HAL Id: hal-01636555
https://hal-univ-rennes1.archives-ouvertes.fr/hal-01636555
Submitted on 16 Nov 2017HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Subtle Steric Differences Impact the Structural and
Conducting Properties of Radical Gold Bis(dithiolene)
Complexes
Agathe Filatre-Furcate, Thierry Roisnel, Marc Fourmigué, Olivier Jeannin,
Nathalie Bellec, Pascale Auban-Senzier, Dominique Lorcy
To cite this version:
Agathe Filatre-Furcate, Thierry Roisnel, Marc Fourmigué, Olivier Jeannin, Nathalie Bellec, et al.. Subtle Steric Differences Impact the Structural and Conducting Properties of Radical Gold Bis(dithiolene) Complexes. Chemistry - A European Journal, Wiley-VCH Verlag, 2017, 23 (63), pp.16004-16013. �10.1002/chem.201703172�. �hal-01636555�
Subtle Steric Differences Impact the Structural and Conducting
Properties of Radical Gold Bis(dithiolene) Complexes
Agathe Filatre-Furcate,[a] Thierry Roisnel,[a] Marc Fourmigué,[a] Olivier Jeannin,[a] Nathalie Bellec,[a] Pascale Auban-Senzier,[b] and Dominique Lorcy*[a]
Abstract
Among single component molecular conductors, neutral radical gold dithiolene complexes [(R-thiazdt)2Au]• derived from the
N-alkyl-1,3-thiazoline-2-thione-4,5-dithiolate (R-thiazdt) ligand provide an extensive series of conducting, non-dimerized, ½-filled band systems. Analogs of the known R = isopropyl derivative were investigated here with R = NMe2, cyclopropyl (cPr) and n-propyl (nPr), aiming
at rationalizing the different solid state structures adopted by these compounds despite very closely related substituents on the heterocyclic nitrogen atom. An original crisscross organization within dimerized chains is observed with R = NMe2,
differing however from the analogous iPr derivative by a 180° rotation of the heterocyclic nitrogen substituent. On the other hand, the cyclopropyl and n-propyl substituents lead to robust, uniform, non-dimerized chains with a strongly 1D electronic structure and a formal ½-filled electronic structure. The semiconducting behaviour of these two radical complexes is characteristic of a Mott insulator, whose sensitivity to external pressure has been evaluated up to 2.5 GPa.
[a]
Dr. A. Filatre-Furcate, Dr. T. Roisnel, Dr. M. Fourmigué, Dr O. Jeannin, Dr. N. Bellec, Prof. D. Lorcy
Institut des Sciences Chimiques de Rennes, UMR 6226 CNRS-Université de Rennes 1, Campus de Beaulieu, Bât 10A, 35042 Rennes cedex (France)
E-mail: [email protected] [b]
Dr. P. Auban-Senzier
Laboratoire de Physique des Solides UMR 8502 CNRS-Université de Paris-Sud, Bat 510, 91405 Orsay cedex (France)
Single component molecular conductors have been defined by opposition to classical charge transfer salts as conducting materials involving only one single molecular entity. Two main classes of compounds respond today to this definition: (i) neutral complexes of electroactive tetrathiafulvalene dithiolene ligands successfully isolated as metallic compounds by Kobayashi et al.[1] and (ii), fully organic neutral radical species such as thiazyl radicals (dithiadiazolyl and dithiazolyl),[2,3,4] spiro-bis(phenalenyl)-boron[5] and tetrathiafulvalene (TTF) based radicals.[6,7] Neutral dithiolene complexes lacking the tetrathiafulvalene backbone were also reported for their metallic conductivity, such as trimetallic molybdenum dithiolene cluster complexes,[8] or more recently [Ni(dmit)2] under
pressure.[9] Neutral gold dithiolene complexes offer another alternative.[10] They are indeed obtained as radical species from the 1e– oxidation of the d8 anionic AuIII [Au(dithiolene)2]– complexes. Strong intermolecular interactions in the solid state between
such delocalized radical species are however not a sufficient condition to observe a sizeable or even metallic conductivity because these radical species most often dimerize to form a 2e– -eventually delocalized- bond.[10] Even the regular stacking of planar π-type radicals leading to a ½-filled conduction band is threatened by the so-called Peierls transition which opens a gap at the Fermi level through a dimerization of the stacks.
We recently prepared a series of neutral radical gold dithiolene complexes, [Au(R-thiazdt)2]• (Chart 1a) based on the N-ethyl-1,3-thiazoline-2-thione-4,5-dithiolate
(Et-thiazdt) ligand with one radical per site,[11] which are not affected by the usual propensity of radical species to dimerize in the solid state, and which behave as Mott insulators. Several modifications of this prototypical [Au(Et-thiazdt)2]• complex have
been reported, aiming at a deeper understanding of the structure-properties relationship within this original single-component-conductor family. For instance, the use of diselenolene ligands, rather than dithiolene ones (Chart 1b), enhances the orbital overlaps and increases the room temperature conductivity by two orders of magnitude.[12] Contrariwise, the replacement of the exocyclic sulfur atom by an oxygen one leads to decreased intra-layer interactions and smaller conductivities (Chart 1b). Such structural modifications do not entail substantial changes in the solid state packing but essentially the strength of the intermolecular interactions through so-called anisotropic chemical pressure effects.[12] The nature of the substituent R on the nitrogen atom in these N-substituted 1,3-thiazoline-2-thione-4,5-dithiolate ligands provides another tool to modify these series in a deeper manner. For instance, introduction of hydroxyethyl substituents revealed solid state structures
with added OH•••S hydrogen bonds (Chart 1c).[13] Also, the higher steric constraints brought by bulkier isopropyl groups (Chart 1c) led to an original crisscross overlap of the radical species within uniform stacks, with a very specific electronic structure, highly resistant to physical pressure effects.[14] It confirms that steric constraints considerably affect the organization of the molecules in the solid state and also the associated physical properties which result from strong intermolecular interactions such as the conducting one.
Chart 1
In this paper, we want to unravel the striking evolutions of structural and electronic properties we observed within an extended series of single component conductors based to these N-isopropyl gold dithiolene complexes, by replacing the isopropyl substituent by either a dimethylamino (NMe2), a cyclopropyl (cPr) or a n-propyl
substituent (nPr). While the dimethylamino group is expected to exhibit steric constraints similar to that of the isopropyl one, the somewhat less bulky but less flexible cyclopropyl group provides another variation, which should also contrast with the linear n-propyl derivative, as highlighted below.
Results and discussion
SynthesesAn efficient strategy for the preparation of 1,3-thiazoline-2-thiones consists in the use of primary amine as starting compound.[15] This chemical route allows the formation of various N-substituted heterocycles depending on the primary amine used in the first step,[16] as here N,N-dimethylhydrazine, cyclopropylamine and
n-propylamine. The preparation of the anionic gold dithiolene complexes 3-NMe2, 3-cPr and 3-nPr follows a described procedure, outlined in Scheme 1.[11] As a first step, the N-alkyl-1,3-thiazoline-2-thione 1 was formed by reacting the dithiocarbamate salt, obtained by adding carbon disulfide and triethylamine to a solution of the primary amine, with chloroacetaldehyde followed by cyclisation and dehydration in the presence of sulfuric acid. Bis-metallation of 1-R (R = NMe2, cPr,
nPr) with LDA followed by the addition of S8 and bromopropionitrile led to the
formation of the protected dithiolate ligands 2-R (Scheme 1).
