The Detection of Immortal DNA Strand Co-segregation
as a Method of Adult Stem Cell Identification
By
Jennifer J. Cheng B.E. Biomedical Engineering
Vanderbilt University, 2001
Submitted to the Division of Bioengineering in partial fulfillment of the requirements for the degree of
Master of Science in Bioengineering at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY June 2004
© 2004 MIT
All rights reserved. MASSACHUSETTS NST'lrTE;OF TECHNOLOGY
JUL 2 2 2004
LIBRARIES
Signature of Author: Certified by: Accepted by: I,K
6
V Division of Bioengineering May 24, 2004 James L. SherleycJ
o
Associate Professor Division of Bioengineering n,-- x In Thesis Supervisor / ''r '" c ' ... Alan J. GrodzinskyProfe of Electri Me cal, and Biological Engineering Chair, BE Graduate Committee
Abstract
The study of stem cells is one of the most fascinating topics in biology. Adult stem cells (ASC), which play the prime role in the maintenance and restoration of tissues, are thought to hold great potential for the advancement of medicine. It has been postulated that adult stem cells are able to retain "immortal" DNA template strands over successive generations by non-random chromosome co-segregation, and in so doing, to protect the long-term genomic fidelity of whole tissue compartments. The investigation of this theory may yield insights into areas such as the development of cancer and the process of aging. In addition, it may lead to the discovery of an effective method for the unique identification of adult stem cells, the study of which has thus far suffered from the lack of unique identifiers. Thus, the goal of this research was to develop an assay for the
detection of immortal DNA strand co-segregation that could be applied to the detection and analysis of adult stem cells. It is proposed that such an assay may in itself serve as a unique identification method for adult stem cells. In this thesis, the development of such an assay is described. This assay, referred to as the label release assay, has provided further evidence for the existence of immortal strand co-segregation in model cell lines, and will potentially be useful in the study of adult stem cells in tissues.
Table of Contents
List of Figures ... 4
Chapter 1: Introduction ... 5
Stem Cell Definitions ... 5
Stem Cell Kinetics ... 9
Im m ortal Strand Co-segregation ... 12
Purpose ... 17
Chapter 2: Assay Developm ent ... 18
M odel Cell System s ... 18
Label Retention A ssay ... 19
Label Release A ssay ... 21
Chapter 3: M ethods ... 26
Cell Culture ... 26
Label Retention Assay ... 26
Label Release Assay ... 27
Detection ... 27
Chapter 4: Results and Discussion ... 28
Label Retention Assay ... 28
Label Release Assay ... 30
Chapter 5: Conclusions and Future Directions ... 34
References ... 35
List of Figures
1. Stem Cell Division Kinetics ... 9
2. Chrom osom e Segregation ... 14
3. Label Retention A ssay Schem atic ... 19
4. Label Release A ssay Schem atic ... 21
5. Label Retention A ssay Results ... 29
Chapter 1: Introduction
Recently, there has been an explosion of interest in the field of stem cell research. The study of stem cells holds enormous potential both for the further development of medical therapies and for the advancement of our knowledge of the biological sciences. It is envisioned that stem cells, perhaps in conjunction with tissue engineering and gene therapy, will someday be used to regenerate and repair tissues and organs of every kind. The elucidation of the mechanisms by which stem cells function will provide further insight into our understanding of how humans develop, grow, function, heal, age, and develop cancer.
Stem Cell Definitions
Stem cells are cells that exhibit both self renewal and the ability to give rise to
differentiated progeny. A stem cell can be classified as either an embryonic stem cell (ESC) or an adult stem cell (ASC). Embryonic stem cells are primordial, undifferentiated cells derived from the developing embryo. Specifically, they are isolated from the inner cell mass of an embryo in the blastocyst stage, at which point the embryo has not yet implanted in the uterine lining. 1'2 The inner cell mass is the portion of the blastocyst that
develops into the fetus. Thus embryonic stem cells are pluriplotent - that is, they are able to generate all of the types of differentiated cells that make up the body. Significantly, they are able to generate progeny representing each of the three germ layers of the developing fetus - endoderm, mesoderm, and ectoderm. 1'4
Adult, or somatic stem cells, by contrast, are undifferentiated cells found in more mature tissues. Although their exact point of origin is unclear, it is presumed that adult stem cells arise at some point in fetal development from embryonic stem cells. Adult stem cells reside in different tissue compartments throughout the body, each filling its own stem cell "niche".5 They are self renewing and either unipotent or multipotent, giving
rise to cell types specific to each tissue compartment. For instance, hematopoietic stem cells (HSCs),, which reside in the bone marrow, produce all the different types of blood cells that make up the hematopoietic or blood system.6 There has been some controversy
surrounding the plasticity of adult stem cells - that is, their ability to give rise to multiple cell lineages, including those lineages that have been thought to be distinctly divergent from the lineages to which the stem cells are seemingly committed to. There have been reports, for instance, of skin-derived stem cells generating cells such as neurons and adipocytes (fat cells), 7 9and of bone marrow-derived stem cells generating multiple
non-hematopoietic cell types.'0 However, it is still unclear whether these observations truly
represent adult stem cell transdifferentiation events. As such, their significance - either as an indication of normal adult stem cell function or as a potential technology for the manipulation of adult stem cells - remains to be determined.' 1-13
Although there has also been debate concerning the relative merits of studying adult and embryonic stem cells, both are thought to have great potential for the illumination of our biological understanding. 14 Embryonic stem cells appear to have the greater potential to
virtually unlimited self renewal capacity.1 5 However, it is not yet clear whether these self
renewal and differentiation properties can be effectively controlled such that ESCs can safely be used therapeutically. Embryonic stem cells have been shown to form tumor-like masses called teratomas when injected into mice. 1-3,16, 17 Because the more mature,
post-natal tissues of the body do not represent normal embryonic stem cell environments, ESCs may need to be properly differentiated or perhaps converted into adult stem cells prior to their clinical use in vivo. Moreover, the use of human embryonic stem cells is laden with ethical concerns regarding their derivation from human embryos. At present, they have been most commonly obtained from unused embryos from in vitro fertilization
clinics or from electively aborted fetuses (embryonic germ cells).16-18 There has also been a recent report of human embryonic stem cells being derived from an embryo created by the nuclear-transfer cloning of a somatic cell.19 This report has in itself generated even more controversy because it involves the issue of human cloning.20
Regardless, the use of either pre-existing or newly created human embryos for the purpose of scientific research, even if it leads to beneficial therapies, will continue to encounter strong opposition from many.21
The research discussed herein has been focused solely upon the study of adult stem cells. The use of adult stem cells avoids many of the ethical issues raised above concerning the use of ESCs, because ASCs can be directly obtained from adult tissues. In addition, adult stem cells may be used in autologous therapy - that is, a patient may be treated with ASCs obtained from his or her own body. Because these cells are recognized as "self', autologous therapy avoids provoking immune reactions that occur when cells from donor,
non-self sources are introduced into the body. Adult stem cells are an attractive source for cell therapy, as they occur naturally in the various tissue compartments of the body, and they already function to maintain and replenish those tissues. Thus in contrast to embryonic stem cells, adult stem cells already have the precise self-renewal and differentiation properties that are needed in vivo. However, the use of adult stem cells presents a different set of barriers - namely, their identification, isolation, and expansion. Adult stem cells are very rare in the body. For instance, it has been estimated that only 1 in every 10,000 cells in the bone marrow is a hematopoietic stem cell.22 23 Methods have
been devised to enrich the proportion of HSCs in a given cell population,24 26but no set
of markers has been found that uniquely identifies an adult stem cell and enables the perfect purification of a stem cell population.27 28 Some types of adult stem cells, such as
those found in the small intestine, have a precisely defined physical location within the tissue architecture.2 9 For other tissues, the location or characteristics of the adult stem
cell niche have yet to be definitively established. For these reasons, adult stem cells are difficult to identify and isolate. The barrier to adult stem cell expansion is a direct consequence of an adult stem cell characteristic termed asymmetric cell kinetics (ACK), which is discussed below.
