Biological and Medical Implications of Discordance
in G
etyltin
ASACin CpG Methylation
P
MASSACOFby
MA
Mark Kendell Clement
LI
Submitted to the Harvard-MIT Program in Health Sciences and
Technology
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Medical Engineering and Medical Physics
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
February 2017
Massachusetts Institute of Technology 2017. All rights reserved.
X7 , /*
Author...
Certified by..
Signature redacted
Harvard MIT Progt "in Health Sciences and Technology
Feb 6, 2017
I
Signature redacted
Alexander Meissner, PhD
Professor, Harvard University, Department of Stem Cell and
Regenerative Biology
Thesis Supervisor
Accepted by
.r
...
Emery N. Brown, MD, PhD
Director, Harvard-MIT Program in Health Sciences and Technology
Professor of Computational Neuroscience and Health Sciences and
Technology
HS S INSTITUTE TECHNO LOGYR 0
/
2017
BRARIES
..Biological and Medical Implications of Discordance in CpG
Methylation
by
Mark Kendell Clement
Submitted to the Harvard-MIT Program in Health Sciences and Technology on Feb 6, 2017, in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy in Medical Engineering and Medical Physics
Abstract
DNA methylation is an important epigenetic mark that is linked to the regulation of
gene expression. It is a critical part of controlling cellular identity and is essential for normal development.
DNA methylation is generally studied by comparing methylation levels at
indi-vidual cytosines or computing region-level averages to identify differential methyla-tion. Here we extended this classic viewpoint by capitalizing on a unique feature of next-generation sequencing, which provides the methylation status of CpGs that are located on the same sequencing read and hence originate from the same DNA molecule.
When comparing methylation states of CpGs on the same read, we observed dif-ferent levels of discordant methylation, defined as molecules where the methylation of neighboring cytosines are not correlated. We quantified the proportion of discor-dantly methylated reads (PDR) in normal and cancer samples, and found that global PDR levels were elevated in cancer, suggesting widespread epigenetic deregulation. While we have not yet established the mechanistic contribution of this feature, we find that discordant methylation is linked to higher genetic diversity, greater cell-to-cell transcriptional heterogeneity, and adverse clinical outcome in chronic lymphocytic leukemia (CLL).
Our analytic approach introduces a novel perspective on utilizing epigenomic se-quencing data, which we anticipate will be a valuable tool in understanding the reg-ulation of DNA methylation and its contribution to cellular identity.
Thesis Supervisor: Alexander Meissner, PhD
Title: Professor, Harvard University, Department of Stem Cell and Regenerative Biology
Acknowledgments
I would like to acknowledge all of those who have contributed to my graduate
ex-perience. Although it has little to do with epigenetics or DNA methylation, one of the most important things I have learned during my tenure as a graduate student has been the importance of learning from others and of recognizing the support from others. On that note, I would like to acknowledge the contribution of the following people:
o Alexander Meissner for his example of commitment to scientific discovery, for his role in introducing me to pivotal collaborators and his motivating office discussions.
o My thesis committee, Mary Gehring, Gad Getz, and Rafael Irizarry. I appreci-ate the time they have taken to meet with me and for their support, suggestions, and insightful questions. Thank you to Mary for her careful reading of this doc-ument and for her helpful suggestions. Gaddy played a pivotal role in the development of the analysis techniques discussed in this thesis, as well as in the development of many additional metrics, algorithms, and models that may or may not ever enter the realm of broader academic discourse.
o The members of the Meissner Lab with whom I have worked over the years. Thank you, Christoph Bock, for introducing me to DNA methylation analysis, and for several of the datasets you passed on to me. Thank you, Michael Ziller, for guidance and inspiration through the majority of my doctoral experience, and also for your chair and its associated good thesis-writing karma. Thank you to those who have worked with me on projects, entrusted me with your data, attended otherwise unbearable department retreats with me, or made lab an enlightening place to be: Casey Gifford, Yingying Zhang, Jing Liao, Jamie Webster, Julie Donaghey, Alex Tsankov, Zack Smith, Camille Sindhu, Christina Galonska, Rahul Karnik, and Jocelyn Charlton.
espe-cially Andreas Gnirke, Hongcang Gu, and Patrick Boyle who have generated thousands of methylation libraries for this and other analyses.
" Cathy Wu and the members of her lab. Cathy for her scientific insights and
motherly advice, Dan Landau with whom I worked closely on the CLL project described in this thesis, Michaela Gruber for her involvement with an ongoing followup project to this work, and Donna Neuman for sage advice and statistical significance.
" My parents and siblings: my mom for her example of persistence and dedication
to improvement, as well as for her perpetual support and inquiry regarding my health and vegetable consumption; my dad for introducing me to the science of computers and for helping me see my potential and not give up; and my brothers and sisters: Nathan, Spencer, Sarah, Rebecca, Josh, Zach, Hannah and Rachel.
" My intelligent, loyal, and beautiful wife, Brenda. Thank you for your constant
encouragement and support, and for bearing the brunt of parenting while I finished up this thesis (thank you Maddie for being patient).
Contents
1 Introduction
1.1 Epigenetics . . . .
1.2 DNA methylation . . . .
1.3 Regulation of DNA methylation . . . . .
1.3.1 De novo methylation . . . .
1.3.2 Maintenance methylation . . . . .
1.3.3 Active demethylation . . . .
1.3.4 Passive demethylation . . . .
1.4 Region-based methylation characteristics
1.4.1 Promoters . . . . 1.4.2 Intragenic regions . . . . 1.4.3 Enhancers . . . . 1.4.4 Repetitive elements . . . . 1.4.5 Pericentromeric heterochromatin 1.4.6 X-chromosome inactivation . . . . 1.4.7 Imprinted genes . . . .
1.5 The cancer methylome . . . .
1.5.1 Global hypomethylation . . . . .
1.5.2 Punctate hypermethylation .
1.6 Thesis Outline and Overview . . . .
in normal tissues
2 Methylation Discordance
2.1 DNA methylation is a binary feature . . . .
and cancer 15 16 17 18 19 22 24 26 27 27 29 30 32 33 34 35 36 37 38 41 43 44
2.2 Assaying DNA methylation . . . .
2.2.1 Endonuclease digestion . . . . 2.2.2 Affinity enrichment . . . .
2.2.3 Bisulfite conversion . . . .
2.3 Bisulfite-sequencing methods for assaying methylation . . . . 2.4 Processing and analysis of bisulfite sequencing data . . . .