! " #$ %
& ' ( & ' (
Scheme 1. Synthetic route toward the neutral radical gold complexes 4-R (R =
NMe2, cPr, nPr)
The dithiolene ligands were generated by adding sodium methanolate to 2-R and subsequent addition of KAuCl4 and PPh4Cl to the medium led to monoanionic AuIII
complexes [PPh4][Au(R-thiazdt)2] 3-R (R = NMe2, cPr, nPr) which were recrystallized
in CH3CN. According to this procedure, crystals amenable for X-ray diffraction were
obtained for two monoanionic complexes 3-NMe2 and 3-nPr as
tetraphenyphosphonium salts. The monoanionic complex 3-cPr was also prepared with the Et4N+ counter ion, [NEt4][Au(cPr-thiazdt)2], allowing us to obtain crystals
suitable for X-ray diffraction analyses. The description of the molecular structures of these three monoanionic species is detailed below. Due to the dissymmetrical character of the dithiolate ligand, these monoanionic complexes 3-R are susceptible to exist in two configurations, the trans and cis ones. 1H NMR spectra exhibit at room
temperature one set of signals indicating a fast exchange in solution between the cis and trans isomers. In order to generate the neutral radical complexes, we focused on the electrocrystallization approach as it generally allows the formation of good quality crystals at the anode.[17] Upon application of a constant current intensity of 0.3-0.5 µA to a solution of [PPh4][Au(R-thiazdt)2] 3-R (R = NMe2, cPr, nPr) in CH3CN
containing NBu4PF6 as supporting electrolyte, crystals of the three neutral species
4-R were obtained on the electrode.
Electrochemical properties
The redox properties of the three monoanionic complexes [PPh4][Au(R-thiazdt)2]
3-R were investigated by cyclic voltammetry carried out in CH2Cl2 using NBu4PF6 as
supporting electrolyte. The redox potentials are gathered in Table 1 together with those of analogous complexes for comparison purposes and a representative CV is given in Figure 1 for 3-nPr. For all these complexes, the shapes of the CV are very similar. They exhibit two reversible oxidation waves assigned to the successive formation of the neutral and the monocationic complex and one irreversible reduction wave corresponding to the reduction to the dianionic species (Figure 1). Whatever the nature of R, the redox potentials are very close to those found for analogous complexes with ethyl and isopropyl substituents [Au(R-thiazdt)2] R = Et, iPr. Note the
sharp potential responses on the reduction processes from cation-to-neutral and neutral-to-anion that reveal adsorption phenomena for all these complexes.
Figure 1 Cyclic voltamogramm of [Au(nPr-thiazdt)2]1– 3-nPr in CH2Cl2 with NBu4PF6
0.1 M as electrolyte, E in V vs SCE, Pt electrode, scan rate 100 mV.s–1.
Table 1. Redox potentials of [Au(R-thiazdt)2]–1 3-R complexes (E in V vs. SCE) in
CH2Cl2, NBu4PF6, Pt, scan rate 100 mV.s–1.
R Ered*
Epa1/Epc1–
1/0 Epa2/Epc2 0/+1
Ref.
NMe2 –0.96 0.51/0.43** 0.66/0.57 this work
cPr –0.95 0.52/0.46** 0.65/- this work
nPr –0.85 0.56/0.53** 0.73/0.64 this work
iPr –0.95 0.56/0.50** 0.69/0.59 14
Et –0.90 0.55/0.49 0.70/0.61 11
*
Irreversible process. ** Adsorption
Molecular structures
The monoanionic gold dithiolene complex 3-NMe2 crystallizes as Ph4P+ salt in the
monoclinic system, space group P21/n. Two types of crystals were obtained for the
monoanionic 3-nPr complex, the n-propyl derivative was isolated either as acetonitrile solvate, formulated as (Ph4P)(3-nPr)•(CH3CN)2 or as the pure compound
(Ph4P)(3-nPr), both crystallizing in the monoclinic system, space group C2/c while
the Et4N+ salt of 3-cP crystallizes in the triclinic system, space group P–1. The
molecular structures of the complexes are presented in Figure 2. All complexes, with such dissymmetrically substituted ligands, adopt a trans configuration with a square planar geometry around the gold atom. The only exception is (Ph4P)(3-nPr) which
adopts also a cis configuration (Figure 2d). All the metallacycles are slightly distorted along the S•••S axis with angles from 2 to 5° (Table 2). Selected bond lengths are collected in Table 2 and are similar in the four structures.
Figure 2. Detail of the monoanionic complexes 3-R showing the orientation of the
nitrogen substituents. (a) R = NMe2; (b) R = cyclopropyl (cPr); (c) R = nPr in the
solvate (Ph4P)(3-nPr)•(CH3CN)2; (d) R = nPr in (Ph4P)(3-nPr).
Table 2. Selected bond lengths (Å) torsion angle )S1…S2 (°) within monoanionic and
neutral gold dithiolene complexes trans-3-R and 4-R (R = NMe2, cPr, nPr).
3-NMe2 4-NMe2 3-cPr 4-cPr 3-nPr 4-nPr
Charge monoanion neutral monoanion neutral monoanion neutral Au–S1 2.3310(9) 2.3355(10) 2.3218(10) 2.3242(11) 2.3167(8) 2.3189(9) 2.3182(14) Au–S2 2.3263(11) 2.3201(10) 2.3159(11) 2.3208(16) 2.3206(36) 2.3239(7) 2.3206(16) S1–C1 1.740(4) 1.739(4) 1.714(5) 1.732(6) 1.714(14) 1.739(3) 1.710(6) S2–C2 1.749(4) 1.738(4) 1.714(3) 1.752(4) 1.725(14) 1.747(4) 1.735(6) C1–C2 1.330(5) 1.344(5) 1.365(6) 1.347(8) 1.344(19) 1.331(5) 1.369(9) C1–S3 1.746(4) 1.758(4) 1.748(4) 1.748(4) 1.750(14) 1.748(4) 1.737(6) C2–N3 1.410(5) 1.404(5) 1.384(6) 1.397(7) 1.414(17) 1.408(4) 1.388(8) N3–C4 1.352(5) 1.358(5) 1.374(6) 1.366(5) 1.361(17) 1.357(5) 1.366(8) S3–C4 1.747(4) 1.760(4) 1.752(6) 1.742(7) 1.725(14) 1.733(4) 1.748(7) C4–S4 1.672(4) 1.656(4) 1.648(5) 1.659(5) 1.671(14) 1.667(4) 1.646(6) )S1•••S2 4.81(8) 4.96(16) 2.63(2) 1.06(30) 3.42(7) 2.14(12) Au S2C2 C1 S1 N3 C4 S3 S4 R
Despite having very similar size, the orientation of the R substituent of the heterocyclic nitrogen atom with regard to the thiazoline ring strongly differs from one complex to the other. The syn disposition of the methyl groups in 3-NMe2 (Scheme
2), with one methyl above and one methyl below the plane of the thiazoline-2-thione core and the lone pair oriented towards the metallacycle, recalls the syn conformation observed for the methyl groups in the isopropyl (iPr) analogous complex 3-iPr.[14] Besides, the cyclopropyl groups in 3-cPr are located above and below the thiazoline plane (Figure 2b) and the n-propyl substituents are strongly disordered in the cis 3-nPr complex (Figure 2d).
Scheme 2.
