs travaux ont montré un rôle crucial du couple Flt3L/Flt3 dans la différenciation des pDC à partir des CSH, in
HSC CLP MPP CMP MDP CDP pDC cDC Moelle osseuse
vitro et in vivo, aussi bien chez l’homme que chez la souris. Les cultures de moelle osseuse n’aboutissent à la génération de pDC qu’en présence de Flt3L116–118. In vivo, l’administration de Flt3L à des sujets sains volontaires provoque une augmentation importante du nombre de pDC dans le sang périphérique119. De la même manière, l’induction d’expression de Flt3L dans des souris transgéniques mène à une forte augmentation du taux de pDC dans le sang périphérique et différents tissus (moelle osseuse, ganglion lymphatique mésentérique, rate et thymus)120. Inversement, une
diminution drastique du taux des pDC est observée dans les rates de souris KO pour Flt3L en comparaison avec des souris WT121,122.
2.3.1.2 TPO
!
En plus du Flt3, la thrombopoïétine (TPO) semble jouer un rôle dans le développement des pDC. La TPO ajoutée dans des cultures de progéniteurs hématopïétiques (HPC) humains agit en synergie avec le Flt3L pour induire une forte expansion des HPC et générer ainsi de grande quantité de pDC in vitro123.
2.3.1.3 M-CSF
!
Le facteur de croissance Macrophage-Colony Stimulating Factor (M-CSF), connu pour son rôle dans le développement des monocytes et des macrophages, est capable d’induire la génération de pDC et cDC à partir de cultures de moelle osseuse murine en l’absence de Flt3L. Néanmoins, la quantité de pDC est considérablement inférieure à celle obtenue en présence de Flt3L124.
2.3.2 Les facteurs de transcription
!
2.3.2.1 La protéine E2-2
!
Au niveau transcriptionnel, le développement des pDC est principalement contrôlé par le gène TCF4 codant pour la protéine E2-2. Ce facteur de transcription appartient à la famille des protéines E à motif hélice-tour-hélice (basic helix-loop-helix bHLH) jouant un rôle central dans le développement et le fonctionnement des lymphocytes125. L’activité de ces protéines est antagonisée par les inhibiteurs de la famille Id. Les travaux de Spits et al. ont démontré l’inhibition du développement des lymphocytes humains et des pDC (pas des cDC), in vitro, par la surexpression des protéines Id (Id2 et 3)103. Il s’agit d’un des premiers arguments en faveur de l’implication des protéines E dans le développement des pDC. Parmi la famille des protéines E, E2-2 a été identifié comme le facteur de transcription essentiel contrôlant spécifiquement le développement des pDC. Il est fortement exprimé dans les pDC murines et humaines, plus faiblement dans les LB et presque absent dans les cDC et autres types cellulaires. La délétion constitutive ou inductible de E2-2 bloque le développement des pDC uniquement et abolit la sécrétion d’IFN-α en réponse à une stimulation par un ligand ADN hypométhylé. La perte de fonction monoalléique ou la délétion d’un seul allèle du gène TCF4 provoque une haploinsuffisance de E2-2 causant le syndrome de Pitt-Hopkins (PHS) chez l’homme. Les pDC de patients souffrant du syndrome de Pitt-Hopkins ont un phénotype de surface aberrant et sont incapables de produire de l’IFN-I126. L’inhibition de E2-2 ou sa surexpression dans les progéniteurs hématopoïétiques humains abolit ou stimule, respectivement, le développement in vitro des pDC127.
En outre, E2-2 contrôle l’expression d’autres facteurs de transcription impliqués dans le développement des pDC (Spi-B, IRF8, IRF7…) et constitue alors un réseau transcriptionnel responsable du programme d’expression génétique spécifique des pDC126,128,129.
2.3.2.2 Autres facteurs de transcriptions
!
En plus de E2-2, d’autres facteurs de transcription non spécifiques des pDC sont impliqués dans leur développement, tels que STAT3118,130, Ikaros131 et IRF8132,133. Il a
été montré que STAT3 s’active par la signalisation Flt3 pour contribuer au développement Flt3L-dépendant de pDC humaines et murines in vitro. Quant à Ikaros et IRF8, leur implication dans le développement des pDC a été étudiée uniquement dans des modèles murins. Il a été démontré que les pDC sont absentes du sang périphériques de souris exprimant de faibles taux d’Ikaros ou de souris déficientes pour IRF8.