2.4.1 A lignm ent . . . .
2.4.2 CpG methylation calling . . . .
2.4.3 Differential methylation analysis . . . .
2.5 Phasing is an important readout of sequencing-based methylation
2.6 Sequencing assays uncover underlying discordance . . . .
2.7 Measuring methylation discordance: PDR . . . .
2.8 A stochastic model of discordance . . . . 2.9 Biological interpretation of discordance . . . . 2.10 Discordance is an useful measure in cancer . . . . 2.10.1 Sample purity . . . . 2.10.2 Intratumor heterogeneity . . . . 2.10.3 Intermediate methylation . . . . . . . 44 . . . 45 . . . 46 . . . 46 . . . 47 . . . 49 49 . . . 50 . . . 50 assays 51 . . . 53 . . . 54 . . . 55 . . . 57 . . . 58 . . . 58 . . . 58 . . . 59 3 Methylation Discordance in CLL 3.1 Disease Background . . . . 3.1.1 Genomic variability in CLL . . . . 3.1.2 Epigenetic variability in CLL . . . . 3.2 M otivation . . . . 3.3 R esults . . . .
3.3.1 Increased intra-sample DNA methylation heterogeneity in CLL
arises from locally disordered methylation . . . .
3.3.2 Locally disordered methylation broadly affects the CLL genome
3.3.3 Locally disordered methylation appears to be a largely
stochas-tic process . . . . 63 63 64 64 65 66 66 68 69
3.3.4 Increased susceptibility to locally disordered methylation in
gene-poor regions and silent genes . . . . 71
3.3.5 Locally disordered methylation and gene expression . . . . 72
3.3.6 Higher intra-tumoral gene expression heterogeneity in genes with disordered promoter methylation . . . . 74
3.3.7 Locally disordered methylation impacts stem cell genes and may facilitate leukemic evolution . . . . 74
3.3.8 Locally disordered methylation impacts clinical outcome . . . 76
3.4 D iscussion . . . . 76
3.5 Experimental Procedures . . . . 79
3.6 Acknowledgments . . . . 80
3.7 Extended Experimental Procedures . . . . 81
3.8 Figures . . . . 95
3.9 Supplementary Figures . . . 102
3.10 Supplementary Table . . . . 112
4 Conclusion and future directions 115 4.1 Open Questions . . . 116
4.1.1 Extension of discordance measures to other methylation assays 116 4.1.2 Finding the biological cause of epigenetic discordance . . . . . 117
4.1.3 Measuring methylation discordance at a cellular level . . . . . 118
List of Figures
2-1 Schematic of RRBS . . . . 48
2-2 Phasing of CpGs with intermediate methylation values . . . . 52
2-3 Phasing in a region with an intermediate methylation value . . . . 53
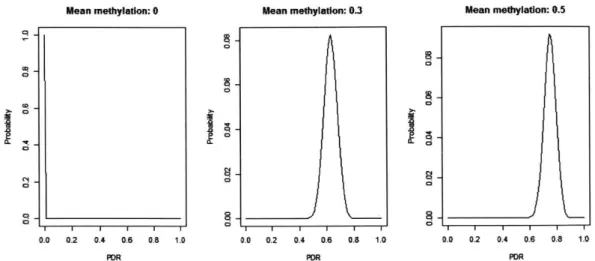
2-4 Distribution of PDR under a stochastic model. . . . . 57
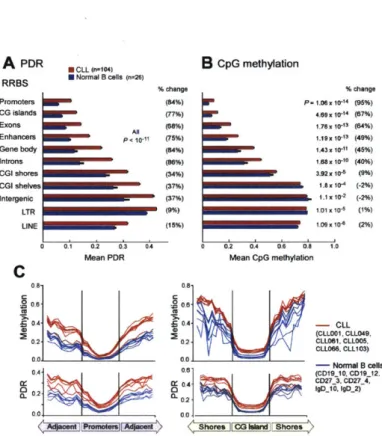
3-1 Higher DNA methylation intra-sample heterogeneity in CLL arises from locally disordered methylation . . . . 95
3-2 Locally disordered methylation affects all genomic regions in CLL, in-cluding CpG islands (CGIs) and repeat regions. . . . . 96
3-3 Locally disordered methylation in CLL is consistent with a stochastic process. . . . . 97
3-4 Locally disordered methylation affects preferentially gene-poor regions and can be traced back to non-expressed genes in normal B cells. . . 98
3-5 Locally disordered methylation is associated with transcriptional vari-ation . . . . 99
3-6 Locally disordered methylation may interact with evolution through drift towards a stem-like state. . . . . 100
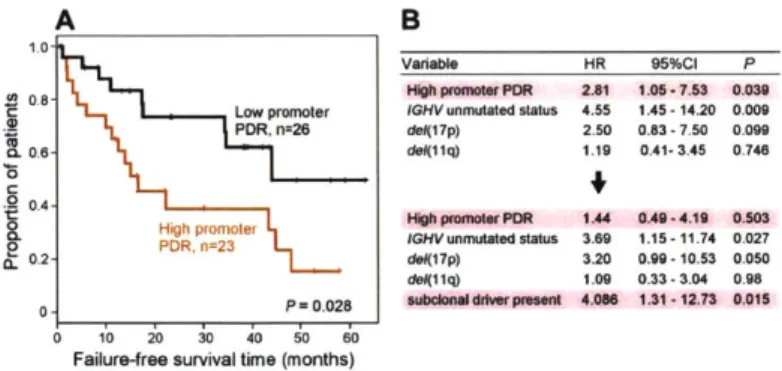
3-7 Locally disordered methylation is associated with adverse clinical out-com e . . . . 101
3-8 Proposed interaction between methylation disorder and clonal evolution.101 3-9 WGBS and RRBS data from CLLs and normal B cells shows higher Intratumoral DNA methylation heterogeneity that arises from locally disordered methylation. . . . . 103
3-10 Intermediate methylation in WGBS and RRBS data from CLLs and
norm al B cells. . . . 104
3-11 Discordant methylation is not the result of allelic or epiallelic
methy-lation patterns. . . . 105
3-12 Genomic characterization of locally disordered methylation including
analysis of CpG island subtypes and repeat elements based on WGBS. 106
3-13 Locally disordered methylation in CLL is consistent with a stochastic
process . . . . ... . . . 107
3-14 Locally disordered methylation affects many regions. . . . . 108
3-15 The association between PDR and distance from somatic mutation is
similar for clonal and subclonal mutations. . . . . 109
3-16 Locally disordered methylation is linked to transcriptional variation. . 110
3-17 Increased locally disordered methylation involves differentially
List of Tables
3.1 Characteristics and mean promoter PDR of the 104 CLL patients
Chapter 1
Introduction
In 2016, approximately 1 in 4 deaths in America will result from cancer. Currently, the lifetime risk of developing cancer is 42% in men and 38% in women, and cancer is the second leading cause of death in America after heart disease [6].
Cancer is a disease of proliferation of abnormal cells in the body [129]. Historically, the transformation from a normal cell to an abnormal cancer cell has been studied in the context of genetic mutations to DNA that disrupt proper cellular function. For example, genes that control cellular replication rate-such as Ras genes-are frequently mutated, resulting in cells with uncontrolled proliferation [325]. However, the lack of unifying genetic mechanisms between cancers [58] and especially within cancers of a single subtype indicates that additional factors influence the disease [231].