All the neutral complexes 4-R (R = NMe2, cPr, nPr) crystallize as the
trans-isomers and they all exhibit a planarization of the metallacycles upon oxidation leading to quasi planar skeletons. Geometrical modifications between the monoanionic and neutral species are those generally observed in gold bis(dithiolene) complexes such as, within the metallacycles, a shortening of the C–S bonds and an elongation of the C=C bond (Table 2). This phenomenon is due to the non-innocent character of the dithiolene ligand. Within the thiazoline-2-thione ring, the modification of the bond lengths is less pronounced. Focusing now on the alkyl group, we note that its orientation with respect to the thiazoline ring is the same as the one observed for the monoanionic species for all the three complexes. For instance, the syn configuration of the NMe2 (Scheme 2) maintains the orientation of the lone pair
towards the metallacycle, a possible consequence of a stabilizing interaction between the lone pair of the nitrogen atom and the closest sulfur atom of the metallacycle. This orientation contrasts with the anti conformation observed for the methyl groups in the isopropyl (iPr) analogous complex 4-iPr (Scheme 2).[14]
Structural and electronic properties of the radical complexes
Transport measurements were carried out on crystals of the neutral radical species 4-R along the long axis of the crystals which correspond in all cases to the stacking axis of the complexes 4-R. Conductivity values obtained at room temperature are collected in Table 3 together with those of some analogous complexes with R = Et, iPr for comparison. For the three novel complexes, the temperature dependence of the resistivity at ambient pressure shows a semiconducting behavior with an activation energy in the range of 0.07–0.18 eV, as deduced from the fit of the data with a law of the type ρ = ρ0 exp(Ea/kT) (Figures S2 &
S3). Temperature dependence of the magnetic susceptibility was measured for both
4-nPr and 4-cPr, revealing a very weak, temperature independent response.
Table 3. Conductivity values at room temperature and ambient pressure for the
radical complexes 4-R together with energy activation (Ea). 4-R *RT (S.cm–1) Ea (eV) Ref
R = NMe2 0.045 0.18 this work
R = cPr 0.037 0.14 this work R = nPr 0.36 0.07 this work
R = iPr 5 0.06 14
R = Et 0.33 0.12 11
R = EtOH 0.05-0.07 0.14 13
We will see below that the large differences of conductivity and activation energies are intimately correlated to the details of the solid state arrangement adopted by these complexes, which furthermore strongly differ from what was observed earlier in 4-Et or 4-iPr. The highly conducting 4-Et species adopts indeed a non-dimerized, uniform chain structure with strong inter-chain interactions. It even becomes metallic under pressure.[11] On the other hand, the larger steric constraints of the iPr groups, which adopt an anti conformation (See Scheme 2) led to an
original crisscross staking of the radicals and, despite a uniform stack, the apparition of a small direct gap in the band structure due to an avoided band crossing.[14]
If we consider now the NMe2-subsituted complex 4-NMe2, it crystallizes in the
orthorhombic system, space group I222 (n° 23), with the gold complex on a two-fold axis. The neutral radical molecules do form stacks along the a axis, with a crisscross orientation comparable to that in 4-iPr (Figure 3). However, at variance with 4-iPr which exhibits an apparently similar structure, the stacks are not uniform any more, as shown for example from the Au•••Au distances at 3.4660(6) Å and 3.5395(6) Å, indicating that the complexes are actually dimerized along the stacking axis.
Figure 3. (a) Projection view along a of the unit cell of 4-NMe2. (b) Details of the
overlap interactions a1, a2, b1 and b2. The dotted lines are only a guide to the eyes to visualize interacting molecules.
The calculated overlap energies β associated with these two intra-stack interactions, a1 and a2 (Figure 3b) amount to +a1 = 0.8 and +a2= –0.357 eV
respectively. Two other interactions b1 and b2 are found between the stacks,
associated with short S•••S inter-columnar contacts, with associated overlap energies of βb1 = 0.213 eV and βb2 = 0.036 eV. The consequences of this
dimerization on the electronic structure are clearly seen in the calculated band structures of 4-NMe2 and 4-iPr shown in Figure 4. With four different molecular
orientations in the unit cell, each molecular energy level gives rise to four bands. As a consequence of the stack dimerization in 4-NMe2, a large gap is observed within
the four SOMO bands marked with an asterisk (*), with the two lower bands filled and the two upper bands empty. On the other hand, in 4-iPr, an avoided crossing at Fermi level generates a much smaller gap. The direct consequence is a room temperature conductivity in 4-NMe2 decreased by two orders of magnitude and an
activation energy multiplied by 3 (Figure S2). The origin of these striking differences between the two complexes despite the very similar shape and volume of the iPr and NMe2 substituents can be traced back to the details of their relative orientation. We
have already noted the anti conformation of the iPr group in 4-iPr at variance with the syn orientation of the NMe2 group in 4-NMe2 (Scheme 2). As shown in Figure 5,
it brings a small but notable difference in the dihedral angle between complexes along the chains, which then can contribute to the observed differences in the staking patterns, and the resulting electronic structures.
Figure 4 Calculated band structure of the gold complexes a) 4-NMe2 and b) 4-iPr
with the HOMO-1(4 bands), SOMO (4 bands marked with red asteriks) and LUMO (4 bands) where , = (0, 0, 0), X = (½, 0, 0), Y = (0, ½, 0), Z = (0, 0, ½), and N = (½, 0, ½) of the Brillouin zone of the triclinic lattice. The dashed line in 4-iPr refers to the Fermi level assuming a metallic filling of the levels.
Figure 5. Comparison of overlap patterns with the chains in 4-iPr and 4-NMe2.
The crystal structures of the n-propyl and cyclopropyl derivatives (4-nPr, 4-cPr) are closely related and will be described together in the following. Albeit both compounds are semiconductors, 4-nPr exhibits a higher conductivity and smaller activation energy than 4-cPr (Table 2). The analysis and comparison of both structures can provide a rationale for these differences. As shown in Figure 6, 4-nPr crystallizes in the triclinic system, space group P–1, and 4-cPr in the monoclinic system, space group P21/c. In both structures, the radical gold complexes are
located on inversion centers, and stack into uniform chains running along a axis. Much weaker inter-stack interactions further develop in both structures along b axis, generating (a,b) conducting layers, isolated from each other along c axis. A detail of the interactions within each layer is given in Figure 7. The associated βHOMO-HOMO
overlap interactions amount to +a = –0.4067 eV, +b1 = –0.0078 eV and +b2 = 0.0383
eV in 4-nPr, and to +a = –0.1975 eV, +b1 = 0.0314 eV and +b2 = 0.0080 eV in 4-cPr.
The resulting band structure calculations shown in Figure 8 reflect this structural organization. With only one molecular orientation per unit cell, it is composed of one band per molecular energy level. The βa value twice as large in 4-cPr than in 4-nPr
gives rise to a much larger band dispersion along the stacking Γ-X direction in the former complex, which also explains its higher conductivity and smaller activation energy (see above, Figure S3).
Figure 6. Projection views along a axis of the unit cells of (a) 4-nPr and (b) 4-cPr.
Figure 7. The overlap interactions a, b1 and b2 in the layers (left), and detail of the
intra-stack overlap a (right) in (a) 4-nPr and (b) 4-cPr. The dotted lines are only a guide to the eyes to visualize interacting molecules.