Stem Cell Kinetics
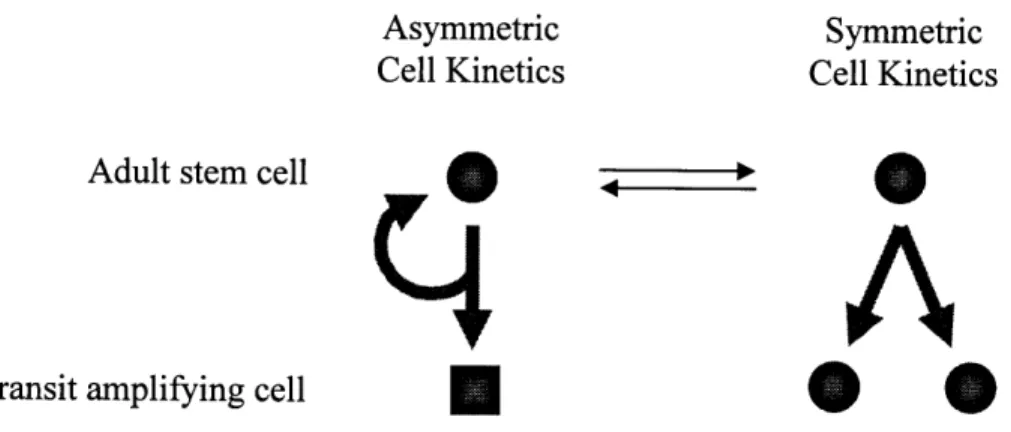
It has been proposed that adult stem cells accomplish their dual tasks of self-renewal and generating differentiating progeny under the paradigm of asymmetric cell kinetics. That is, when an adult stem cell divides, it produces an identical adult stem cell, and a transit amplifying cell (Figure 1). This division is asymmetric because the two resulting cells have unequal kinetic and differentiating potentials. The adult stem cell daughter will continue to self-renew, whereas the transit amplifying daughter undergoes expansion and differentiation. Eventually it will terminally differentiate into the specialized cell types that make up that particular tissue compartment. In this way, a relatively small and
constant number of stem cells can generate the multitude of cells needed to maintain a tissue.
Asymmetric Symmetric
Cell Kinetics Cell Kinetics
Adult stem cell m ,
4
A
Transit amplifying cell * * c
Figure 1. Stem cell division kinetics.
The term "symmetric cell kinetics", on the other hand, denotes situations in which a cell divides to produce two functionally identical daughter cells with equal proliferative potential (Figure 1). Embryonic stem cells divide with symmetric cell kinetics, whereas
adult stem cells normally divide with asymmetric cell kinetics. However, adult stem cells may reversibly switch to symmetric cell kinetics during times in which the stem cell pool needs to be expanded or replenished, such as during body growth or wound healing. Asymmetric cell kinetics, combined with symmetric cell kinetics induction when needed, allow adult stem cells to maintain homeostasis within a tissue unit throughout the lifetime of an organism. In tissues that experience high cell turnover rates, such as the intestinal epithelium, terminally differentiated cells are continually lost from the system, and they are continually replenished by adult stem cells cycling asymmetrically.3 0
Evidence that adult stem cells divide with asymmetric cell kinetics has been demonstrated in many different contexts. Through the use of retroviral marking to conduct clonal lineage analyses in mice, neural stem cells (NSCs) have been found to divide asymmetrically in order to self renew and generate differentiating progeny in
vivo.31 It has also been inferred from the analysis of methylation patterns in human colon
crypts that intestinal stem cells exhibit ACK in vivo.3 2 Several studies have provided
evidence that explanted hematopoietic stem cells have ACK, using various methods including time-lapse microscopy, cell growth analysis, and functional analyses including repopulation and colony formation assays.33 38 Explanted rat liver stem cells have also
been demonstrated to have ACK through time-lapse microscopy, colony formation, and daughter pair analyses.39
From in vitro investigations of model cell systems, it has been found that asymmetric cell kinetics is dependent upon intracellular guanine nucleotide levels.40 42 Specifically, it
was shown that ACK can be activated in symmetrically cycling cells by the induction of the tumor suppressor gene p53. This occurs through p53's down regulation of inosine-5'-monophosphate dehydrogenase (IMPDH), an enzyme that is rate-limiting for the
formation of guanine ribonucleotides. Conversely, it was found that constitutive over-expression of IMPDH induces symmetric cell kinetics, even when p53 is expressed.4 0 44
As mentioned above, asymmetric cell kinetics also create a barrier to the expansion of adult stem cells.4 5' 46 Because adult stem cells occur rarely in the body and may be
difficult to obtain in many tissues, a small number of ASCs may need to be expanded in order to be useful for tissue and organ replacement therapies. Moreover, in order to simply study adult stem cells in vitro, the need arises to expand and propagate them over long periods of time. However, adult stem cells cycling with asymmetric cell kinetics will become progressively diluted in a cell population due to the continuous production
of transit amplifying cells and terminally differentiated cells. While the total number of adult stem cells remains constant, the number of non-stem cell progeny produced
increases. Over successive passages of a cell culture, the ASC fraction steadily decreases, until eventually the culture undergoes senescence, appearing to stop dividing.4 5' 46 Thus,
in order to effectively culture, study and utilize adult stem cells, it will be necessary to devise ASC expansion methods which can overcome the barrier of asymmetric cell kinetics.