As opposed to genetic mutations, epigenetic modifications do not alter the DNA sequence but instead regulate the function of the DNA by controlling its accessibil-ity and abilaccessibil-ity to interact with other cellular components. These modifications are a key component that enables the diversity of cellular identities from the same ge-netic template. Epigege-netic changes that accompany the transition from normal cells to abnormal cancer cells have been studied for decades and are further increasing our molecular understanding of the origins of cancer, while also presenting highly attractive therapeutic targets due to their reversibility [21, 2871.
Indeed, broad themes in epigenetic dysregulation are seen in many different cancer subtypes [187, 3571 and bring clarity to the origin and mechanism of cancer even in
the absence of genetic mutations [3801. Individual cancers may show preference for mutations in a specific pathway, but as our understanding of traditional oncogenes and tumor suppressor genes matures, it appears that one feature that is consistently perturbed in all cancers is the regulation of epigenetics [95].
1.1
Epigenetics
DNA is organized and packaged into structural units collectively called chromatin,
in the cell which can either be in a dense, inaccessible state called heterochromatin, or in a relatively open, accessible state called euchromatin. The basic structural unit of chromatin is the nucleosome which is composed of an octamer of four core histone proteins, around which 147bp of DNA are wound [229]. Each of the four core histones-named H3, H4, H2A, and H2B-have long N-terminal tails that can be modified in a variety of ways [190].
At least eight different modifications on more than 60 distinct residues of the various histone tails have been identified, and the presence or absence of these mod-ifications in different combinations affect chromatin architecture by altering DNA accessibility or by recruiting other non-histone proteins [190, 210]. Within euchro-matin, the transcription of genes has been associated with histone marks on histone
3 (H3): H3K4, H3K36, and H3K79. Repressed genes as well as heterochromatin are
marked by H3K9, H3K27 and H4K20 methylation.
Methylation of cytosines in the DNA sequence itself is an extensively studied epigenetic mark. DNA methylation is generally seen as a repressive mark, and is frequently associated with heterochromatin and other epigenetic modifications linked to transcriptional repression. Regions with low DNA methylation are associated with active gene promoters and regulatory elements, correlating with an accessible or transcriptionally permissive environment.
DNA methylation and histone modifications tightly cooperate in genome
regula-tion [45, 173, 289, 125]. For example, the methyl-CpG binding protein MeCP2 binds methylated CpGs as well as Suv39h1/2 histone methyltransferases [105], which leads
to H3K9 methylation at regions of DNA methylation. Conversely, parts of the DNA methylation machinery are strongly inhibited by methylation of H3K4 [257] which prevents DNA methylation at those regions. On an allosteric level, when DNA is wrapped around the histone core, de novo DNA methylation is severely inhibited
[97, 56].
1.2
DNA methylation
DNA methylation refers to the presence of a methyl group on the 5' carbon of cytosine. DNA methylation in humans is found on around 4-5% of cytosines. In most somatic
cells, methylated cytosines are found almost exclusively in the CpG context, when the cytosine is followed by a guanine residue [224]. Although non-CpG methylation has been observed in some cell types 1298, 224, 275, 390, 127], CpG methylation is more widely studied, and has been shown to be of particular biological significance
[29, 350].
In normal tissues, dense clusters of CpGs, or CpG Islands (CGIs), are largely unmethylated, while CpGs in low-CpG-density regions are generally methylated [9]. CGIs tend to occur near gene promoters and other regions of regulatory importance
[296, 207]. DNA methylation at promoters is generally associated with repressed gene
expression[282, 96, 174].
In cancer, DNA methylation has been observed to drive tumor progression in much the same way as mutations to DNA, for example by acting as a silencer at tumor suppressor genes or other targets of genetic mutation [141]). However, most of these apparently targeted methylation changes are actually part of a global epigenetic dysregulation of normal methylation patterns. The cancer methylome is defined by global hypomethylation [83, 341] and localized hypermethylation, which often includes hypermethylation of tumor suppressor gene promoters [163, 120, 34, 143, 240].
Further emphasizing the importance of faithful methylation regulation, genetic mutations have been observed to occur at genes involved in the regulation of DNA methylation [307, 3611. In addition, the mutation targets observed in cancers with few
genetic alterations such as acute myeloid leukemia (AML) are frequently epigenetic modifiers [78, 249]. However, dysregulation of the cancer methylome appears to be independent of individual mutations in the methylation regulatory machinery, as it is observed even in patients with no genetic mutation in the methylation machinery
[177].
1.3
Regulation of DNA methylation
In mammals, DNA methylation is added to the genome by enzymes called DNA methyltransferases (DNMTs). There are three catalytically-active DNMTs in human cells: DNMT1, DNMT3A, and DNMT3B [71]. Methylation proceeds in one of two main ways: 1) de novo methylation of an unmethylated CpG, and 2) maintenance methylation, or copying methylation at a hemimethylated CpG to the unmethylated
CpG.
There are also two principle ways in which DNA methylation can be removed. Active demethylation involves a complex multistep enzymatic reaction at the methy-lated cytosines. Alternatively, passive demethylation involves preventing the copying of CpG methylation from a parent DNA strand to the newly synthesized strand. If CpGs remain hemimethylated until the next cycle of DNA replication, future DNA strands derived from the unmethylated strand will remain unmethylated, resulting in consecutive dilution of methylation.
Generally somatic methylation patterns are relatively stable, in contrast to two major reprogramming events that occur in early development and the germline where methylation patterns are globally erased and then re-established [314].
The first reprogramming event happens immediately after fertilization in the zy-gote, during which the paternal genome is rapidly demethylated. Passive, replication-dependent demethylation continues to demethylate the paternal and maternal genomes until a low methylation state is achieved at the morula/blastocyst stage. Next, de novo methylation proceeds to rapidly methylate the genome. The second phase of global reprogramming occurs as primordial germ cells (PGCs) emerge from the
epi-blast, and undergo further reprogramming including demethylation, erasure of im-prints, and X-chromosome reactivation. PGCs are then methylated to gametic levels
by de novo methylation [300].
1.3.1
De novo methylation
DNA Methyltranfserases 3A and 3B (DNMT3A and DNMT3B) are de novo
methyl-transferases and methylate unmethylated CpGs [256, 255]. DNMT3A and DNMT3B are highly homologous and share greater thin 80% sequence homology in the C-terminal part of the proteins which contains canonical methyltransferase motifs and is responsible for methylation. The N-terminal domain of these enzymes contain recognition domains that have been shown to associate with DNA 126, 273, 170].