Figure 8. Band structures of the (a) 4-nPr and (b) 4-cPr gold complexes. The
SOMO bands are marked with a red asterisk. , = (0, 0, 0), X = (½ , 0, 0), Y = (0, ½, 0), and R = (½, – ½, 0) of the Brillouin zone of the monoclinic lattice. The dashed line refers to the Fermi level assuming a metallic filling of the levels.
The stronger overlap interaction within the stacks in 4-cPr, together with weak inter-stack interactions give rise in both compounds to a strongly one-dimensional band structure, as evidenced from calculated open Fermi surfaces (Figure S4) for both compounds. It follows that the observed semiconducting properties cannot be attributed to an energy gap as described above in 4-NMe2 or 4-iPr, but is rather the
consequence of strong electron localization. We are faced here with two rare examples of so-called molecular Mott insulators, an electronic ground state where the radical species are neither strongly delocalized in a ½-filled conduction band, nor paired two-by-two, eventually in a dimerized chain (Peierls transition). Such an electronic state was already observed in 4-Et and opens very interesting perspectives. Indeed, this electronic structure is particularly sensitive to external stimuli such as pressure or electric field pulses. In 4-Et, application of external pressure leads indeed to the stabilization of a metallic state (above 1.3 GPa),[11,12] while electric field pulses were used to generate a remnant and reversible highly conducting state, of interest toward their use in Resistive Random Access Memories (RRAM).[18] Along these lines, we therefore investigated the pressure dependence of the most conducting Mott insulator system, i.e. 4-nPr, up to 2.5 GPa. The RT conductivity increases by two orders of magnitude from 0.36 S cm–1 at ambient pressure, up to 60 S cm–1 at 2.5 GPa, following an exponential law characteristic of semiconductors up to 1.8 GPa (Figure S5). At higher pressure, the increase of the conductivity is slowed down but also perturbed by the freezing of the pressure transmitting medium around 2.1 GPa. Although the conductivity at 2.5 GPa is similar to 4-iPr value, it is not clear if the conducting regime is reached for 4-nPr as observed in 4-iPr. The temperature dependence of the resistivity at different pressures (Figure 9) shows that 4-nPr keeps its semi-conducting behavior, at least up to 2.5 GPa. The activation energy deduced from the Arrhenius fit of the data in the temperature range 80-150 K decreases continuously from 73 meV at ambient pressure down to 28 meV at 2.5 GPa but does not vanishes (Figure S6 and S7), at variance with the behavior reported for 4-iPr.[14] Again the difference between the two complexes finds its origin in their structural organization of the two complexes. The crisscross overlap stacking in 4-iPr and the numerous inter-stack interactions in the perpendicular plane allowed indeed for a three-dimensional overlap pattern while, as already mentioned, the calculated Fermi surface for 4-nPr (Figure S4), with
its strong one-dimensional nature does not favor the metallic state to the same extend.
Figure 9. Temperature dependence of the resistivity of 4-nPr at different pressures.
The continuous lines are the Arrhenius fit to the data giving the activation energy.
Conclusion
In the search for novel single-component conductors, we have shown here how the use of sterically demanding, but closely related substituents such as R = NMe2,
cyclopropyl and n-propyl on the heterocyclic nitrogen atom of the N-alkyl-1,3-thiazoline-2-thione-4,5-dithiolate (R-thiazdt) ligand give rise, in the corresponding radical gold dithiolene complexes [(R-thiazdt)2]•, to varied solid state structures. The
relative orientation of the methyl groups in the isopropyl and NMe2 groups vs. the
thiazoline heterocycle is the only difference between both complexes but completely modifies their structural and electronic properties, despite an apparently comparable original crisscross stacking. On the other hand, the R = cyclopropyl and R= n-propyl radical gold complexes avoid this dimerization and organize into uniform chains with strong one-dimensional character and semiconducting behaviour. The reluctance of these radical complexes toward usual dimerization makes them attractive Mott insulator systems and the evolutions of their conductivity under pressure (up to 2.5 GPa) demonstrate that other stimuli such as electric field pulses could induce a transition to a metallic state. Further investigations along these lines are currently underway, as well as other chemical modifications, with even smaller (R = Me) or larger (R = tBu) substituents.
Experimental section
General. NMR spectra were recorded at room temperature using CDCl3 unless
otherwise noted. Chemical shifts are reported in ppm and 1H NMR spectra were referenced to residual CHCl3 (7.26 ppm), 13C NMR spectra were referenced to CHCl3
(77.2 ppm). Mass spectra were recorded with a Bruker MaXis 4G for 3-nPr and
4-cPr, with a Thermo Fisher Q-Exactive for 3-4-cPr, 3-NMe2 and 4-nPr, and with Waters
Q-Tof 2 instrument for all the other derivatives by the Centre Régional de Mesures Physiques de l'Ouest, Rennes. CVs were carried out on a 10-3 M solution of complex in CH2Cl2 with NBu4PF6 0.1 M with a Pt working electrode. Potentials were measured
versus Saturated Calomel Electrode (SCE). Chemicals and materials from commercial sources were used without further purification.
N-alkyl-1,3-thiazoline-2-thiones (1): To a solution of primary amine (50 mmol,
propylamine: 4.1 mL, cyclopropylamine: 3.5 mL, N,N-dimethylhydrazine: 3.8 mL) and triethylamine (7.8 mL, 56.0 mmol) in 250 mL of diethylether, CS2 (3.4 mL, 56.0 mmol)
was slowly added. The reaction mixture was stirred for 30 minutes at 0°C. The white solid was filtered, washed with diethyl ether and dried under vacuum. The dithiocarbamate salts were obtained as white powders and used in the next step without further purification.
Triethylammonium propyldithiocarbamate: white powder. Yield: 9.6 g (81%); mp =
136 °C. 1H NMR (D2O, 300 MHz) . (ppm) : 0.79 (t, 3H, CH3, 3JHH = 7.2 Hz), 1.15 (t,
9H, CH3, 3JHH = 7.4 Hz), 1.51 (sext, 2H, CH2, 3JHH = 7.2 Hz), 3.06 (q, 6H, CH2, 3JHH =
7.4 Hz), 3.34 (t, 2H, CH2, 3JHH = 7.2 Hz).13C NMR (D2O, 75 MHz) . : 8.3 (CH3), 10.7
(CH3), 21.3 (CH2), 46.6 (N-CH2), 49.7 (N-CH2-), 209.9 (C=S).
Triethylammonium cyclopropyldithiocarbamate: Yield: 9.7 g (83%); mp = 118 °C. 1
H NMR (D2O, 300 MHz) . (ppm) : 0.56 (m, 2H, CH2), 0.69 (m, 2H, CH2), 1.17 (t, 9H,
CH3, 3JHH = 7.3 Hz), 2.83 (m, 1H, CH), 3.09 (q, 6H, CH2, 3JHH = 7.3 Hz). 13C NMR
(D2O, 75 MHz) . (ppm) : 6.2 (CH3), 8.2 (CH2), 29.9 (CH), 46.6 (CH2), 212.9 (C=S).
Triethylammonium 2,2-dimethylhydrazine-1-carbodithioate: Yield: 4.1 g (35%);
mp =144°C. 1H NMR (D2O, 300 MHz) . (ppm) : 1.16 (t, 9H, CH3, 3JHH = 7.3 Hz), 3.08
(q, 6H, CH2, 3JHH = 7.3 Hz), 3.14 (s, 6H, CH3).