Immortal Strand Co-segregation
As discussed, adult stem cells are self-renewing, capable of generating differentiating progeny, and cycle with asymmetric cell kinetics. It has been proposed that adult stem
cells also possess a fourth characteristic - namely, immortal DNA strand co-segregation. This theory is described below.
Assuming that asymmetrically cycling adult stem cells are responsible for replenishing the tissues of the body throughout the lifetime of an organism, the issue of maintaining genomic fidelity arises. It has been estimated that for the intestinal epithelia alone, normal tissue maintenance requires roughly 1013 cell divisions in the lifespan of a rat, and 106 cell divisions in a human.4 7 Spontaneous mutations are known to occur at a rate of
at least 10-8 per nucleotide per generation in humans.48' 49 Given these figures, it is
expected that at least 108 spontaneous mutations will occur over the lifetime of a human being, again in intestinal epithelial tissues alone. Similar numbers of mutations are expected to occur in other tissues with similar turnover rates. One might further predict that this overwhelming number of spontaneous mutations would inevitably lead to the development of cancer, and the serious malfunction of the cells of the body. However, a correspondingly high incidence of cancer has not been empirically observed. For
instance, in the United States, the lifetime risk for the development of stomach cancer is around 1%, and under 1% for esophageal cancers.50 Cancers of the small intestine are even less prevalent, being diagnosed 20 times less frequently than stomach cancer in the US.51
In 1975, John Cairns formulated the immortal strand co-segregation theory to explain why this high rate of spontaneous mutation does not lead to a correspondingly high incidence of cancer. He proposed that adult stem cells protect their genomic code by selectively retaining chromosomes containing "immortal" DNA template strands over successive generations.4 7 This nonrandom segregation of chromosomes ensures that
newly synthesized DNA strands are always passed onto transit amplifying daughter cells within two divisions, while the original templates, or immortal DNA strands, remain perpetually in the stem cell. By using this mechanism, adult stem cells avoid the accumulation of spontaneous mutations arising from unrepaired replication errors.52
Instead, the spontaneous mutations, which occur on newly synthesized strands, are passed onto transit amplifying daughter cells, and eventually lost from the system due to
terminal differentiation and turnover. Thus, by maintaining the genomic fidelity of adult stem cells, the overall integrity of the tissue compartment is protected.
Figure 2 shows a schematic example of immortal strand co-segregation for an adult stem cell with three chromosomes. For each chromosome, one strand, depicted in gray, is the immortal template strand. Following S phase, each of the original strands has been replicated and is paired with a newly synthesized strand. During mitosis, the theory dictates that the chromosomes containing immortal strands are non-randomly co-segregated to the adult stem cell daughter, whereas the other chromosomes go to the transit amplifying daughter. Note that in the next asymmetric cell division, the stem cell daughter will lose the rest of the strands that were synthesized in the previous cycle.
Immortal strand co-segregation
t
ii
X~
vs.
Adult stem cell Mitosis
Random strand segregation
Figure 2. Chromosome segregation. Immortal strands are depicted in gray.
Squares represent transit amplifying daughters.
Also shown in Figure 2 is one possible result of random chromosome segregation between the two daughter cells. The conventional view is that chromosomes are
randomly segregated when cells undergo mitosis. We and others propose that this is not true for adult stem cells. When considering the existing evidence that immortal strand co-segregation does occur, it is useful to note the following. There are only 8 possible ways in which the three chromosomes can be divided amongst the two cells. There is a
1:4 probability that the three immortal strand-containing chromosomes will co-segregate by chance alone, and a 1:8 probability that they will co-segregate specifically to the adult stem cell daughter. Similarly, the probability that a cohort of n chromosomes will specifically co-segregate by chance in a given cell division is 1 :2, or about 1 in 7x10 13
for the 46 chromosomes in a human cell. Consider that the probability that this would occur by chance repeatedly over x successive divisions, decreases to 1:( 2n)X. Clearly,
Nonrandom chromosome segregation has been observed in various forms and contexts, since before Cairns' formulation of the immortal strand hypothesis. Some of the first reports of nonrandom chromosome segregation came from studies of bacteria and plants, as well as mammalian cells in vitro.53 54 It has been suggested that some mutant strains
of yeast, as well as certain wild-type strains, also exhibit nonrandom chromosome segregation. 55 56 Others have searched for immortal strand co-segregation in vivo, in
studies of tissues ranging from the intestinal crypt to the tongue papilla. 57 61 Some of the
clearest evidence for immortal strand co-segregation has been found in the study of model cell lines with inducible asymmetric cell kinetics, as described above.6 2
All of the studies mentioned are similar in that they entail the labeling of DNA according to specific regimens, followed by the retrospective analysis of the cells or tissues
involved. Generally, they have attempted to label immortal DNA strands, in order to use this label to observe whether the immortal DNA strands are retained in adult stem cells. The most common method of permanently labeling DNA involves the incorporation of a detectable thymidine analogue such as bromodeoxyuridine (BrdU) or tritiated thymidine (3H-thymidine) into new strands during DNA synthesis. However, by definition, "new"
immortal DNA template strands are not synthesized during normal asymmetric adult stem cell cycling. In order to observe labeled immortal DNA strands, one must first label the DNA during symmetric cell division, and then study the cells as they begin to cycle asymmetrically. When the cells switch from symmetric cell kinetics to ACK, they will begin to retain "immortal" strands, some of which will already be labeled. In the model
cell systems, this assay is made possible through the precise control of cell kinetics, as will be discussed further.6 2 However, in vivo, adult stem cells should only cycle
symmetrically when the stem cell pool needs to be expanded - namely, during
development and wound repair. Thus in order to capture immortal strand co-segregation
in vivo using this method, scientists have either subjected animals to injury first, or
studied young, developing animals.5 7 61 It is both desirable and fundamental, however, to
also be able to study adult stem cell function during normal tissue maintenance, without causing these perturbations, and in mature tissue compartments as well as in ex vivo cell preparations.