Much of our understanding of the function of the de novo methyltransferases comes from experiments that modulate methyltransferase activity in mouse embryonic stem
(ES) cell lines [222]. ES cells can be cultured indefinitely in an undifferentiated state
and De novo transferases are highly expressed in ES cells [256]. De novo methylation
by DNMT3A and DNMT3B is the principle means of methylation of repetitive
ele-ments in mouse ES cells, and even in the presence of active methylation maintenance machinery, these elements loose methylation without de novo methyltransferase ac-tivity [220, 54]. Knocking out both Dnmt3a and Dnmt3b in mouse ES cells results in a gradual reduction of overall methylation by around 90% over 70 passages. No methylation change was seen in wildtype or cells lacking either Dnmt3a or Dnmt3b. When Dnmt3a and Dnmt3a were expressed in unmethylated mouse ES cells, genomic methylation levels (except for imprinted genes) were restored to wildtype levels [54]. De novo transferases play a somewhat different role in differentiated cells. DNMT3A and 3B are highly expressed in mouse embryonic stem cells, but at low levels differen-tiated cells and in adult tissues [256]. Mouse embryonic fibroblasts (MEFS) are a cell type that are used to model a more differentiated cell type. In MEFs, Dnmt3b dele-tion resulted in loss of some genomic DNA methyladele-tion, while no apparent loss was observed in Dnmt3a deletion [76]. Additionally, when only Dnmt3b was knocked out in MEFs, methylation levels progressively decreased, which was not observed in ES
cells [761. In addition to their direct roles in the addition of methylation, DNMT3A and DNMT3B play a role in maintenance of genomic stability [172]. MEFs in which Dnmt3b was deleted showed premature senescence or spontaneous immortalization, as well as chromosomal abnormalities. Whereas 70% of wildtype MEFs had the ex-pected 40 chromosomes, less than 50% of Dnmt3b deletion MEFs maintained a normal karyotype [76].
DNA methylation established by DNMT3A and DNMT3B activity is necessary for
cellular differentiation. DNA methylation has been shown to increase at promoters of key pluripotency genes during differentiation, signaling the transition from a stem-like pluripotent state to a differentiated state [96]. Indeed, differentiation appears to be inhibited without proper de novo methylation activity. In mouse ES cells, cells lacking Dnmt3a and Dnmt3b gradually lost methylation and showed a concurrent loss of the ability to form teratomas. After 10 passages without de novo activity, small teratomas were formed, but after 70 passages, no palpable teratomas were observed [96], linking the ability to differentiate with methylation level and de-novo methylation activity. This was also observed in induced pluripotent stem cells (iPSCs) lacking Dnmt3a
and Dnmt3b, which showed restricted developmental potential. Restoring functional
DNMT3A and DNMT3B restored developmental potential in these cells[262].
Despite the complexity of living organisms, many of the experimentally-learned roles of de novo transferases can be seen in mice, particularly the reduced developmen-tal potential of cells lacking de novo transferases. In mice, Dnmt3a deletion results in death of embryos at around 4 weeks of age, and Dnmt3b deletion is embryonic lethal
[2551. Methylation levels in Dnmt3a knockout mouse embryos are normal, and only a
small reduction in overall methylation level is observed in Dnmt3b knockout embryos. In contrast, when both Dnmt3a and Dnmt3b are knocked out, methylation levels are significantly reduced, almost to levels seen in Dnmtl knockout embryos [255]. This suggests that some redundancy in de novo methyltransferases exists, although unique roles for DNMT3A and DNMT3B have been described [179, 48, 18].
In humans, DNMT3A is moderately expressed in most adult tissues, and at a higher level in placental tissue and embryonic stem cells. DNMT3B is lowly expressed
in adult tissues and highly expressed in embryonic stem cells [298]. Both DNMT3A and DNMT3B can be knocked out in human ES cells with no change in morphology or differentiation potential. When both DNMT3A and DNMT3B are knocked out, global methylation decreases gradually over time ( 10% reduction over 22 passages) [221].
Mutations in the DNMT3B gene are associated with the rare autosomal recessive disorder associated with immunodeficiency, centromeric instability, and facial
anoma-lies - called ICF syndrome [367, 134]. Mutations in DNMT3B occur frequently at
the catalytic domain, where they affect catalytic activity of DNMT3B
1244].
Whereaspericentromeric satellites 2 and 3 are methylated in normal patients, they are almost completely unmethylated in patients with ICF syndrome [367, 169]. Demethylation of pericentromeric repeats resulting from inadequate DNMT3B activity may contribute to chromosomal instability and the other symptoms associated with ICF.
In cancer, DNMT3A is mutated in about 20-30% of AML cases [216, 156, 288], and mutations in DNMT3A are associated with significantly worse overall survival [283]. Interestingly, sites in DNMT3A that are frequently targeted by genetic mutations have been found to be enriched for methylation changes between normal and can-cer AML [176], suggesting that even in tumors with wild-type DNMT3A sequence, epigenetic modifications may still affect the function of DNMT3A. DNMT3A mu-tant AML cells show a 9% decrease in average methylation and hypomethylation affects the leukemogenic HOX cofactor MEISi, which is expressed at a higher level in DNMT3A-mutant AML patients [98].
Cofactors of de novo activity
DNMT3A and DNMT3B are often associated with a catalytically-inactive regulatory
factor called DNMT3L which recognizes unmodified H3K4 histone tails and associates with DNMT3A [257, 384, 171]. The DNMT3A-DNMT3L complex appears to have a preference for targeting CpGs spaced 8bp apart, a periodicity that is frequent at known targets of DNMT3A, including imprinted regions [171] and retroelements
activity by 15-fold [119].
In mice, the chromatin remodeler LSH protein (HELLS in human) has been shown
to be required for maintaining global methylation levels [72]. Because nucleosome-bound DNA is a poor substrate for de novo methylation [97], it is proposed that LSH may play a role in repositioning chromosomes for access by de novo DNMTs [3311. Lsh knockout mice show 50% reduction of global methylation [72], particularly affecting minor satellite repeats, IAP retrotransposons and LINE elements [151]. In the absence of LSH, histones in these regions accumulate H3K4me2 and H3K4m3 [374]. LSH was shown to interact with either DNMT3A or DNMT3B in methylating a retroviral reporter vector, and in the absence of LSH, vector methylation by DNMT3A and
DNMT3B was impaired by 25-30% [387], confirming its role in de novo methylation.
Re-expression of wild-type Lsh in Lsh-null MEFs restores global methylation levels to 52% - 75% of wild-type levels [331]. This is surprising because it suggests a model
in which the targeting (e.g. to repetitive elements) of de novo methyltransferases is performed by other chromatin indicators, and de novo methylation activity proceeds in the absence of other developmental cues.