To a solution of the triethylammonium dithiocarbamate salts prepared above (54 mmol, 12.8 g for R = nPr, 12.6 g for R = cPr and 12.8 g for R = NMe2) in
acetonitrile (100 mL) chloroacetaldehyde (7.2 mL, 56.7 mmol, 50% in water) was slowly added. The pale yellow solution was stirred at room temperature overnight except for R = NMe2 which was refluxed overnight. The volume was reduced to about
10 mL by rotary evaporation and the brown residue was slowly added to 25 mL of concentrated sulfuric acid cooled at 0° C. The reaction mixture was stirred during 20 minutes, hydrolyzed with ice water and extracted with dichloromethane. The organic phase was washed with water, dried over MgSO4 and the solvent was removed under
reduced pressure to give thiazoline-2-thione 1 which was purified by column chromatography using dichloromethane as eluent for 1-nPr and 1-cPr and using dichloromethane /diethyl ether (9.9/0.1) as eluent for 1-NMe2.
1-nPr, orange oil; 76 % yield (6.5 g); Rf = 0.55 (SiO2, CH2Cl2); 1H NMR (300 MHz)
0.92 (t, 3H, CH3, 3JHH = 7.4 Hz), 1.80 (sext, 2H, CH2, 3JHH = 7.4 Hz), 4.07 (t, 2H, CH2, 3
JHH = 7.4 Hz), 6.56 (d, 1H, =CH, 3JHH = 4.6 Hz), 7.00 (d, 1H, =CH, 3JHH = 4.6 Hz). 13C NMR (75 MHz) . 10.8 (CH
3), 21.5 (CH2), 51.2 (CH2), 110.8 (=C), 131.3 (=C),
186.9 (C=S). HRMS (ASAP) calcd. for C6H10NS2+: 160.02492, found: 160.0249.
1-cPr, brown oil; 72 % yield (6.1 g); Rf = 0.58 (SiO2, CH2Cl2); 1H NMR (300 MHz) .
0.97 (m, 2H, CH2), 1.18 (m, 2H, CH2), 3.42 (m, 1H, CH), 6.51 (d, 1H, =CH, 3JHH = 4.7
Hz), 6.93 (d, 1H, =CH, 3JHH = 4.7 Hz); 13C NMR (CDCl3, 75 MHz) . (ppm) : 7.6 (CH2),
32.3 (CH), 110.6 (=C), 131.1 (=C), 188.9 (C=S). HRMS (ASAP) calcd for C6H8NS2+:
158.00982, found: 158.0098; Anal. Calcd. for C6H7NS2: C, 45.83 ; H, 4.49 ; N, 8.91.
Found : C, 45.96 ; H, 4.35 ; N, 8.84.
1-NMe2, orange solid; 30% yield (2.6 g); mp = 102°C. Rf = 0.53 (SiO2, CH2Cl2/Et2O
(9.9-0.1)). 1H NMR (CDCl3, 300 MHz) . (ppm) : 3.02 (s, 6H, CH3), 6.53 (d, 1H, =CH, 3
JHH = 4.9 Hz), 7.16 (d, 1H, =CH, 3JHH = 4.9 Hz).13C NMR (CDCl3, 75 MHz) . (ppm) :
44.2 (CH3), 109.9 (=C), 128.4 (=C), 185.0 (C=S). HRMS (ASAP) calcd for
C5H8N2S2Na+: 183.00266, found: 183.0028. Anal. Calcd for C5H8N2S2: C, 37.47, H,
5.03, N, 17.48. Found : C, 37.12, H, 5.06, N, 17.38 .
4,5-bis(cyanoethylthio)-1,3-thiazoline-2-thiones 2: Under inert atmosphere, at -10
1,3-thiazoline-2-thione 1 (8.0 mmol 1.27 g for 1-nPr, 1.26 g for 1-cPr, 1.28 g for 1-NMe2) in 40 mL of
anhydrous THF. After stirring for 30 minutes at -10°C, sulfur S8 (0.38 g, 12.0 mmol)
was added and the solution was stirred for an additional 30 minutes. A solution of 16.0 mmol of LDA was added to the medium. The reaction mixture was stirred for 3 hours, at -10°C, and sulfur was added (0.51 g, 16.0 mmol) followed 30 min later by the addition of 3-bromopropionitrile (6.6 mL, 80.0 mmol). The temperature was allowed to rise to room temperature and the reaction was stirred overnight. The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane. The organic phase was washed with water and then dried over MgSO4. The 4,5-biscyanoethylthio-1,3-thiazoline-2-thione 2 was purified by column
chromatography using dichloromethane as eluent for 2-nPr and 2-cPr and using dichloromethane /diethyl ether (9.9/0.1) as eluent for 2-NMe2.
2-nPr, brown powder; 46 % yield (1.21 g); mp = 103 °C; Rf = 0.75 (SiO2, CH2Cl2). 1H
NMR (300 MHz) . 1.02 (t, 3H, CH3, 3JHH = 7.5 Hz), 1.78 (sext, 2H, CH2, 3JHH = 7.5
Hz), 2.75 (t, 4H, CH2, 3JHH = 7.0 Hz), 3.10 (t, 2H, CH2, 3JHH = 7.0 Hz), 3.15 (t, 2H,
CH2, 3JHH = 7.0 Hz), 4.35 (m, 2H, CH2). 13C NMR (75 MHz) . 10.7 (CH3), 18.3 (CH2),
20.9 (CH2), 31.3 (S-CH2), 31.9 (CH2), 50.7 (CH2), 117.0 (CN), 125.1 (=C), 135.6
(=C), 186.9 (C=S). HRMS (ASAP) calcd for C12H16N3S4+: 330.02216, found:
330.0220; Anal. calcd for C12H15N3S4: C, 43.74, H, 4.59, N, 12.75. Found: C,
43.98, H, 4.74, N, 13.03.
2-cPr, brown powder; 75 % yield (1.96 g); mp = 113°C; Rf = 0.57 (SiO2, CH2Cl2); 1H
NMR (300 MHz) . 1.21 (m, 2H, CH2), 1.36 (m, 2H, CH2), 2.72 (t, 2H, CH2, 3JHH = 6.8
Hz), 2.74 (t, 2H, CH2, 3JHH = 6.8 Hz), 2.94 (m, 1H, CH), 3.08 (t, 2H, CH2, 3JHH = 6.8
Hz), 3.19 (t, 2H, CH2, 3JHH = 6.9 Hz); 13C NMR (75 MHz) . 10.6 (CH2), 18.6 (CH2),
31.4 (CH2), 31.9 (CH), 117.1 (CN),126.3 (=C), 137.5 (=C), 189.9 (C=S); HRMS
(ASAP) calcd for C12H14N3S4+: 328.00706, found: 328.0070; Anal. calcd for
C12H13N3S4: C, 44.01, H, 4.00, N, 12.83. Found: C, 43.80, H, 3.80, N, 12.59.
2-NMe2, orange-brown oil; 49 % yield (1.30 g); Rf = 0.53 (SiO2, CH2Cl2/Et2O, 9.9/0.1); 1
H NMR (300 MHz) . 2.74 (t, 4H, CH2, 3JHH = 6.9 Hz), 3.06 (t, 2H, CH2, 3JHH = 6.9
Hz), 3.18 (t, 2H, CH2, 3JHH = 6.9 Hz), 3.25 (s, 6H, CH3); 13C NMR (CDCl3, 75 MHz) .