2.4 Caractérisation phénotypique des pDC
!2.4.1 Profil d’expression des marqueurs de surface
!
Malgré les ressemblances morphologiques et fonctionnelles entre les pDC humaines et les pDC murines, leur profil d’expression des marqueurs de surface est différent. Par souci de clarté, nous ne décrirons que le phénotype des pDC humaines.
Chez l’homme, les pDC n’expriment aucun marqueur spécifique de lignée (lin) des cellules immunitaires connues, notamment : les immunoglobulines (Ig) membranaires et cytoplasmiques et le CD19 des LB, le TCR et le CD3 des LT, le CD14 des monocytes, le CD16 et le CD56 des cellules NK. Elles sont négatives pour la plupart des marqueurs exprimés par les cellules myéloïdes, entre autres le CD11b, CD13, CD14 et CD33134. Elles sont HLA-DR+CD123(IL-3Rα)+ et contrairement aux mDC n’expriment pas le CD11c. Quant aux marqueurs spécifiques, Dzionek et al. ont identifié deux antigènes de surface nommées Blood Dendritic Cell Antigen-2 et -4 (BDCA-2 et BDCA-4) spécifiquement exprimés par les pDC du sang périphérique135. Cao et al. ont également
2.4.2 Morphologie des pDC
!
Observée en microscopie optique après coloration au Giemsa, la pDC humaine non activée ressemble au plasmocyte, légèrement plus petite qu’un monocyte mais plus grosse qu’un lymphocyte au repos. Son noyau est excentré, réniforme et son cytoplasme basophile est caractérisé par une zone de Golgi pâle.
En microscopie électronique à balayage, la pDC présente une morphologie lymphoïde, ronde et lisse avec un diamètre de 8 à 10 µm. Plus précisément, en microscopie électronique à transmission, la pDC montre un noyau avec une hétérochromatine marginale et un cytoplasme contenant un réticulum endoplasmique rugueux bien développé, un appareil de golgi relativement petit et de nombreuses mitochondries. Une fois activée, la pDC adopte une morphologie dendritique134 (figure 3). Les pDC murines isolées de la moelle osseuse ou de la rate, ont une morphologie plasmacytoïde homogène ressemblant à celle retrouvée avec les pDC humaines137.
!
Figure 3. Morphologie des pDC humaines
pDC humaines observées en microscopie électronique à transmission (a) et en microscopie électronique à balayage, au repos (b) et après activation (c). Grossissement Å~7,000 (a) et Å~3,000 (b, c).
(D’après Grouard, G. et al., J. Exp. Med. 1997 et Colonna, M. et al., Nature Immunol. 2004)
2.5 Localisation et migration des pDC
2.5.1 Dans les conditions homéostatiques
Dans les conditions homéostatiques, les pDCs sont constamment produites dans la moelle osseuse. Elles sont ensuite libérées dans le sang puis migrent vers les organes lymphoïdes secondaires [ganglions lymphatiques, rate et tissus lymphoïdes associés aux muqueuses (MALT)]138. Ce profil de migration similaire à celui décrit pour les LB et les LT est remarquablement différent de celui des mDC qui migrent d’abord vers les tissus non-lymphoïdes avant leur activation139. Dans les ganglions lymphatiques, les
pDC arrivent dans les zones riches en LT (zones T) via les veinules post-capillaires à endothélium épais (HEV)100,102,140 (figure 4).
!
Figure 4. Localisation des pDC dans les ganglions lymphatiques humains
(a) Représentation schématique de la localisation des pDC dans les compartiments du ganglion lymphatique (zones bleues) à proximité des HEV (high endothelial venules), des nodules T (T) dans la zone paracorticale et des follicules de LB (B) dans la zone corticale. (SCS: subcapsular sinuses, MC: medullary cord; MS: medullary sinuses). (b) pDC marquées par un anti-BDCA2, observées sur une coupe histologique de ganglion lymphatique humain. La flèche de l’encadré pointe vers des pDC localisées dans la lumière d’une veinule à endothelium épais HEV.