HELLS has been implicated in very few cancers. HELLS overexpression has been
found in nasopharyngeal carcinoma [1381 where silencing of the gene fumarate hy-dratase was reported to lead to cancer progression. HELLS expression was reported to be downregulated in leukemia [213]. In 34 non-small cell lung cancers, HELLS ex-pression was decreased in 9 tumors (26%), showed no change in 18 tumors (53%), and increased in 7 tumors (21%). In the cancer context, repression attributed to HELLS may be more associated with its regulation of histone methylation than DNA methy-lation [3741, although nucleosome repositioning has been shown to precede promoter hypermethylation in cancer [144].
1.3.2
Maintenance methylation
Because CpGs are symmetric in double-stranded DNA, meaning that a CpG on a positive strand is always accompanied by a CpG on the negative strand at the same position. This enables methylation marks to be copied from parent to daughter strand
during DNA replication [1471. DNA methyltransferase 1 (DNMT1) is the principle enzyme that is associated with copying methylation information from the parent DNA strand to the daughter DNA strand.
DNMT1 is comprised of a catalytic domain at the C-terminus, and a regulatory
domain at the N-terminus. The catalytic domain is homologous to bacterial DNA
methyltransferases
125],
and the regulatory domain contains sequences domains, whichdirect DNMT1 to replication foci [228], to hemimethylated CpGs [63], to interact with other proteins [181, 142], and to regulate the methylation of the catalytic domain
[91, 317]. The role and function of each of these domains is being actively investigated [91, 109, 88]. The copying efficiency of DNMT1 in humans has been estimated to be
between 99.7 and 99.97%, and DNMT1 can faithfully maintain methylation patterns at most genomic regions in the absence of de novo methyltransferases for several generations [221].
In mouse embryonic stem cells, Dnmtl knockout results in a decrease from 78% in wildtype cells to less than 20% in Dnmtl knockout cells. When all three methyltrans-ferases are knocked out, the methylation average drops to nearly undetectable levels. Loss of methylation does not appear to be specific to promoters, gene bodies, or in-tergenic regions [219]. DNMT1 plays an integral role in embryonic development and cellular function. Dnmtl knockout in mouse embryos results in general hypomethyla-tion [220, 215] and a lethal phenotype, with embryos not surviving past mid-gestahypomethyla-tion
[217].
In humans, DNMT1 is expressed in all dividing adult tissues as well as in embry-onic stem cells [298], and deletion of DNMT1 results in cell death [221, 53].
DNMT1 has been reported to be overexpressed in leukemia [241] and colon
can-cer [162], as well as hepatocarcionma [291], and mutations in DNMT1 are rare [177]. Decreasing Dnmt1 expression levels in mice by a combination of knockout and hypo-morphic alleles results in genome-wide hypomethylation, as well as the development of T-cell lymphomas within 4-8 months [110].
UHRF1 is an essential cofactor in the maintenance of methylation, and colocalizes and physically interacts with DNMT1 [38]. Even in the presence of DNA
methyltrans-ferases, knockout of Uhrfl in mice is lethal and results in a loss of methylation similar to that resulting from Dnmtl knockout [227].
1.3.3
Active demethylation
The major players in the active demethylation of CpGs are members of the Ten-Eleven Translocate (TET) protein family, called TETI, TET2, and TET3. They contain a catalytic domain with a cysteine-rich region and a conserved alpha-ketoglutarate
oxy-genase double-stranded beta-helix core fold [330]. TETI and TET3 also contain a
CXXC zinc finger DNA binding domain [385]. Active demethylation of methylcyto-sine (5mC) has been shown to progress as a stepwise enzymatic process [103]. The first step is oxygenation of 5mC to 5-hydroxymethylcytosine (5hmC) by any of the TET enzymes [164]. It has been suggested that during DNA replication, these modi-fied bases may be complimented with unmethylated cytosines on nascent strands, and these sites are not targeted by Dnmtl methylation maintenance [136]. In addition to replication-dependent TET-mediated demethylation, several other mechanisms for replication-independent demethylation have been proposed [250, 363, 233].
Knockout of either Teti or Tet2 in mouse ESCs results in 5hmC levels to about 40 to 60%, while depleting both results in complete loss of 5hmC. Global 5mC levels increased from approximately 5.3% to 5.7% as a result of knocking out Teti and Tet2 [67], with hypermethylation affecting 3% of CpGs in targeted regions under
500bp long, and located at enhancers [149]. Knocking out either Teti or Tet2 results
in viable offspring, but when both Teti and Tet2 are knocked out, most pups died within the first two days [67].
Teti and Tet2 appear to play a role in demethylation of imprinted regions, and Teti is required for erasure of paternal imprints in the female germ line [371]. Knock-out of Teti and Tet2 results in methylation of repetitive elements [67]. Tet2 has been shown to play an active role in demethylation in normal murine CD4+ T cells, particularly targeting regions of evolutionary conservation and transcription factor binding [153].
responsible for active demethylation of certain loci in the paternal and, to a lesser extent, maternal genome after fertilization [263, 305]. Tet3 is expressed highly in oocytes and zygotes, but is downregulated starting at the two-cell stage, whereas Teti and Tet2 are not expressed at this stage [159]. Deletion of Tet3 in male germ cells does not affect 5hmC or 5mC in male or female proneuclei, but deleting Tet3 from oocytes reduces 5hmC levels in male pronuclei, indicating that maternal Tet3 is responsible for demethylation in the male pronuclei [1231. 5hmC is also lost in
the 13-hour postfertilization female pronuclei
1305].
Recent work has shown that5mC depletion from the paternal genome is concurrent 5hmC accumulation, and that demethylation of the paternal genome proceeds even under the inhibition of Tet activity [7]. This suggests that 5hmC observed after implantation arises from active demethylation of de novo methylation activity on the hypomethylated genome.
TET enzymes, particularly TET2, are frequently mutated in myeloid and lym-phoid malignancies, although they are rare in other cancers [2991. In patients with myeloid malignancies, those with mutant TET2 were associated with low genomic 5hmC [189]. Conflicting reports have suggested that global levels of 5mC increase
[99] or decrease in those with TET2 mutations [189] compared to those without
mu-tations. Recent results indicate that TET2 mutations only have a minor effect (<1%) on global methylation in cancer, and that most methylation changes occur at tran-scription factor binding sites and enhancers [373, 2781. This further supports a role in which de novo methyltransferases and active demethylation machinery are active at a few regulatory regions, which is confirmed by the minimal increase in measurable global methylation after knockout of Teti and Tet2 in mouse ES cells [67].
Mutations in isocitrate dehydrogenases IDH1 and IDH2 commonly allow them to acquire the ability to catalyze alpha-ketoglutarate to 2-hydroxyglutarate. This has been proposed to affect TET function, which relies on alpha-ketoglutarate. In one study of 385 AML patients, TET2 mutations were found in 28/385 patients, and IDH1/2 mutations were found in a mutually exclusive set of 57 patients, indicating that demethylation targets of TET in AML may be hypermethylated as the result of decreasing TET activity [991. Further studies identified the transcription factor WT1
as another interactor of TET, and WT1 mutations were nearly mutually exclusive with TET2 and IDH1/2 mutations in AML [347].