(ppm) : 18.4 (CH2), 30.4 (CH2), 31.1 (CH2), 42.9 (CH3), 117.3 (CN), 121.3 (=C), 138.1
353.0003; Anal. calcd for C11H14N4S4: C, 39.97 ; H, 4.27 ; N, 16.95 ; S, 38.81. Found:
C, 40.05 ; H, 4.37 ; N, 16.95 ; S, 38.60.
Anionic gold dithiolene complexes 3-R: Under inert atmosphere and at room
temperature, to a dry two-neck flask containing 1,3-thiazoline-2-thione 2-R (0.28 g, 0.85 mmol), 5 mL of 1M solution of sodium methalonate in MeOH was added. After complete dissolution of the compound, potassium tetrachloroaurate (III) hydrate (0.19 g, 0.5 mmol) dissolved in 7 mL of dry methanol was added to the reaction mixture followed, 5 h later, by the addition of tetraphenylphosphonium chloride (0.19 g, 0.51 mmol) in 7 mL of anhydrous methanol. The reaction was stirred overnight at room temperature under argon atmosphere. The dark green suspension was filtered under vacuum. The resulting solid was washed with MeOH and recrystallized from acetonitrile.
3-nPr: dark green crystals; 68 % yield (0.28 g); mp = 199 °C; 1H NMR (300 MHz) . 0.95 (t, 6H, CH3, 3JHH = 7.5 Hz), 1.75 (m, 4H, CH2), 3.98 (m, 4H, CH2,), 7.61 (m, 8H,
CHAr), 7.73 (m, 8H, CHAr), 7.89 (m, 4H, CHAr); 13C NMR (CDCl3, 75 MHz) . 11.0
(CH3), 20.6 (CH2), 48.9 (CH2),112.2 (C=C), 116.5 (CAr), 117.7 (CAr), 130.5 (CAr),
132.1 (C=C), 134.1 (CAr), 135.6 (CAr), 191.2 (C=S); UV-vis-NIR (CH2Cl2) / (nm), (0 M -1.cm-1): 364 (5196), 294 (32095), 260 (45449), 234 (79940); HRMS (ESI) calcd for
C60H54N2P2S8Au+: 1317.11881, found: 1317.1192; Anal. calcd for C36H34AuN2PS8: C,
44.16, H, 3.50, N, 2.86, S, 26.20. Found: C, 44.00, H, 3.50, N, 2.70, S, 26.64.
3-cPr: dark green crystals; 52 % yield (0.22 g); mp = 257 °C; 1H NMR (300 MHz) . 1.14 (m, 2H, CH2), 1.23 (m, 2H, CH2), 2.82 (m, 1H, CH), 7.59 (m, 8H, CHAr), 7.76 (m,
8H, CHAr), 7.88 (m, 4H, CHAr). 13C NMR (75 MHz) . 8.7 (CH2), 29.5 (N-CH), 112.4
(C=C), 116.5 (CAr), 117.7 (CAr), 130.5 (CAr), 134.1 (CAr), 134.5 (C=C), 135.5 (CAr),
194.2 (C=S); UV-vis-NIR (CH2Cl2) / (nm), (0 M-1.cm-1): 370 (31377), 396 (25628),
262 (35868), 232 (59101); HRMS (ESI) calcd for C12H10N2S8Au: 634.82808, found:
634.8281; Anal. calcd for C36H34AuN2PS8: C, 44.34, H, 3.10, N, 2.87. Found: C,
43.89, H, 2.94, N, 2.74 .
3-NMe2: dark red crystals; 70 % yield (0.29 g); mp = 238 °C. 1H NMR (CDCl3, 300
MHz) . (ppm) : 3.18 (s, 12H, CH3), 7.61 (m, 8H, CHAr), 7.76 (m, 8H, CHAr), 7.89 (m,
118.6 (CAr), 131.3 (CAr), 134.9 (CAr), 135.2 (C=C), 136.6 (CAr), 194.7 (C=S).
UV-vis-NIR (CH2Cl2) / (nm), (0 M-1.cm-1): 378 (20479), 314 (23592), 232 (49401); HRMS
(ESI) calcd for C12H10N4S8Au-: 640.84988, found: 640.8501. Anal. calcd for
C36H34AuN2PS8: C, 41.62 ; H, 3.29 ; N, 5.71. Found: C, 41.46 ; H, 3.13 ; N, 5.57.
Neutral gold dithiolene complexes 4-R: In a two compartment cell equipped with Pt
electrodes (diameter 1mm, length 2 cm) TBAPF6 (620 mg, 1.6 mmol) dissolved in 16
mL of CH3CN or a mixture of CH3CN/CH2Cl2 was introduced in both compartments
as supporting electrolyte. The monoanionic gold complex (8 mg) was introduced in the anodic compartment. The current intensity was adjusted from 0.3 to 0.5 µA, and the reaction was left during 5 days to 5 weeks. Crystals of the radical neutral complex were collected on the anode.
4-nPr: dark green needles. HRMS (ESI) calcd for C12H14N2S8Au-.: 638.85938, found:
638.8596. Anal. calcd for C12H14AuN2S8: C, 22.53, H, 2.21, N, 4.38. Found: C, 22.06,
H, 1.92, N, 4.33.
4-cPr: dark green needles. mp = 265 °C; HRMS (ESI) calcd for C12H10N2S8Au-.:
634.82808, found: 634.8281.
4-NMe2: black needles; mp = 208 °C; Anal. calcd for C10H12N4S8Au: C, 18.72, H,
1.88, N, 8.73, S, 39.97. Found: C, 18.19, H, 1.81, N, 8.21, S, 40.42.
X-ray crystal structures.
