Homozygous
TBC1D24
mutation
in
two
siblings
with
familial
infantile
myoclonic
epilepsy
(FIME)
and
moderate
intellectual
disability
Anne-Lise
Poulat
a,d,
Dorothée
Ville
a,d,
Julitta
de
Bellescize
b,d,
Nathalie
André-Obadia
c,d,
Pierre
Cacciagli
e,d,
Mathieu
Milh
e,f,d,
Laurent
Villard
e,d,
Gaetan
Lesca
g,h,d,∗aDepartmentofPediatricNeurology,GroupementHospitalierEst,HospicesCivilsdeLyon,Lyon,France bEpilepsy,SleepandPediatricNeurophysiologyDpt.,GroupementHospitalierEst,HospicesCivilsdeLyon, Lyon,France
cDepartmentEpilepsy,SleepandFunctionalNeurologicalExplorations,GroupementHospitalierEst, HospicesCivilsdeLyon,Lyon,France
dNeurophysiologyandEpilepsyUnit,NeurologicalHospitalP.Wertheimer,HospicesCivilsdeLyon,Lyon, France
eINSERM,GMGFUMRS910,13385,AixMarseilleUniversité,Marseille,France
fServicedeNeurologiePédiatrique,HopitaldelaTimone-Enfants,APHM,Marseille,France gDepartmentofMedicalGenetics,HospicesCivilsdeLyon,Lyon,France
hUniversitéLyon1,Lyon,France
Summary MutationsintheTBC1D24genewerefirstreportedinanItalianfamilywithaunique
epilepticphenotypeconsistingofdrug-responsive, early-onsetidiopathicmyoclonicseizures. Patientspresentedwithisolatedbilateralorfocalmyoclonia,whichcouldevolvetolong-lasting attackswithoutlossofconsciousness,withapeculiarreflexcomponent,andwereassociated withgeneralizedtonic—clonicseizures.Thisentitywas named‘‘familialinfantile myoclonic epilepsy’’(FIME).Morerecently,TBC1D24mutationshavebeenshowntocauseavariablerange ofdisorders,includingepilepsyofvariousseizuretypesandseverity,non-syndromicdeafness, andDOORSsyndrome.
We report on the electro-clinical features of two brothers, born to first-cousin parents, affectedwithinfantile-onsetmyoclonicepilepsy.Thepeculiarepilepticpresentationprompted
ustoperformdirectsequencingoftheTBC1D24gene.Thepatientshadveryearlyonsetoffocal myoclonicfitswithvariabletopography,lastingafewminutestoseveralhours,withoutlossof consciousness,whichfrequentlyevolvedtogeneralizedmyoclonusormyoclonicstatus.Reflex myocloniawerenoticedinonepatient.Neurologicaloutcomewasmarkedbymoderate intellec-tualdisability.Despitethehighfrequencyofseizures,repeatedEEGrecordingsshowednormal backgroundrhythmandrareinterictalspikesandwaves.Wefoundahomozygousmissense muta-tion,c.457G>A/p.Glu153Lys,inthetwoaffectedbrothers.Thisobservationcombinedwithrecent datafromtheliterature,suggestthatmutationsinTBCD24causeapathologicalcontinuum,with FIMEatthe‘‘benign’’endandseveredrug-refractoryepilepticencephalopathyonthesevere end.Early-onsetmyoclonicepilepsywithfocalandgeneralizedmyoclonicseizuresisacommon characteristicofthiscontinuum.
©2015ElsevierB.V.Allrightsreserved.
Introduction
Mutations in the TBC1D24 gene (OMIM 613577) have been first reported in an Italian family with autosomal recessive inheritance, including several patients affected with a homogeneous phenotype that was named by the authors: ‘‘familial infantile myoclonic epilepsy’’ (FIME, OMIM605021) (Falaceet al.,2010).The detailed electro-clinical presentation, reported ten years earlier, haslong remainedanisolateddescription(deFalcoetal.,2001).In thepastyear, thankstotheuseofwhole-exome sequenc-ing, additionalmutations in TBC1D24 have been reported inan increasingrangeof developmentaldisorders, includ-ingepilepsyofvariousseverity,non-syndromicdeafnessand DOORSsyndrome(OMIM220500)thatgetsitsnamefromthe acronymofthemainfeatures:deafness,onychodystrophy, osteodystrophy,mental retardation, andseizures (Corbett et al., 2010; Milh et al., 2013; Guven and Tolun, 2010; Rehmanetal.,2014;Campeauetal.,2014;Straˇziˇsaretal., 2014).