Purpose
The purpose of this research is to develop a new assay which can be used to detect immortal strand co-segregation without necessitating the precise control of cell kinetics. This would enable the further investigation of this mechanism in systems such as adult stem cell lines, ex vivo tissue explants, and ultimately, adult stem cells in vivo in their unperturbed, asymmetrically cycling state. Such an assay would serve to substantiate and illuminate the role of immortal DNA strand co-segregation in adult stem cell biology. It would also provide further evidence to support the claim that deterministic asymmetric cell kinetics is the correct model of adult stem cell function. This derives from the fact that immortal strand co-segregation relies upon deterministic asymmetric cell kinetics in order to effectively prevent the accumulation of spontaneous mutations in adult stem cells. An assay for the detection of immortal strand co-segregation could be utilized as a new, unique method of identification of adult stem cells. Not only would this aid in the identification of stem cell niches and further definition of tissue architecture, but it may also enable the more precise isolation and purification of adult stem cell populations. The development of this assay is discussed herein.
Chapter 2 - Assay Development
Model Cell Systems
Two previously developed model cell systems, in which cell division kinetics can be controlled,° 42were used to develop and test the assay for the detection of immortal
strand co-segregation. These were derived from spontaneously immortalized, p53-null murine embryonic fibroblasts, which cycle with symmetric cell kinetics. The first system consists of the Zn-dependent, p53-inducible Ind-8 cell line, and its control cell line Con-3, which lacks the inducible p53 gene. When exposed to zinc, the Ind-8 cell line expresses p53, and begins to cycle with asymmetric cell kinetics. Under the same conditions, the Con-3 cell line, lacking p53, continues to cycle with symmetric cell kinetics.40' 41 The
second model system was derived from the first, by transfection of the p53-inducible cell lines with IMPDH. This resulted in the Zn-dependent, p53-inducible, constitutive
IMPDH expressing tI-3 cell line, and its control, the tC-2 cell line, which lacks the constitutively expressed IMPDH gene. When p53 is induced, the tI-3 cell line continues to cycle symmetrically, due to constitutive IMPDH expression, whereas the tC-2 cell line,
lacking constitutive IMPDH expression, begins to cycle asymmetrically.4 2 It is important
to note that when these model cell lines cycle asymmetrically, each asymmetric division produces one: stem-like daughter cell which has the potential to continue dividing, and one "transit amplifying"-like daughter, which in this system undergoes terminal arrest either immediately or after one more division, as determined by time-lapse microscopy.4
Label Retention Assay
As discussed in the introduction, the method previously used to demonstrate immortal strand co-segregation required the labeling of the immortal strand, followed by the observation that the labeled immortal strands were retained in adult stem cells. This assay, referred to as the label retention assay, can be implemented in the model cell systems as described below (Figure 3).
(4
AI
_-rdl (I CytoDA-I*
5 GT -1 rdU CytoD B 5 GT 0 @Figure 3. The label retention assay.
In the label retention assay, symmetrically cycling cells are labeled with BrdU for one generation time (GT), producing cells with hemi-labeled DNA - that is, for each chromosome, one of the strands is now labeled (depicted in gray). Zinc is then added, inducing p53 expression and asymmetric cell kinetics in the Ind-8 and tC-2 cell lines (pathway A). Simultaneously, the BrdU is removed, and the cells continue to cycle for
i0@+i. rdU (
each chromosome is selected to be the immortal strand. We are currently investigating whether this initial selection of the immortal strand is random, or is always the more recently synthesized (and in this case, labeled) strand. As depicted in Figure 3, it has been shown that at least some of the strands chosen as immortal strands are labeled.6 2
Both labeled and unlabeled immortal strands are retained in the stem-like cell over the 5 successive generations of asymmetric cell division kinetics. Finally, the cells are arrested by the addition of cytochalasin D, an actin inhibitor which prevents cytokinesis, trapping the two daughter cell nuclei in a single cytoplasm. This binucleate will have unequal
fluorescence, because the nucleus that was destined to become the stem cell-like daughter continues to retain some or all of the labeled immortal strands. The other nucleus
contains only strands that were synthesized within the last two divisions, and are thus unlabeled.
Pathway B shows the outcome for cells that continue to cycle symmetrically even when zinc is added - such as Con-3 cells, which lack p53, and tI-3 cells, which constitutively express IMP1)H. Since these cells segregate their chromosomes randomly, the label is geometrically diluted with each successive division. After 6 generations, the cells show a uniformly decreased labeling intensity across the total population, therefore no
Label Release Assay
As discussed, in order to study immortal strand co-segregation in adult stem cells in their native state, we perceived the need for a detection method in which it is not necessary to change or manipulate the cell division kinetics. Instead of trying to label immortal strands and look for their retention, a new strategy was conceived in which non-immortal strands are labeled and observed. In this case, the captured event is the release of these non-immortal strands from the stem cell in the following cycle. This method, termed the label release assay, is shown in Figure 4.
M
( II
f
I 'l +BrC ytBrdU []UNN.- I El14~
~~111 B 0 OFigure 4. The label release assay.
Pathway A illustrates the label release assay as it would occur in a cell that is cycling asymmetrically. For ease of reference, the immortal strands are depicted as striped bars,
)
-BrdUlI1
but note that they are not in fact labeled. Again, the cells are labeled with BrdU for one generation, producing two hemi-labeled cells - the stem cell daughter, and the transit amplifying daughter (square). The newly synthesized labeled strands, depicted as gray bars, are not immortal. In the next generation, the BrdU is chased from the cell and cytochalasin D is added to arrest the cell. During this division, the labeled non-immortal strands generated in the previous cycle are non-randomly co-segregated to the transit amplifying daughter nucleus, while the immortal strands co-segregate to the stem cell daughter nucleus. Each of the chromosomes in either of the 2 cohorts also contains a new, unlabeled strand synthesized in the absence of BrdU during the current cycle. Thus, as depicted, an asymmetric binucleate is produced.
When subjected to the same regimen, cells cycling with symmetric cell kinetics behave in the manner depicted in Pathway B. Following the one generation BrdU labeling period, two equally hemi-labeled daughter cells are produced (only one of these is shown). When the BrdU is removed, and the cells arrested, the previously labeled strands are randomly segregated between the two daughters, producing binucleates with equally fluorescent nuclei.