Single-crystal diffraction data were collected on APEXII, Bruker-AXS diffractometer, Mo-K1 radiation (/ = 0.71073 Å) for all compounds. The structures were solved by direct methods using the SIR97 program[19], and then refined with full-matrix least-square methods based on F2 (SHELXL-97)[20] with the aid of the WINGX[21] program. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions. Details of the data collections and final refinements are given In Table 4. In the crystal structures of (Et4N)(3-cPr), 4-cPr and 4-nPr, relatively large residual densities (2-5 e Å–3)
observed in the last Fourier difference maps are located systematically in the vicinity of the gold atom. CCDC 1559432-1559438 contain the supplementary
crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre
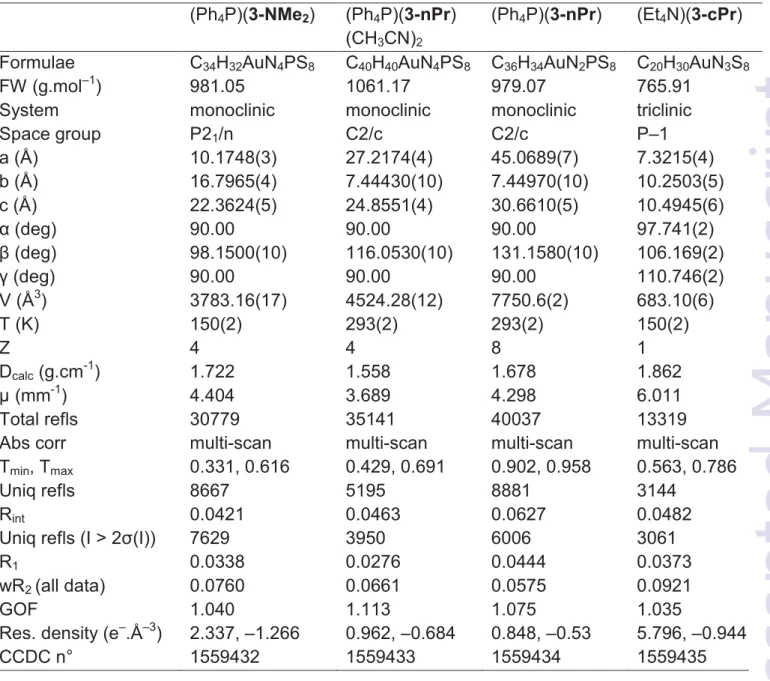
Table 4 Crystallographic data
(Ph4P)(3-NMe2) (Ph4P)(3-nPr)
(CH3CN)2
(Ph4P)(3-nPr) (Et4N)(3-cPr)
Formulae C34H32AuN4PS8 C40H40AuN4PS8 C36H34AuN2PS8 C20H30AuN3S8
FW (g.mol–1) 981.05 1061.17 979.07 765.91 System monoclinic monoclinic monoclinic triclinic
Space group P21/n C2/c C2/c P–1 a (Å) 10.1748(3) 27.2174(4) 45.0689(7) 7.3215(4) b (Å) 16.7965(4) 7.44430(10) 7.44970(10) 10.2503(5) c (Å) 22.3624(5) 24.8551(4) 30.6610(5) 10.4945(6) 1 (deg) 90.00 90.00 90.00 97.741(2) + (deg) 98.1500(10) 116.0530(10) 131.1580(10) 106.169(2) 2 (deg) 90.00 90.00 90.00 110.746(2) V (Å3) 3783.16(17) 4524.28(12) 7750.6(2) 683.10(6) T (K) 150(2) 293(2) 293(2) 150(2) Z 4 4 8 1 Dcalc (g.cm-1) 1.722 1.558 1.678 1.862 µ (mm-1) 4.404 3.689 4.298 6.011 Total refls 30779 35141 40037 13319
Abs corr multi-scan multi-scan multi-scan multi-scan Tmin, Tmax 0.331, 0.616 0.429, 0.691 0.902, 0.958 0.563, 0.786
Uniq refls 8667 5195 8881 3144
Rint 0.0421 0.0463 0.0627 0.0482
Uniq refls (I > 2*(I)) 7629 3950 6006 3061
R1 0.0338 0.0276 0.0444 0.0373
wR2 (all data) 0.0760 0.0661 0.0575 0.0921
GOF 1.040 1.113 1.075 1.035
Res. density (e–.Å–3) 2.337, –1.266 0.962, –0.684 0.848, –0.53 5.796, –0.944
Table 4 Crystallographic data (continued)
4-NMe2 4-cPr 4-nPr
Formulae C10H12AuN4S8 C12H10AuN2S8 C12H14AuN2S8
FW (g.mol-1) 641.68 635.67 639.70 System orthorhombic monoclinic triclinic
Space group I222 P21/c P–1
a (Å) 7.0055(8) 4.7474(5) 4.4422(4) b (Å) 13.7316(14) 6.6311(5) 7.3666(7) c (Å) 20.649(2) 27.607(3) 14.6283(13) 1 (deg) 90.00 90.00 85.148(3) + (deg) 90.00 90.459(3) 88.746(3) 2 (deg) 90.00 90.00 74.489(3) V (Å3) 1986.3(4) 869.05(15) 459.61(7) T (K) 150(2) 150(2) 150(2) Z 4 2 1 Dcalc (g.cm-1) 2.146 2.429 2.311 µ (mm-1) 8.248 9.422 8.908 Total refls 10473 5451 7203
Abs corr multi-scan multi-scan multi-scan Tmin, Tmax 0.538, 0.814 0.623, 0.828 0.660, 0.837
Uniq refls 2283 1951 2044
Rint 0.0370 0.0620 0.0371
Uniq refls (I > 2*(I)) 2029 1479 2015
R1 0.0190 0.0709 0.0380 wR2 (all data) 0.0398 0.1486 0.0960 GOF 1.106 1.153 1.062 Res. density (e–.Å–3) 1.043, –0.681 2.511, –2.438 5.591, –1.542 CCDC n° 1559436 1559437 1559438 Transport measurements.
The resistivity measurements were carried out along the long axis of the needle shaped single crystals which correspond to the stacking axis of the 4-R complexes. Gold pads were evaporated on the crystals in order to improve the quality of the contacts and gold wires were glued with silver paste on those contacts. For 4-NMe2
and 4-nPr, a standard four points technique was used with a low frequency lock-in detection (applied current Iac = 0.1-1µA) for measured resistances below 10 kΩ and
dc measurement for higher resistances (applied current Idc = 0.1-1µA). For 4-cPr,
resistivity was deduced from a two points measurement because of the small size of the crystals. The ρ(T) curve was checked by two different techniques: a dc
measurement with an applied current Idc = 0.1µA and a current measurement with an
applied voltage of 1V using a dc voltage source/picoammeter Keithley 6487.
Resistivity measurements were also performed under high hydrostatic pressure on very thin needles of 4-nPr (section around 50 µm2). A NiCrAl clamped cell was used up to 2.5 GPa with silicon oil (Daphne 7373) as the pressure transmitting medium. The pressure at room temperature was extracted from the resistance of a manganin gauge in the pressure cell and it is this value which is indicated in the figures. However, the loss of pressure during cooling is estimated to 0.2 GPa, slightly decreasing with pressure. The highest pressure ρ(T) curve was measured in 3 points after one contact have been lost during the pressurization. Low temperatures have been provided by a home-made cryostat built around a 4K pulse tube.
Acknowledgements
We thank the CDIFX (Rennes) for access to the X-ray diffraction facilities and the ANR (France) for financial support through project No. 12-BS07-0032.
Conflict of interest
The authors declare no conflict of interest.
Keywords
: S ligands • Gold • Stacking interactions • Conducting materials • Single component conductorsReferences
[1]a) A. Kobayashi, E. Fujiwara, H. Kobayashi, Chem. Rev. 2004, 104, 5243–5264; b) M. Sasa, E. Fujiwara, A. Kobayashi, S. Ishibashi, K. Terakura, Y. Okano, H. Fujiwara, H. Kobayashi, J. Mater. Chem. 2005, 15, 155–163; c) Y. Okano, B. Zhou, H. Tanaka, T. Adachi, Y. Ohishi, M. Takata, S. Aoyagi, E. Nishibori, M. Sakata, A. Kobayashi, H. Kobayashi, J. Am. Chem. Soc. 2009, 131, 7169–7174; d) B. Zhou, Y. Idobata, A.Kobayashi, H. Cui, R. Kato, R. Takagi, K. Miyagawa, K. Kanoda, H. Kobayashi, J. Am. Chem. Soc. 2012, 134, 12724–12731; e) H.Cui, H.
Kobayashi, S. Ishibashi, M. Sasa, F. Iwase, R. Kato, A. Kobayashi, J. Am. Chem. Soc. 2014, 136, 7619–7622.
[2]
J. M. Rawson, A. Alberola, A. Whalley, J. Mater. Chem. 2006, 16, 2560–2575.
[3]
S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley, R. C. Haddon, Science 2005, 309, 281−284.