(D’après Sozzani, S. et al., Trends Immunol. 2010)
HEV SCS
Au contraire, les mDC activées migrent des tissus périphériques vers les ganglions lymphatiques à travers les vaisseaux lymphatiques afférents. En outre, une étude a mis en évidence la migration de pDC via la lymphe afférente, dans deux modèles animaux (mouton et cochon nain)141. Cependant, des études supplémentaires sont nécessaires pour confirmer une telle migration chez l’homme.
Les études concernant les chémokines et les récepteurs spécifiquement impliquées dans la migration des pDCs humaines sont peu nombreuses comparées à celles des mDC et des pDC murines142. Les pDC du sang humain expriment à leur surface différentes molécules d’adhésion et des récepteurs de chémokines de type C-C et C-X-C. Dans les conditions homéostatiques, elles expriment la L-sélectine (CD62L)102,143, le CXCR4144 et le ChemR23145 dont les ligands respectifs GlyCam-1/CD34, CXCL12 et chémérine sont exprimés par les cellules endothéliales des HEV, du côté luminal. Ainsi, ces molécules permettent le recrutement homéostatique des pDC dans les ganglions lymphatiques. De plus, les pDC expriment le récepteur CXCR3 qui, malgré son incapacité à induire leur migration, favorise leur réponse chémotactique au CXCL12144,146,147. Quant au CCR7, le récepteur responsable du recrutement dans les tissus lymphoïdes, son expression sur les pDC non activées reste très controversée. Plusieurs études ont décrit une forte expression du CCR7 sur les pDC non activées purifiées du sang alors que d’autres ont montré une faible voire une absence d’expression sur les « resting » pDC143,146–149. Cependant, après activation notamment
par une stimulation des TLR, les pDC augmentent l’expression de CCR7 et migrent en réponse au CCL21144. Ceci suggère un rôle du CCR7 dans la migration des pDC matures vers les ganglions lymphatiques à travers l’engagement des ligands CCL19 (ELC) et CCL21 (SLC) exprimés par les HEV et les cellules stromales dans les zones T. Dans les conditions inflammatoires, les pDC expriment d’autres molécules impliquées dans le recrutement vers les organes lymphoïdes secondaires, outre les récepteurs déjà cités. A titre d’exemple, elles expriment le PSGL-1 (ligand de la E-sélectine), b1 et b2 intégrines (récepteurs des ICAM-1 et ICAM-2) et le CCR5 (récepteur de CCL3, CCL4 et CCL5)150,151.
2.5.2 Dans les conditions inflammatoires et pathologiques
!
Dans les conditions physiologiques, les pDC sont absentes de la peau et des tissus non lymphoïdes. Toutefois, elles y sont recrutées dans des contextes inflammatoires et pathologiques à travers des récepteurs de chémokines et des signaux associés à l’inflammation et aux lésions tissulaires. En effet, les pDC migrent dans la peau dans le cas d’infections virales143,147,152 ou bien dans des maladies inflammatoires chroniques de la peau, tel que le psoriasis153–155, et dans des maladies auto-immunes avec atteintes cutanées, tel que le SLE71,156. Les pDC ont aussi été retrouvées dans l’environnement tumoral au cours des mélanomes, carcinome basocellulaire, etc157–159. Les mécanismes de recrutement des pDC dans la peau sont imparfaitement compris. Plusieurs études ont montré l’implication de la chémokine chémérine et de son récepteur Chem23R exprimé par les pDC139,145. D’autres signaux induits dans la peau lésée ont été décrits comme ayant un effet chimiotactique sur les pDC : on peut citer, à titre d’exemple, l’adénosine160 et les anaphylayoxines C3a et C5a161. De plus, il a été montré que l’IL-3 produite, entre autres, par les LT dans les organes lymphoïdes régule positivement l’expression de CCR6 et CCR10 (récepteurs respectifs de CCL20 et CCL27) par les pDC induisant ainsi leur migration vers les épithéliums inflammatoires (dans la peau ou les muqueuses)162.
Enfin, dans des modèles murins, il a été démontré que le CCR9 exprimé par les pDC permet leur recrutement dans l’intestin, dans des conditions homéostatiques et inflammatoires163. Ce mécanisme reste à démontrer chez l’homme.
2.6 Rôle physiologique des pDC
!Les pDC, longtemps identifiées comme un des acteurs principaux de l’immunité innée, semblent posséder de plus larges fonctions au sein du système immunitaire.