In the development and optimization of the label release assay in the model cell systems, many variables were considered, including cell density, chase conditions, assay timing, and detection methods. Each of these factors can greatly influence our ability to detect immortal strand co-segregation. For instance, it is known that cell density affects cell division kinetics, both in the model cell systems, and in pre-senescent primary cell
cultures, which are likely to contain adult stem cells.4 1 6 3
, 64 In the model cell systems,
the most efficient induction of asymmetric cell kinetics occurs at low cell densities. At higher cell densities, the cells become resistant to p53-induction and continue to divide exponentially. If the cell density is too low, however, the cells tend to arrest entirely. For the label release assay, the best results were observed at a density of 500 cells / 1.7 cm2, the same density used for the label retention assay.
The next factor mentioned was the chase method. The goal of the chase is simply to halt the incorporation of BrdU into the DNA so that the strands synthesized during this period of the assay are unlabeled, thus allowing us to distinguish between chromosomes
containing immortal strands (unlabeled) and those containing labeled non-immortal strands generated during the first cell cycle. Various chase regimens were attempted, including adding 1 00M deoxythymidine (dT) to compete out the remaining BrdU; adding both 1 00pM dT and 1 00[tM deoxycytidine (dC) in order to ensure that cell kinetics are not affected; and simply changing the media. Each of these methods was seen to be effective in preventing the continued incorporation of BrdU, as determined by in situ immunofluorescence. Changing the media is perhaps the simplest and most obvious method, and empirically, it enabled us to observe the most asymmetric
binucleate events. However, other methods were tested because it has been observed in other contexts that changing the media, even in the continuing presence of zinc, can induce the asymmetrically cycling model cell lines to return to symmetric cell kinetics (J.
Another important variable considered was the timing of the various phases of the assay. In developing the assay, we worked with unsynchronized cell populations because this most closely resembles the systems to be studied in vivo. However, because they are unsynchronized, not all of the cells in the population are expected to produce the same ideal outcome that is depicted in Figure 4. In the current assay configuration, the BrdU labeling period is 24 hours, immediately followed by the simultaneous chase and arrest. For example, cells in mid-S phase at the beginning of the labeling period could partially progress through a second S phase during the labeling period, thus labeling two
successive generations of non-immortal strands and generating binucleate with symmetric fluoresence. In attempting to decrease the observation of such "false negative" symmetric binucleate events, the assay timing was changed in the following ways. The BrdU labeling period was shortened to 8 or 12 hours, followed by a distinct chase period of 12 or 8 hours, respectively, and finally a 16 hour arrest. In each of these attempts, however, the incidence of asymmetric binucleates detected was not increased.
The final experimental feature to be discussed here is the method of detection. The importance of this factor cannot be underestimated, given that one of the ultimate goals of this research is to develop an assay that can be used to identify adult stem cells in heterogeneous populations and tissues, in which they may occur at frequencies as low as
1 in 10,000.22'23 To accomplish this goal, the sensitivity of the detection method must be
improved. Over the course of this research, different detection methods that have been investigated include the use of different BrdU-staining antibody systems, the use of vital DNA dyes as a replacement for BrdU incorporation, the use of flow cytometry to detect
asymmetrically fluorescent binucleates, and the use of two-photon microscopy for automated image collection and data analysis. However at this stage, improved detection methods are still under development. The results presented here were collected as described in Chapter 3, Methods.
Chapter 3 - Methods
Cell Culture
The culture conditions and maintenance of the Zn-dependent, p53-inducible Ind-8 cell line and the control cell line Con-3 have been previously described.4 2 44 Cells were
maintained as subconfluent monolayers in DMEM supplemented with 10% dialyzed fetal bovine serum (JRH Biosciences) and 5,ug/ml puromycin (Sigma), and kept at 370C in
humidified incubators with 5% CO2. The p53-inducible, constitutive IMPDH-expressing
tI-3 cell line and the control cell line tC-2 were maintained in the same way, except that the growth medium was also supplemented with lmg/ml geneticin (Sigma), and for the tI-3 cell line, 45jiM ZnC12.42
Label Retention Assay
Cells were cultured on chamber slides at the density of 500 cells / 1.7cm2. 16-24 hours
post-seeding, BrdU (Sigma) was added to the medium at a final concentration of 20tM for 24 hours. p53 expression was induced by changing the medium to medium
containing 65pgM ZnCl2. After a 96 hour incubation period, cells were arrested by the
Label Release Assay
Cells were cultured on chamber slides at the density of 500 cells / 1.7cm2. 16-24 hours
post-seeding, p53 expression was induced by changing the medium to medium containing 65p1 M ZnCl2. After 24 hours, BrdU (Sigma) was added to the medium at a final
concentration of 20pM for another 24 hours. The cells were then chased and arrested by replacing the media with fresh media containing 65ptM ZnCl2and 2pM cytochalasin D
(Sigma) for 16 hours.
Detection
Cells were fixed in 70% ethanol on ice for 30 minutes and stored at -200C. Staining was
done at room temperature. Slides were washed with PBS, and denatured with 2N HC1 at room temperature for 10 minutes prior to staining. Blocking was performed in PBS containing 0.5% bovine serum albumin and 0.05% Tween-20 for 5 minutes. BrdU incorporation was detected by staining with a FITC-conjugated, mouse monoclonal anti-BrdU antibody (BD Pharmingen) diluted 1:10 in blocking solution for 45 minutes. Cells were counterstained with Hoechst 33258 at the concentration of 0.5pg/ml in PBS for 5 minutes. Fluorescent images were captured using a Zeiss Axiovert 100 inverted microscope, a Zeiss AxioCam CCD camera, and Openlab software (Improvision).
Chapter 4 - Results and Discussion
The primary focus of this research has been the development of the label release assay, in which the release of labeled, non-immortal strands from stem cells is captured. This assay has now been successfully used to detect immortal strand co-segregation in our p53-inducible model cell lines. In addition, new results have been obtained from the label retention assay, in which the retention of labeled immortal strands in stem cells is observed. These results will now be presented and discussed in the context of our current state of understanding.
Label Retention Assay
The label retention assay described above was previously used to demonstrate immortal strand co-segregation in the Ind-8 cell line, which cycles with asymmetric cell kinetics when p53 is induced. In independent experiments, 10% (n=100, S. Ram-Mohan,
unpublished observations) and 22% (n=262)6 2respectively, of binucleates were observed
to have clearly asymmetric BrdU fluorescence in asymmetrically cycling Ind-8 cells. The latter result may have been higher due to the selection of mitotic cells prior to the initiation of the label retention assay. We now report the results of a third independent label retention experiment, in which 13% (n=67) of binucleates were observed to be asymmetric, again in asymmetrically cycling Ind-8 cells (Figure 5). In contrast, clearly asymmetric binucleates were not found in the corresponding label retention experiments performed on symmetrically cycling Con-3 cells (n>400). In these experiments, the level
of BrdU fluorescence is homogeneous across the Con-3 cell populations, in contrast to the Ind-8 cell population, in which fluorescence is heterogeneous.