The expression of all three TET enzymes was decreased in lung and breast cancers, and 5hmC levels were decreased in liver, lung, breast, pancreas, and prostate cancers
[376]. However, low 5hmC levels may be due to the rapid proliferation of cancer
cells [279]. TET expression levels appear to be correlated with function and clinical outcome. TETI and TET2 were significantly downregulated in breast cancer (DCIS) vs corresponding adjacent normal tissue, and low 5hmC and 5mC were associated with poorer outcomes [336]. In hepatocellular carcinoma, low 5hmC levels were associated with larger tumor size and with shorter overall survival [2261.
1.3.4
Passive demethylation
Passive demethylation is marked by the dilution of methylation with each progressive cell division when DNA methylation marks are not copied from the parent strand to the nascent daughter strand. Methylation may not be transmitted due to im-paired methylation maintenance machinery (DNMT1) function, or because methyla-tion marks on the parent strand have been modified (e.g. by TET enzymes) and are not recognized by methylation maintenance machinery [300].
On a global level, passive methylation manifests as a reduction methylation by
50% with each round of replication. After one round of DNA replication without
methylation maintenance, DNA will be hemimethylated - composed of the
methy-lated parent strand and the unmethymethy-lated daughter strand. In mice, methylation was measured in cells with a partially inactivated Dnmtl. Hemimethylation was prominent especially at CpG-poor regions [82].
Because DNA replication is necessary for passive demethylation, demethylation in non-dividing cells has been attributed to active demethylation. Demethylation that happens too rapidly to be attributed to cell division has also been attributed to active demethylation. Classically, the rapid demethylation of the paternal genome has been
attributed mostly - if not entirely - to active DNA demethylation [258] because of the
of Tet3 demethylation activity (5hmC) in the male pronucleus [362]. However, recent studies have suggested that the majority of methylation loss during reprogramming of the paternal methylome are due to passive demethylation [300, 158, 71, citing hemimethylated DNA and a decrease of methylation in the paternal genome after the first cell cycle [12].
1.4
Region-based methylation characteristics in
nor-mal tissues and cancer
Mechanisms of de novo methylation, methylation maintenance, and active and passive demethylation act in concert to direct methylation levels across the genome. In most normal tissues, these regulatory patterns are well-defined, can be observed across cell types. In cancer, however, these patterns of normal methylation regulation are disrupted, contributing to the cancer phenotype.
1.4.1
Promoters
The promoter region describes the region immediately distal to the site of transcrip-tion initiatranscrip-tion. Promoters have been divided into functranscrip-tional classes based on CpG content [3491. CpGs are not evenly distributed across the genome. Because of spon-taneous deamination of methylated cytosines to thymine, the majority of remaining CpGs are found in unmethylated clusters called CpG Islands (CGI) which have a GC content of greater than 50% and an observed vs expected ratio CpG content of at least 0.6 [1081. Initial sequencing of the human genome revealed 50,267 CGIs [204], most of which were less than 1kb long. CGIs are frequently located at the promoters of genes, and approximately two-thirds of known promoters are associated with CGIs
[207, 154]. CGIs not associated with a known promoter may indicate an alternative
transcription start site only expressed in certain developmental lineages, or may have some other regulatory significance as a binding site for transcription factors [154].
HCPs are associated with housekeeping genes that are constitutively expressed [349], and are generally referred to as broad promoters because transcription initiation can start at any of several transcription start sites within a 50-100bp window. Addition-ally, HCP are frequently the site of bidirectional transcription, with genes transcribed in either direction originating from the CGI promoter [431. HCP are generally un-methylated, regardless of whether the associated genes are expressed or not expressed [349]. At these promoters, expression is regulated by other mechanisms including si-lencing by H3K27me3.
Genes with intermediate-CpG promoters (ICP) and low-CpG promoters (LCP) are infrequently expressed, and these promoters are associated with genes with selec-tive expression, such as lineage-specific genes. Some ICPs contain CGIs, but the CpG islands are shorter, and less CG dense than those found in HCPs. The methylation state of CGIs at ICPs is more dynamic, and methylation correlates with repression of transcription. Silent ICPs are more frequently methylated than silent HCPs. LCPs are associated with tissue-specific genes, and are generally methylated, although ex-pression does not correlate with methylation at these genes [3491. LCPs are usually associated with a single, well-defined transcription initiation site [2931.
The role of promoter methylation in the regulation of the pluipotency gene Oct4 demonstrates the canonical relationship between promoter methylation and gene ex-pression. Oct4 is unmethylated and expressed in pluripotent cells, including those of the early embryo [260, 114]. During gastrulation, the repressor germ cell nuclear fac-tor (GCNF) binds to the Oct4 promoter, silencing the gene [104, 3451. GCNF recruits de novo methyltransferase Dnmt3a [295] as well as MBD2 and MBD3, a member of the nucleosome remodeling and deacetylase (NuRD) complex [122], to irreversibly methylate and silence the locus [96]. Three enhancer regions upstream from the tran-scription start site are also methylated. These enhancers and the promoter region remain methylated and not expressed after 6.5 days post-coitum and in differentiated cells [114]. These epigenetic changes happen within only a few days. In in vitro dif-ferentiation of mouse embryonic stem cells with retinoic acid, Oct4 silencing occurs after 1.5 days, and methylation marks are established within 3 days [1221. Similar
dynamics have been reported in human [188].
Methylation at gene promoters - especially tumor suppressor genes - has been
noted to be a conspicuous characteristic of the cancer methylome. Early studies of promoter methylation in cancer led researchers to ask whether the promoter changes seen in cancer were similar to mutations in genomic DNA, in which the cancer progres-sion could be attributed to a mutation in a single key gene. Melki et al. studied eight tumor-related genes in 20 AML patients, and found that 15 (75%) of them showed aberrant hypermethylation in more than one of the eight genes. They concluded that the methylation of promoters was the result of genetic dysregulation than the result of targeted methylation modifications at specific gene promoters [2401.
An increasing number of methylation datasets from different types of cancer are becoming available, which enables researchers to study patterns of promoter methy-lation across different cancer types [357]. Analysis in ten cancer types revealed that certain subsets of promoters tended to be differentially methylated across cancers. Genes frequently hypermethylated in cancer were those involved in early develop-ment and morphogenesis and embryonic developdevelop-ment. The majority of genes were hypermethylated in cancer, but the genes that were hypomethylated in cancer were related to immune function or ectoderm development, which may reflect themes of cell invasion and cancer growth [187].