[4]
a) A. Mailman, J. W. L. Wong, S. M. Winter, R. C. M. Claridge, C. M. Robertson, A. Assoud, W. Yong, E. Steven, P. A. Dube, J. S. Tse, S. Desgreniers, R. A. Secco, R. T. Oakley, J. Am. Chem. Soc. 2017, 139, 1625−1635; b) D. Tian, S. M. Winter, A. Mailman, J. W. L. Wong, W. Yong, H.Yamaguchi, Y. Jia, J. S. Tse, S. Desgreniers, R. A. Secco, S. R. Julian, C. Jin, M. Mito, Y. Ohishi, R. T. Oakley, J. Am. Chem. Soc. 2015, 137, 14136−14148; c) J. W. L. Wong, A. Mailman, K. Lekin, S. M. Winter, W. Yong, J. Zhao, S. V. Garimella, J. S. Tse, R. A. Secco, S. Desgreniers, Y. Ohishi, F. Borondics, R. T. Oakley, J. Am. Chem. Soc. 2014, 136, 1070−1081.
[5]
a) S. K. Pal, P. Bag, M. E. Itkis, F. S. Tham, R. C. Haddon, J. Am. Chem. Soc.
2014, 136, 14738−14741; b) S. K. Mandal, S. Samanta, M. E. Itkis, D.W. Jensen,
R. W. Reed, R.T. Oakley, F. S. Tham, B. Donnadieu, R. C. Haddon, J. Am. Chem. Soc. 2006, 128, 1982−1994.
[6]
M. Souto, H Cui, M. Peña-Álvarez, V. G. Baonza, H. O. Jeschke, M. Tomic, R. Valenti, D. Blasi, I. Ratera, C. Rovira, J. Veciana, J. Am. Chem. Soc. 2016, 138, 11517−11525.
[7]
T. Isono, H. Kamo, A. Ueda, K. Takahashi, A. Nakao, R. Kumai, H. Nakao, K. Kobayashi, Y. Murakami, H. Mori, Nat. Commun., 2013, 4, 1344–1349; (b) T. Isono, H. Kamo, A. Ueda, K. Takahashi, M. Kimata, H. Tajima, S. Tsuchiya, T. Terashima, S. Uji and H. Mori, Phys. Rev. Lett., 2014, 112, 177201.
[8]
a) R. Llusar, S. Uriel, C. Vicent, J. M. Clemente-Juan, E. Coronado, C. J. Gomez-Garcia, B. Braïda, E. Canadell, J. Am. Chem. Soc. 2004, 126, 12076–12083; b) R. Llusar, S. Triguero, V. Polo, C. Vicent, C. J. Gomez-Garcia, O. Jeannin, M. Fourmigué, Inorg. Chem. 2008, 47, 9400–9409.
[9]
H. Cui, T. Tsumuraya, T. Miyazaki, Y. Okano, R. Kato, Eur. J. Inorg. Chem. 2014,
24, 3837–3840. [10]
a) N. C. Schiødt, T. Bjørnholm, K. Bechgaard, J. J. Neumeier, C. Allgeier, C. S. Jacobsen, N. Thorup, Phys. Rev. B 1996, 53, 1773-1778; b) D. Belo, H. Alves, E.
B. Lopes, M. T. Duarte, V. Gama, R. T. Henriques, M. Almeida, A. Pérez-Benítez, C. Rovira, J. Veciana, Chem. Eur. J. 2001, 7, 511−519; c) O. J. Dautel, M. Fourmigué, E. Canadell, P. Auban-Senzier, Adv. Funct. Mater. 2002, 12, 693−698; d) T. Higashino, O. Jeannin, T. Kawamoto, D. Lorcy, T. Mori, M. Fourmigué, Inorg. Chem. 2015, 54, 9908−9913; e) A. Filatre-Furcate, T. Roisnel, P. Auban-Senzier, M. Fourmigué, D. Lorcy, Dalton Trans. 2015, 44, 15683–15689; f) D. G. Branzea, F. Pop, P. Auban-Senzier, R. Clérac, P. Alemany, E. Canadell, N. Avarvari, J. Am. Chem. Soc. 2016, 138, 6838–6851; g) M. M. Andrade, R. A. L. Silva, I. C. Santos, E. B. Lopes, A. Rabaça, L. C. J. Pereira, J. T. Countinho, J. P. Telo, C. Rovira, M. Almeida, D. Belo, Inorg. Chem. Front., 2017, 4, 270-280.
[11]
N. Tenn, N. Bellec, O. Jeannin, L. Piekara-Sady, P. Auban-Senzier, J. Íñiguez, E. Canadell, D. Lorcy, J. Am. Chem. Soc. 2009, 131, 16961–16967.
[12] G. Yzambart, N. Bellec, N. Ghassan, O. Jeannin, T. Roisnel, M. Fourmigué, P.
Auban-Senzier, J. Íñiguez, E. Canadell, D. Lorcy J. Am. Chem. Soc. 2012, 134, 17138−17148.
[13] Y. Le Gal, T. Roisnel, P. Auban-Senzier, T. Guizouarn, D. Lorcy Inorg. Chem. 2014, 53, 8755−8761.
[14]
A. Filatre-Furcate, N. Bellec, O. Jeannin, P. Auban-Senzier, M. Fourmigué, J. Íñiguez, E. Canadell, B. Brière, V. Ta Phuoc, D. Lorcy, Inorg. Chem. 2016, 55, 6036−6046.
[15]
a) W. J. Humphlett, R. W. Lamon, J. Org. Chem. 1964, 29, 2146−2148; b) C. M. Roussel, R. Gallo, M. Chanon, J. Metzger, Bull. Soc. Chim. Fr. 1971, 5, 1902−1907.
[16]
a) N. Bellec, D. Lorcy, A. Robert, Synthesis, 1998, 1442−1446; b) N. Bellec, D. Lorcy, K. Boubekeur, R. Carlier, A. Tallec, S. Los, W. Pukacki, M. Trybula, L. Piekara-Sady, A. Robert. Chem. Mater. 1999, 11, 3147−3153; c) N. Bellec, D. Lorcy, A. Robert, R. Carlier, A. Tallec, C. Rimbaud, L. Ouahab, R. Clérac, P. Delhaes, Adv. Mater. 1997, 9, 1052−1056. d) D.Guérin, R. Carlier, D. Lorcy, J. Org. Chem. 2000, 65, 6069−6072.
[17] P. Batail, K. Boubekeur, M. Fourmigué, J. C. Gabriel, Chem. Mater. 1998, 10, 3005-3015.
[18]
P. Stoliar, P. Diener, J. Tranchant, B. Corraze, B. Brière, V. Ta-Phuoc, N. Bellec, M. Fourmigué, D. Lorcy, E. Janod, L. Cario, J. Phys. Chem. C. 2015, 119, 2983−2988.
[19]
A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori, R. Spagna, J. Appl. Cryst. 1999, 32, 115−119.
[20]
G. M. Sheldrick, Acta Cryst. 2008, A64, 112−122.
[21]
L. J. Farrugia, J. Appl. Crystallogr. 1999, 32, 837–838.
Graphical Abstract
![Figure 1 Cyclic voltamogramm of [Au(nPr-thiazdt) 2 ] 1– 3-nPr in CH 2 Cl 2 with NBu 4 PF 6](https://thumb-eu.123doks.com/thumbv2/123doknet/14448394.518173/6.892.115.386.801.1015/figure-cyclic-voltamogramm-au-npr-thiazdt-npr-nbu.webp)
![Table 1. Redox potentials of [Au(R-thiazdt) 2 ] –1 3-R complexes (E in V vs](https://thumb-eu.123doks.com/thumbv2/123doknet/14448394.518173/7.892.110.542.171.454/table-redox-potentials-au-r-thiazdt-r-complexes.webp)