In the present paper, we report on two brothers presenting withearly-onset myoclonicepilepsywith spon-taneous and reflex myoclonia predominant in the face, frequentmyoclonicstatus,moderatedevelopmentaldelay, andnormalinterictalEEG,orshowingonlyfewparoxysmal abnormalities.Onthebasisofthisparticularphenotype,we performedfocusedgeneticanalysistoTBC1D24.
Patients
and
methods
Patients
The parents (I-1 and I-2) were healthy first cousins of Algerian origin. There wasno additionalfamily historyof epilepsyor other neurodevelopmental disorders.The kin-ship is composed by twoaffected (II-2 and II-3) boys and twohealthygirls(II-1andII-4).Thepedigreeofthefamily isshowninFig.1A.
PatientII-2
This 10-year-old boy was born in Algeria, at term, by C-section.Birthparameterswerenormal.Epilepsystartedat 24h oflifewithfrequent episodesofrapideyelid myoclo-nia. Neurological examination revealed global hypertonia
andincessantcrying.Hehaddailymyoclonicseizures involv-ingtheface andthe eyelids,sometimes extending tothe limbsor thewholebody.Therewasnoobvious precipitat-ingfactor.Myocloniaoccurreddaily,inclustersorinstatus episodes,refractorytovalproate,carbamazepine, lamotrig-ine,phenobarbital,andpartiallycontrolledwithphenytoin. Prolonged seizures could only be stopped by intravenous clonazepam.RepeatedEEGrecordingsshowedapreserved background rhythm and multiple diffuse spikes and slow waves.CerebralMRI,attheageof11months,wasnormal. Whenhewasfirstreferredtoourneuropediatricsunit, at age of 3 years and 8 months, he had daily myoclonic seizures withvariabletopographyand weekly episodesof generalizedmyoclonicseizures,without loss of conscious-ness,whichlastedseveralhoursandrequiredhighdosesof benzodiazepines.OneEEGrecording,severalhoursaftera prolongedmyoclonicseizurethatstartedintheleftupper limb,showedabasic thetarhythmof 6—7Hz.Therewere neither light-sensitive seizures nor EEG photoparoxysmal response.Noictal EEGcouldbeperformed. He had mod-eratedevelopmentaldelaywithoutepisodesofregression. Sittingwasachievedat9monthsandwalkingat2years.He couldsayafewsinglewords.Hecouldnoteatbyhimself. Hehadself-injuriousbehaviorandsleepdisturbance.Hehad microcephaly(48cm,−2.5SD)andalargebulbousnosewith flatnasalroot.Therewerenofingerabnormalities(Fig.2). At6yearsand10months,hehadacardiac arrest dur-inganEpstein—Barrvirus infection,followedbyprolonged myoclonicstatus(treatedwithclonazepam,phenobarbital and phenytoin) that led to anoxic—ischemic neurological sequelae. Brain MRI, one month later, showed hyper-intensity of the lentiform nuclei, moderate ventricular dilatation, and white matter rarefaction, on the T2 and FLAIRsequences.Auditoryandvisualevokedpotentialsand electroretinogramwerenormal.Metabolicscreening includ-inglactate/pyruvate,bloodandurinaryaminoacid,creatine and guanidoacetate, transferrin electrophoresis, cerebral spinal fluid analysis, and mitochondrial respiratory chain analysisinmusclewere normal.Array-CGHandmolecular screeningofPOLGgenedidnotshowanyanomaly.
At last follow-up, the control of myoclonic seizures wasmostlyachievedwithphenytoin,levetiracetam, topira-mate,clonazepam,andvitamins(B1,B2,B6,andbiotin). Myoclonic status persisted only during febrile episodes. Weobserveddailyerraticmyoclonia,triggeredbyacoustic stimuli.Thepatienthadspastictetraparesisandmoderate
Figure1 Pedigreeandgeneticfindings.(A)Pedigreeofthepresentfamily.m=mutation,—=normalallele.(B)Electropherogram showingthec.457G>Amutation(arrow)inpatientII-2(homozygous)andthemother(heterozygous).(C)Localalignmentofthe TBC1D24proteinsequenceamongdifferentspeciesshowinghighconservationofthemutatedglutamine.