In addition, promoter hypermethylation is the oncogenic side effect of the common cancer methylation pattern called CIMP, discussed in subsection 1.5.2.
1.4.2
Intragenic regions
Presence of DNA methylation at promoters is generally thought to block the initi-ation of transcription, while DNA methyliniti-ation in intragenic regions has been asso-ciated with transcript elongation. There is generally a positive correlation between gene body methylation and gene expression levels [195, 2241, and genes are more methylated than flanking genomic regions [11].
During transcript elongation, the RNA polymerase II complex recruits the histone methyltransferase SETD2, which leads to the addition of H3K36me3 histone marks
at transcribed regions [378]. De novo methyltransferases read the H3K36me3 mark, and add methylation to these transcribed regions [75].
In addition, intragenic methylation is thought to suppress alternative promoters, non-coding RNAs, and transposable elements within gene bodies[195. Further, DNA methylation in gene bodies has been associated with alternative splicing. CTCF was shown to bind to unmethylated DNA, causing PolII pausing and inclusion of an upstream exon of CD45 [3111. Recently, it was shown that TET-mediated oxidation
of 5mC accommodates CTCF binding at this locus [2351.
Mutations in SETD2 were found in 10-15% of clear cell renal cell carcinomas (ccRCCs), and biallelic inactivation was associated with a reduced survival time and earlier time to recurrence. In patients with SETD2 mutations, H3K36me3 was lost over gene bodies, and paradoxical hypermethylation was observed primarily at intra-genic regions. Hypermethylation was also observed to be a feature in kidney (KIRP) and lung (LuCa) samples with SETD2 mutations [332].
1.4.3
Enhancers
Transcription factors bind at specific regulatory regions called enhancers to regulate gene expression programs [321]. Enhancers are defined by a 100-300bp region free of nucleosomes and enriched for the histone acetyltransferase p300 and low methylation. They are associated with several chromatin marks, including H3K4mel and H3K27ac, and a lack of H3K4me3 [41, 340, 3221. While all enhancers are associated with the H3K4me1 mark, H3K27ac marks active enhancers. Enhancers without H3K27ac (only H3K4me1) have been shown to be in a poised state, protected from DNA methylation and ready for activation at a future time in differentiation [62].
Super enhancers are clusters of enhancers that are expected to have a greater effect on development and cellular identity. They were originally identified by the presence of the Mediator protein complex and H3K27ac, and were noted be associated with genes that were expressed at higher levels than other enhancers [351, 356]. It has been proposed that the histone mark H3K27ac identifies super enhancers and not regular enhancers [146]. These regions are generally more unmethylated than traditional
enhancers, particularly at the borders of the enhancer regions where transcription factors are thought to bind and define the boundaries of the super enhancer region [1451.
Enhancer methylation has been shown to be dynamic - the target of de novo
methylation and active demethylation. Enhancers are enriched for 5hmC, the byprod-uct of TET activity [3261, and knockout of Teti and Tet2 in mouse ES cells results in hypermethylation at these enhancers. Methylation was especially gained at en-hancers with high 5hmC in wildtype cells, and at those with low transcription factor occupancy [1491.
Methylation changes have been observed to target intragenic enhancer regions, and in some cases methylation dynamics at enhancers may influence gene expression more than promoter methylation [101. Deaton et al. [691 studied methylation changes at CGIs between differentiated cells of the hematopoetic lineage. They observed that although the majority of genomic CGIs occur at transcription start sites, differential methylation was enriched at intragenic CGIs. Of particular interest for this research, Deaton et al. also attempted to discriminate between allele-specific or strand-specific methylation by performing targeted bisulfite sequencing of DMRs. Sequencing re-vealed disordered methylation at DMRs, with low correlation of methylation states between neighboring CpGs [691.
The methylation of super enhancers is also modulated in cancer. A comparison between five normal and eight cancer methylomes showed that 727/5111 (14%) of detected super enhancers showed dynamic methylation between normal and cancer tissue. Loss of methylation was observed in 75.4% of super enhancers, whereas 24.6% gained methylation in the cancer. Colorectal tumors showed hypomethylation of super enhancers, but the remainder of the hypomethylation events appeared to be the result of global hypomethylation, and not targeted hypomethylation at super enhancer regions. Hypermethylated enhancers in cancer were associated with shutting down genes that were highly expressed in associated normal tissues [145].
1.4.4
Repetitive elements
More than half of human DNA is made of up repetitive elements. In addition to simple repeats of nucleotides of DNA sequence, some of these repetitive elements encode retrotransposons which are capable of moving to new locations in the genome. The only active retrotransposon in humans belongs to the Long Interspersed Nucleotide Element (LINE) family, and is 6-8kb long [1181. Our genome is littered with extinct versions of these retrotransposons, and is a record of the evolution of these elements as they have copied and pasted themselves to new locations and evolved to withstand genomic suppression. Based on phylogenetic analysis, some elements are estimated to be up to 600 million years old [234]. The youngest and most active LINEs are in the Li family and DNA methylation is thought to be a main factor responsible for silencing these elements [301]. Li element expression is elevated in mouse ES cells deficient in all three methyltransferases [337]. In mice, an additional class of repetitive element, Intracisternal A Particle (IAP) retroviruses, is regulated by DNA methylation [74]. Around half of CpGs are located at repetitive elements [92].
In mouse embryonic stem cells, the knockout of all three active methyltransferases does not transcriptionally activate Li retrotransposons, but IAP elements are acti-vated. In stem cells, which are generally hypomethylated, histone modifications or other epigenetic control mechanisms are used to control retrotransposon expression [343]. In mice deficient in Dnmtl, IAP transcript levels are highly elevated [342]. The chromatin modeler LSH is also necessary for proper Li and IAP methylation in mice
[151].
In mice, genes with an LAP within 10kb of the gene were found to be associ-ated with cancer development, cell growth and proliferation, and other cancer-relassoci-ated functions. Upregulation of IAPs in cancer could be associated with an upregulation of these genes [2721.
Li hypomethylation is frequently observed in cancer [2611, and Li hypomethyla-tion has been linked with poor prognosis in many cancers [253, 3061. A meta-analysis of 46 cancer studies totaling to 2554 cancer patients and 3553 control samples showed
that overall, Li methylation levels were significantly lower (6.4%) in cancer patients than in control patients, although analysis of specific cancer types showed that Li methylation levels were significantly lower in colorectal and gastric patients, and that no difference was seen in hepatocellular cancer [14].
The link between Li hypomethylation and cancer is only beginning to be under-stood [366]. Li methylation could be a good marker for cancer progression because it is a surrogate for global methylation status [3751, and global hypomethylation is associated with genomic instability, chromosome rearrangements, and mutations
[297, 52, 237]. Additionally, demethylation and retrotransposition of these elements
has been shown to insert and interfere with tumor suppressor gene function. In 2/19 hepatocellular carcinoma genomes, Li insertions into the MCC tumor suppressor disrupted MCC expression [3101.