Hoescht BrdU Phase
Figure 5. Example asymmetric Ind-8 binucleate
seen in the label retention assay.
In order to further interrogate the relationship between asymmetric cell kinetics and immortal strand co-segregation, the label retention assay was also previously used to study the constitutive IMPDH expressing tI-3 cell line. No clearly asymmetric binucleates were observed for the symmetrically cycling tI-3 cells, even under the induction of p53 (S. Ram-Moham, unpublished results). The same result has now been obtained in an independent tI-3 label retention experiment. The tI-3 cell line continues to cycle symmetrically under the induction of p53 due to constitutive expression of IMPDH, and it is therefore predicted that immortal strand co-segregation does not occur. By confirming this, we have provided further evidence that immortal strand co-segregation is dependent upon asymmetric cell kinetics, and is not merely an artifact of p53-induction.
Label Release Assay
The label release assay has now been tested on all four of the model cell lines discussed. When p53 was induced, asymmetric binucleates were observed in both the Ind-8 and tC-2 cell lines, at the frequencies of 8% (n=107) and 15% (n=26), respectively (Figure 6). Asymmetric binucleates were not observed, however, in the Con-3 (n=178) and
p53-induced tI-3 cell lines (n=135), which cycle symmetrically. As may be noted in Figure 6A, in some instances, although asymmetry is clearly observed, some label also appears
Hoescht BrdU Phase
A
B
C
Figure 6. Examples of asymmetric binucleates seen in the
label release assay.
in the other daughter nucleus. It is hypothesized that these events may arise from cells which completed portions of two successive S phases during the BrdU labeling period, thus segregating some labeled strands with their immortal strands to the stem cell-like nucleus. An analogous feature, illustrated in Figure 6B, is that sometimes the label is not uniformly spread across the labeled nucleus, but is instead localized to a confined region. Similar labeling patterns were frequently observed in the label retention assays (J. Merok, and S. Ram-Mohan, unpublished observations), and they may have implications with regards to the mechanism by which immortal strand co-segregation is accomplished. For instance, this intranuclear co-localization or clustering of the labeled strands may imply that non-random strand co-segregation is achieved through the stable attachment of the strands to some cellular structure, such as microtubules. One must be careful to note, however that the localized staining patterns have appeared in both assays, indicating that the behavior, if it exists, may occur in both immortal and non-immortal strands.
The above results confirm that the label release assay may be used as a new method for the detection of immortal strand co-segregation without perturbing asymmetric cell kinetics. Using this novel approach, it has again been demonstrated that immortal strand co-segregation occurs in the Ind-8 cell line, and this observation has been extended to the independently-derived tC-2 cell line. To clarify, this cell line was transfected with an empty vector to serve as the control for the IMPDH-transfected tI-3 cell line. It has the same p-53 inducible asymmetric cell kinetics as the Ind-8 cell line. The results obtained from the tI-3 experiments also again reinforce the claim that immortal strand
co-segregation depends upon asymmetric cell kinetics, as distinct from p53 expression in general.
It must be noted, however, that the label release assay in its present form is not as robust as the label retention assay. Multiple label retention experiments conducted by multiple investigators have produced similar results. The results obtained from the label release assay, on the other hand, have not thus far been reproducible. Even when asymmetric binucleates have been observed, the frequency of detection has been quite low. Out of the many possible explanations for this, perhaps the most readily apparent one is that there are still technical shortcomings in the assay. Technical problems such as those involving staining and detection, the induction of asymmetric cell kinetics, BrdU
incorporation, and cell arrest may be ruled out, since most of these are common elements shared with the label retention assay. The mostly likely culprits appear to be those of timing of the assay regimen, and chase conditions, two of the most important factors studied during assay development. It is still possible that chasing by changing the media disrupts the asymmetric cell kinetics, or that we have not yet found the optimal timing regimen for capturing the largest cohort of cells undergoing the label release event. Perhaps we only stumbled upon the original asymmetric binucleates events by the slimmest of margins, and the assay needs to reconfigured once again. This also suggests that the assay may require re-optimization for its use in other cell lines and systems.
Leaving technical difficulties aside, one must also consider the distinct possibility that the cells do not behave exactly as supposed. Even the results obtained from the label
retention analyses fall short of expected detection levels, if it is assumed that most cells are labeled during the labeling period and that the majority of the binucleates arise from asymmetrically cycling stem-like cells. Perhaps on the individual cellular level,
asymmetric cell kinetics are not always uniformly preserved, or perhaps the immortal strand co-segregation itself is an imperfect process. Considering biology as a whole, it is quite likely that the process contains its own imperfections. However, the evidence obtained thus far from these and other analyses, including the direct physicochemical analysis of BrdU labeled immortal DNA strands (J. Lansita, unpublished observations),6 2
still strongly suggests that immortal strand co-segregation does occur, imperfections and all.
Chapter 5 - Conclusions and Future Directions
In summary, a new assay has been developed to study immortal strand co-segregation without necessitating the control of cell division kinetics. This label release assay has provided further evidence of immortal strand co-segregation in model cell systems, substantiating this in a novel way, and in an additional cell line. Although the label release assay, has not yet achieved the desired robustness, it has already demonstrated that it is possible to study immortal strand co-segregation by capturing the release of labeled, non-immortal strands from stem like-cells. Now that the concept has been established, all that remains is the further optimization of the assay and its application to other
systems. Indeed, preliminary investigations into the application of the label release assay to the study of pre-senescent cell cultures, adult stem cell lines, and tissues in vivo, have already begun. Specifically, the label release assay has already been applied to pre-senescent wild-type murine embryonic fibroblasts (wt MEFs), pre-pre-senescent WI-38 human diploid fibroblasts, and early passage Lig 8 rat liver adult stem cells grown in the absence of xanthosine to stimulate asymmetric cell kinetics. Thus far, however, the wt MEFs and WI-38s have appeared to be highly sensitive to the assay conditions, and have not shown BrdU incorporation, perhaps due to growth arrest. In the Lig 8s, on the other hand, only symmetrically fluorescent binucleates have been observed (n=92). Again, further assay optimization is necessary before it can be determined whether these results are truly indicative of chromosome segregation behavior in these cell lines. Nevertheless, the label release assay is a promising candidate for a method of immortal strand co-segregation detection that can be used to study adult stem cells.
