Synthesis and characterization of diblock and statistical copolymers based on hydrolyzable siloxy silylester methacrylate monomers
Texte intégral
Figure
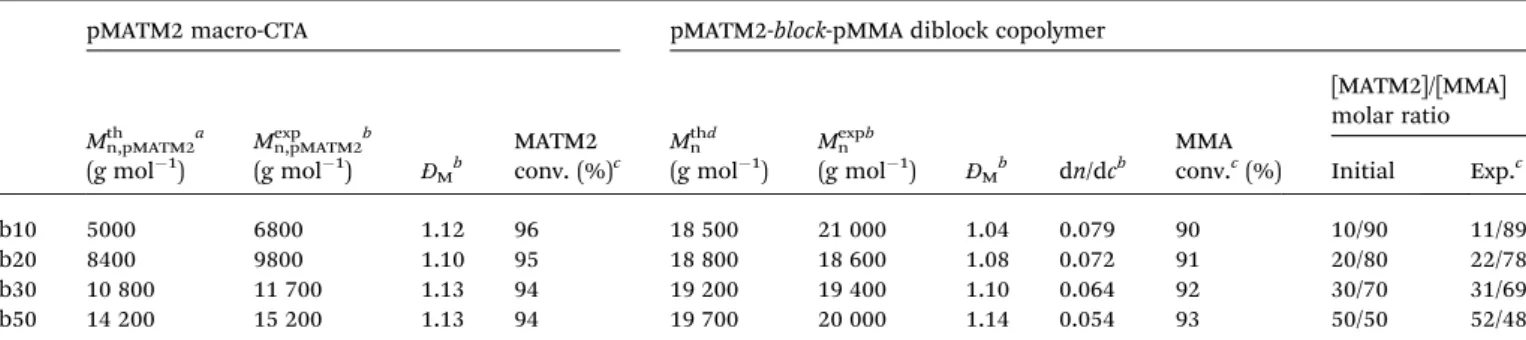
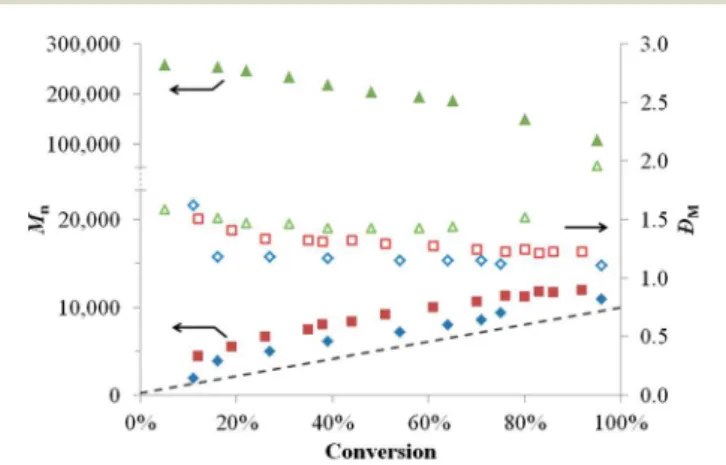

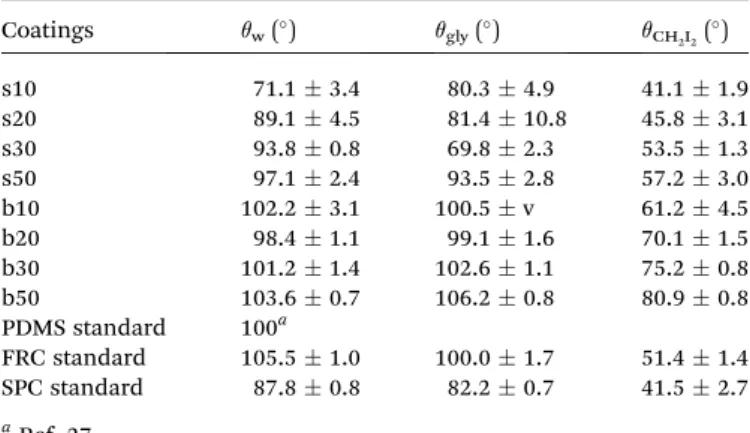
Documents relatifs
The influence of the macro-RAFT agent architecture on the morphology of the self-assemblies obtained by aqueous RAFT dispersion polymerization in PISA is studied by
Based on combined analyses of sample 10 by SEC, DLS, TEM and turbidimetry, we can reasonably assume that amphiphilic copolymers are actually formed that assemble during
In this work, a detailed investigation is performed on the synthesis, characterization, self-assembly and drug release behavior of PLLA/P(NIPAAm-co-DMAAm) and
Using an ATRP initiator containing a functional group such as epoxide, carbonate, NHS-ester, azlactone is an easy way to target reactive polymers towards amines.
Myrcene / Isobornyl Methacrylate SG1 Nitroxide-Mediated Controlled Radical Polymerization: Synthesis and Characterization of Gradient, Diblock and Triblock copolymers). Despite
strongly segregated limit, the lowest free energy state for the pure diblock copolymers, is the lamellae phase up until E = 0.28, according to a recent calculation by
1,2 Polyelectrolytes have been widely used in many applications such as ionic conductors in lithium‐ion batteries 3‐5 , as a dopant
axis correspond to metustable state, with one preferential state n * for each parity. In this example, the lowest chemical potential is obtained for ,I* even, which