1.4.5
Pericentromeric heterochromatin
Functional centromeres are enriched with repetitive regions that assist in proper chro-mosome segregation during cell division [212]. Centromeric regions contain minor satellite repeats, while pericentromeric regions contain major satellite repeats that
are normally maintained in a heterochromatic state
12921.
Immediately afterfertil-ization, DNA at the centromere of the paternal genome lacks heterochromatin, and is instead enriched for H3K27me3 and members of the Polycomb repressive complex PRCI [271]. At this point, pericentromeric repeats are expressed, and resulting satel-lite RNA proliferation that acts in a similar manner to Xist, stabilizing the region and ushering in constitutive heterochomatinization [268]. The addition of H3K9me3
by Suv39h [214] is recognized by heterochromatin HP1 proteins [198], which recruit
DNMTs [3131. These changes to chromatin ensure that pericentromeric regions are maintained in a state of heterochromatin [292].
Pericentromeric satellite repeats show loss of methylation when knocking out methyltransferases in mouse ES cells, although pericentromeric heterochromatin is maintained even in the absence of DNA methylation [3371.
is designated as constitutive heterochromatin, as opposed to the facultative hete-rochromatin found at silenced genomic regions which is instead marked with proteins of the Polycomb group. In cancers and some diseases, constitutive heterochromatin switches to facultative chromatin, and acts as a sink for Polycomb that are displaced from other genomic regions. This is proposed to affect the epigenetic landscape and promote genome instability in caner [70].
Hypomethylation and upregulation of satellite transcripts is a common theme in cancer. Hypomethylation of satellite regions was seen in hepatocarcinoma, even though expression levels of all methyltransferases were elevated in the hepatocar-cinoma cells vs normal controls. In addition, satellite hypomethylation was seen in noncancerous hepatocarcinoma precursors, suggesting a functional role for peri-centromeric demethylation in cancer transformation [291]. In glioblastoma (GBM), satellite repeat HSATII was reported to be demethylated with no mutations in DNA methyltransferases, with upregulation of DNMT1 [891. In comparison to normal pan-creas, pancreatic ductal adenocarcinomas (PDACs) show a 21-fold increase of all satellite transcripts, and the pericentromeric satellite HSATII was undetectable in normal tissue but 131-fold upregulated in PDACs. This trend was confirmed in lung, kidney, ovarian, and prostate cancers [334].
1.4.6
X-chromosome inactivation
In female cells with two X chromosomes, one X chromosome is silenced so that proper gene dosage is maintained. DNA methylation plays an important role in the estab-lishment and maintenance of silencing on the inactive X chromosome.
X chromosome silencing begins at the X-inactivation center, which includes the lncRNA Xist which is necessary for inactivation [265]. Methylation of the Xist pro-moter is associated with its expression, and the Xist propro-moter is demethylated on the inactive X chromosome [22]. Transcription of Xist produces RNA that is local-ized to the X-inactivation center, then spreads to silent, gene-dense regions, and then coats the remainder of the genome in cis [85]. This triggers a cascade of events that decrease accessibility by transcription factors and transcription machinery, including
the removal of euchromatic marks H3K4me2/3, H3K9Ac and H4Ac. The Xist-coated regions interact with Polycomb repressive complexes and other enzymes, and become enriched with repressive H3K27me3, H3K9me2, H2Aub1 and H4K20mel 1641. DNA methylation is added to further silence genes on the inactive X chromosome [46, 47]. Improper inactivation of the X chromosome, and abnormalities involving the X chromosome have been associated with cancer. Deleting the Xist gene in hematopoi-etic stem cells in mice resulted the development of leukemia in female mice. While male mice remained healthy, female heterozygous or homozygous Xist mutant mice began to die as early as 1.5 months, and by 2 years only 10% remained alive. Multi-ple X-linked genes were upregulated in the disease mice, including Gatal and Kif4. Interestingly, many autosomal genes were also upregulated [377].
The X chromosome contains many genes involved in growth regulation and signal-ing that may contribute to cancer development and progression [3191. Although the exact mechanisms remain to be elucidated, many cancer show bias in gender distri-bution, and these biases may be a reflection of X chromosome involvement [319, 791.
In a study of children with acute lymphoblastic leukemia (ALL), an acquired X chromosome was present in 33/45 (73%) patients [1401. The gain of an additional X chromosome in a male with chronic neutrophilic leukemia was the only abnormality noted in the disease [3721.
The loss of the inactive X chromosomes has long been associated with breast can-cer, and is correlated with poor prognosis and poor response to therapy. Additionally, some breast and ovarian cancer cells have two active X chromosomes. An interaction between BRCA1 and XIST has been suggested to contribute to this observation [2591.
1.4.7
Imprinted genes
Every cell contains two copies of each gene - one maternal allele and one paternal
allele. Imprinted genes are those in which one parental allele is marked with DNA methylation and is not expressed. Most imprinted genes are clustered in an imprinted locus containing 3-12 imprinted genes and an imprinting control region which exhibits allele-specific methylation. These methylation marks are is established during
ooge-nesis for maternal imprints and during spermatogeooge-nesis for paternal imprints [151. There are approximately 150 imprinted genes currently identified in mice, and
many of them are conserved in humans [17, 161. Some of these imprinted genes have
been implicated in the regulation of cell proliferation and growth [167, 1261, and the decline in expression of these genes has been shown to correlate with the organismal decline in growth rate from birth until reaching adulthood [230]. De novo transferases
DNMT3A and DNMT3L are required for establishment of most imprinting control
regions [1791.
Expression and methylation of imprinted genes are dysregulated in cancer. Im-printed genes were more often downregulated in cancer, and expression was variable in the majority of imprinted genes in breast (BRCA), kidney (KICH), and lung (LUAD) cancers. Imprinted genes, and particularly imprinting control regions are more often methylated in cancer, however maternal and paternal imprinting control regions were targeted in a balanced manner [1861.
1.5
The cancer methylome
Early studies in cancer pointed to genetic lesions in the form of gain-of-function mu-tations at oncogenes or loss-of-function mumu-tations at tumor suppressor genes that led to uncontrolled growth and proliferation. Likewise, in the study of cancer epigenetics,
both hypermethylation - associated with repression - and hypomethylation -
asso-ciated with genomic accessibility - are associated with the dysregulation of normal
cellular function leading to cancer progression.
Generally, the methylome of normal cells is methylated except at regions such as active promoters and enhancers that are in an open state. In cancer, the clear methylation pattern observed in normal cells is eroded, so global methylation levels decrease, and methylation increases at previously-unmethylated regions, referred to
as punctate hypermethylation [1931.
It is interesting to note while both global hypomethylation and punctate hyper-methylation are observed together in many cancers, some cancer types show either