References
1. Martin GR. 1981. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad
Sci ULSA. 78(12):7634-8.
2. Evans MJ, Kaufman MH. 1981. Establishment in culture of pluripotential cells from mouse embryos. Nature. 292(5819): 154-6.
3. Smith AG. 2001. Embryo-derived stem cells: of mice and men. Annu Rev Cell
Dev Biol. 17:435-62.
4. Bradley A, Evans M, Kaufman MH, Robertson E. 1984. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature.
309(5965):255-6.
5. Fuchs E, Tumbar T, Guasch G. 2004. Socializing with the neighbors: stem cells and their niche. Cell. 116(6):769-78.
6. Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, et al. 2003. Biology of hematopoietic stem cells and progenitors: implications for clinical application
Annu Rev Immunol. 21:759-806.
7. Toma JG, Akhavan M, Fernandes KJ, Barnabe-Heider F, Sadikot A, et al. 2001. Isolation of multipotent adult stem cells from the dermis of mammalian skin Nat
Cell Biol. 3(9):778-84.
8. Jahoda CA, Whitehouse J, Reynolds AJ, Hole N. 2003. Hair follicle dermal cells differentiate into adipogenic and osteogenic lineages. Exp Dermatol. 12(6):849-59. 9. Chunmeng S, Tianmin C. 2004. Skin: a promising reservoir for adult stem cell
populations. Med Hypotheses. 62(5):683-8.
10. Herzog EL, Chai L, Krause DS. 2003. Plasticity of marrow-derived stem cells.
Blood. 102(10):3483-93.
11. Raff M. 2003. Adult stem cell plasticity: fact or artifact? Annu Rev Cell Dev Biol. 19:1-22.
12. Wagers AJ, Weissman IL. 2004. Plasticity of adult stem cells. Cell. 116(5):639-48. 13. Camargo FD, Chambers SM, Goodell MA. 2004. Stem cell plasticity: from
14. Czyz J, Wiese C, Rolletschek A, Blyszczuk P, Cross M, et al. 2003. Potential of embryonic and adult stem cells in vitro. Biol Chem. 384(10-11): 1391-409. 15. Odorico JS, Kaufman DS, Thomson JA. 2001. Multilineage differentiation from
human embryonic stem cell lines. Stem Cells. 19(3):193-204.
16. Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. 2000. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat
Biotechnol. 18(4):399-404.
17. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, et al. 1998. Embryonic stem cell lines derived from human blastocysts. Science. 282(5391):1145-7.
18. Shamblott MJ, Axelman J, Wang S, Bugg EM, Littlefield JW, et al. 1998. Derivation of pluripotent stem cells from cultured human primordial germ cells.
Proc .NatlAcadSci USA. 95(23):13726-31.
19. Hwang WS, Ryu YJ, Park JH, Park ES, Lee EG, et al. 2004. Evidence of a pluripotent human embryonic stem cell line derived from a cloned blastocyst.
Science. 303(5664):1669-74.
20. Normile D. 2004. RESEARCH ETHICS: South Korean Cloning Team Denies Improprieties. Science. 304(5673):945.
21. Chu (;. 2003. Embryonic stem-cell research and the moral status of embryos.
Intern Med J. 33(11):530-1.
22. Boggs DR, Boggs SS, Saxe DF, Gress LA, Canfield DR. 1982. Hematopoietic stem cells with high proliferative potential. Assay of their concentration in marrow by the frequency and duration of cure of W/Wv mice. J Clin Invest. 70(2):242-53.
23. Spangrude GJ, Heimfeld S, Weissman IL. 1988. Purification and characterization of mouse hematopoietic stem cells. Science. 241(4861):58-62.
24. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. 1996. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. JExp Med. 183(4): 1797-806.
25. Spangrude GJ, Brooks DM, Tumas DB. 1995. Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood. 85(4): 1006-16.
26. Osawa M, Hanada K, Hamada H, Nakauchi H. 1996. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 273(5272):242-5.
27. Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, et al. 2002. A stem cell molecular signature. Science. 298(5593):601-4.
28. Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. 2002. "Stemness": transcriptional profiling of embryonic and adult stem cells. Science. 298(5593):597-600.
29. Bach SP, Renehan AG, Potten CS. 2000. Stem cells: the intestinal stem cell as a paradigm. Carcinogenesis. 21(3):469-76.
30. Marshman E, Booth C, Potten CS. 2002. The intestinal epithelial stem cell.
Bioessays. 24(1):91-8.
31. Morshead CM, Craig CG, van der Kooy D. 1998. In vivo clonal analyses reveal the properties of endogenous neural stem cell proliferation in the adult
mammalian forebrain. Development. 125(12):2251-61.
32. Yatabe Y, Tavare S, Shibata D. 2001. Investigating stem cells in human colon by using methylation patterns. Proc Natl Acad Sci US A. 98(19):10839-44.
33. Brummendorf TH, Dragowska W, Zijlmans J, Thombury G, Lansdorp PM. 1998. Asymmetric cell divisions sustain long-term hematopoiesis from single-sorted human fetal liver cells. JExp Med. 188(6):1117-24.
34. Brummendorf TH, Dragowska W, Lansdorp PM. 1999. Asymmetric cell divisions in hematopoietic stem cells. Ann N YAcad Sci. 872:265-72; discussion 272-3. 35. Huang S, Law P, Francis K, Palsson BO, Ho AD. 1999. Symmetry of initial cell
divisions among primitive hematopoietic progenitors is independent of ontogenic age and regulatory molecules. Blood. 94(8):2595-604.
36. Punzel M, Zhang T, Liu D, Eckstein V, Ho AD. 2002. Functional analysis of initial cell divisions defines the subsequent fate of individual human
CD34(+)CD38(-) cells. Exp Hematol. 30(5):464-72.
37. Punzel M, Liu D, Zhang T, Eckstein V, Miesala K, et al. 2003. The symmetry of initial divisions of human hematopoietic progenitors is altered only by the cellular microenvironment Exp Hematol. 31(4):339-47.
38. Takano H, Ema H, Sudo K, Nakauchi H. 2004. Asymmetric division and lineage commitment at the level of hematopoietic stem cells: inference from
differentiation in daughter cell and granddaughter cell pairs. JExp Med. 199(3):295-302.
39. Lee HS, Crane GG, Merok JR, Tunstead JR, Hatch NL, et al. 2003. Clonal expansion of adult rat hepatic stem cell lines by suppression of asymmetric cell kinetics (SACK). Biotechnol Bioeng. 83(7):760-71.
40. Sherley JL, Stadler PB, Johnson DR. 1995. Expression of the wild-type p53 antioncogene induces guanine nucleotide-dependent stem cell division kinetics.
Proc Natl Acad Sci US A. 92(1):136-40.
41. Rambhatla L, Bohn SA, Stadler PB, Boyd JT, Coss RA, et al. 2001. Cellular Senescence: Ex Vivo p53-Dependent Asymmetric Cell Kinetics. JBiomed
Biotechnol. 1(1):28-37.
42. Liu Y, Bohn SA, Sherley JL. 1998. Inosine-5'-monophosphate dehydrogenase is a rate-determining factor for p53-dependent growth regulation. Mol Biol Cell. 9(1):15-28.
43. Sherley JL. 1991. Guanine nucleotide biosynthesis is regulated by the cellular p53 concentration JBiol Chem. 266(36):24815-28.
44. Liu Y, Riley LB, Bohn SA, Boice JA, Stadler PB, et al. 1998. Comparison of bax, wafl, and IMP dehydrogenase regulation in response to wild-type p53 expression under normal growth conditions. J Cell Physiol. 177(2):364-76.
45. Sherley JL. 2002. Asymmetric cell kinetics genes: the key to expansion of adult stem cells in culture. Stem Cells. 20(6):561-72.
46. Merok: JR, Sherley JL. 2001. Breaching the Kinetic Barrier to In Vitro Somatic Stem Cell Propagation JBiomed Biotechnol. 1(1):25-27.
47. Cairns J. 1975. Mutation selection and the natural history of cancer. Nature. 255(5505): 197-200.
48. Crow JF. 1995. Spontaneous mutation as a risk factor. Exp Clin Immunogenet. 12(3): 121-8.
49. Kondrashov AS. 2003. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat. 21(1):12-27.
50. Ries L, Eisner M, Kosary C, Hankey B, Miller B, et al. (eds). 2004. SEER Cancer
Statistics Review, 1975-2001. National Cancer Institute. Bethesda, MD.
http://seer.cancer. gov/csr/1975 2001/.
51. Young J, Roffers S, Liu G, Fritz A. 2004. Introduction to UGI Tract Cancer. in
SEER Web-based Training Module. National Cancer Institute. Bethesda, MD.
http:'Ytraining.seer.cancer.gov/ss module07 ugi/unit0l secOl ugi cancer.html. 52. Cairns J. 2002. Somatic stem cells and the kinetics of mutagenesis and
carcinogenesis. Proc Natl Acad Sci US A. 99(16): 10567-70.
53. Lark KG, Consigli RA, Minocha HC. 1966. Segregation of sister chromatids in mammalian cells. Science. 154(753): 1202-5.
54. Lark KG. 1967. Nonrandom segregation of sister chromatids in Vicia faba and Triticum boeoticum. Proc Natl Acad Sci USA. 58(1):352-9.
55. Dalgaard JZ, Klar AJ. 2001. Does S. pombe exploit the intrinsic asymmetry of DNA synthesis to imprint daughter cells for mating-type switching? Trends Genet.
17(3):153-7.
56. Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, et al. 2002. Evidence that the Ipl l-Sli 15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 108(3):317-29. 57. Potten CS, Hume WJ, Reid P, Cairns J. 1978. The segregation of DNA in
epithelial stem cells. Cell. 15(3):899-906.
58. Bickenbach JR, Mackenzie IC. 1984. Identification and localization of label-retaining cells in hamster epithelia. JInvest Dermatol. 82(6):618-22.
59. Bickenbach JR, Holbrook KA. 1987. Label-retaining cells in human embryonic and fetal epidermis. JInvest Dermatol. 88(1):42-6.
60. Morris RJ, Potten CS. 1999. Highly persistent label-retaining cells in the hair follicles of mice and their fate following induction of anagen. J Invest Dermatol.
112(4):470-5.
61. Potten CS, Owen G, Booth D. 2002. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci. 115(Pt 1 1):2381-8. 62. Merok JR, Lansita JA, Tunstead JR, Sherley JL. 2002. Cosegregation of
chromosomes containing immortal DNA strands in cells that cycle with asymmetric stem cell kinetics. Cancer Res. 62(23):6791-5.
63. Hayflick L, Moorhead PS. 1961. The serial cultivation of human diploid cell strains. Exp Cell Res. 25:585-621.
64. Todaro GJ, Green H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 17:299-313.
Acknowledgements
First and foremost I would like to thank my advisor, James Sherley, for the guidance he has provided me throughout my time at MIT. He has been a caring, wonderful mentor and teacher, and his enthusiasm and excellence as a scientist has truly been inspiring. I would also like to thank him for encouraging and helping me to achieve my dream of going into medicine. James, I would just like to add, GO HOPKINS!!
I would also like to acknowledge all of the members of the Sherley laboratory. I feel extremely grateful to have been a part of this group. They have been with me through thick and thin, and have always given me their support, advice, and friendship. We make a great team! In particular, I would like to thank Krisha Panchalingam, Minsoo Noh, Rouzbeh Taghizadeh, Janice Lansita, Jean-Francois Pare, and Gracy Crane, for always making the time to help me. You have been as much a part of my training as a scientist
as anyone. I would especially like to thank Sumati Ram-Mohan, for blazing the trail in learning the techniques required in the experiments discussed above, and for taking the time to help me learn them as well. Thanks also to Michal Ganz, Amy Shi, Johnathon
King, Jason Ellis, and Heelo Sudo (you're a member too!) for always being there for me. Last but not least, Ashley Rothenberg, thank you for being a wonderful UROP student!
I would also like to acknowledge the following people, who have each helped me a great deal at various stages of my research. Nicki Watson of the Whitehead Institute and Elena Gostjeva of the Center for Environmental Health Sciences both provided invaluable training in fluorescence microscopy. Glenn Paradis of the Center for Cancer Research, was a wonderful resource for flow cytometry, and Tim Ragan helped me with two-photon
imaging.
Finally, I would like to acknowledge my family and all of my friends for their
unconditional love and support. Thank you especially to Mom, Dad, Vince, and Nasir. I could never have done it without you.