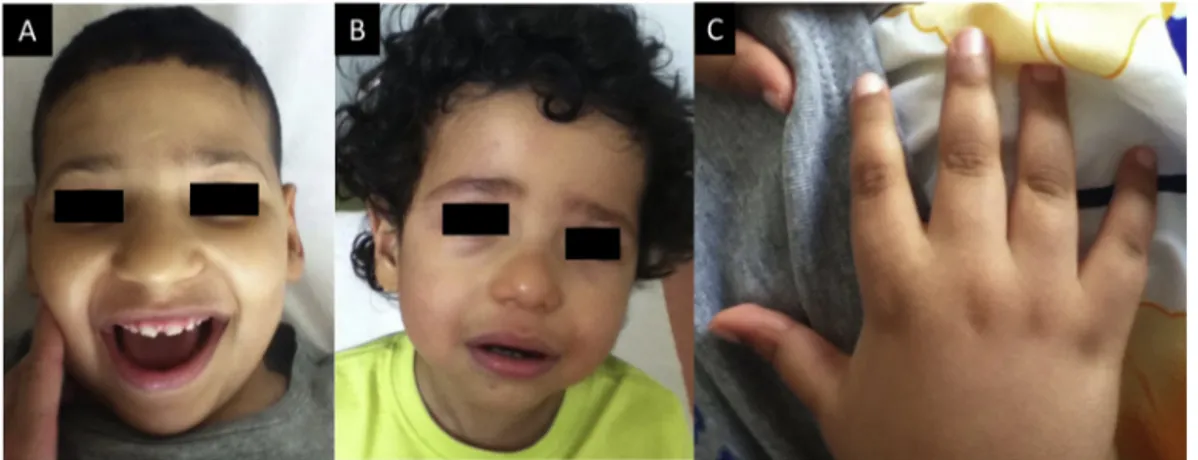
Figure2 MorphologicalfeaturesofpatientII-2andII-3.(A,B)FlatnasalrootandbulbousnasaltipinpatientsII-2(A)andII-3 (B).PatientII-3alsohasdown-slantingpalpebralfissures.(C)RighthandofpatientII-2withnormalfingersandnails.
intellectual disability. Hehad noverballanguage buthad minimalsocialinteractionwithayes—nocode.EEGshowed amoderateandbilateralslowingofthebackgroundrhythm, 6-Hzthetaandslowspikesonthevertex,aswellasspike andwavesincentralregions.
PatientII-3
This 3.5-year-old male patient was born in France at 37 weeksof gestation,byC-section. Birth weightwas3140g (mean), height was 52cm (mean), and head circumfer-ence was 35.5cm (mean). Hyperexcitability was noticed
soonafterbirth,characterizedbyepisodesofopistothonus, startles, and incessant crying. Neonatal EEGwas normal. Myoclonicepilepsystartedat2.5monthsoflife.Thepatient had long-lastingdiffuse or generalized myoclonicseizures anddailyfocalmyoclonicepisodesofvariabletopography, involving the eyelids, the peri-oral region, or the whole face,withoutlossofconsciousness.Facialmyocloniawere increasedbyfeeding. Myoclonicseizures occurredin clus-ters, lasting from a few minutes to several hours, with seizure-freeintervalsnotlongerthan15days,exacerbated byfebrileepisodes.InterictalEEGrecordingswerenormalat ageof2.5and3months.A24h-EEGrecording,performedat 4monthsoflifefailedtorecordastatus.Anictalrecording
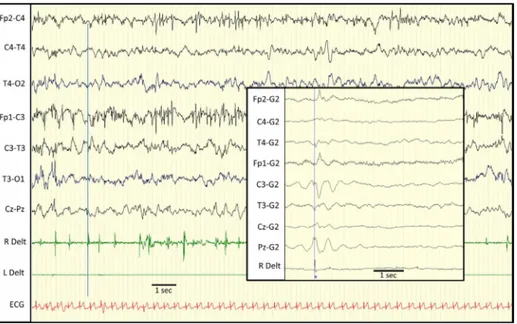
Figure3 EEGrecordingofpatient2.Myoclonicpartialstatusinvolvingpredominantlytherightarm(RDelt:rightdeltoid).Some ofthemyoclonicjerksaretimelockedtolowvoltagevertexspikesandwavesseenonvertex(artifactsinfrontalcentralregions). Framedtraces(rightpanel):EEGback-averagingof196rightdeltoidmyoclonicjerksshowsthatmyoclonusisprecededbyacortical potentialmaximalatC3derivation.
wasfinallyobtainedseveraldayslater:thepatienthada 20-minlongmyoclonusepisodeoftherightarm,associatedwith sporadic myocloniathatextended tothehead,the chest, andtheotherlimbs.Seizuresstoppedspontaneouslyduring sleep.IctalEEGwascharacterized by verylow amplitude spikes,mostlyonthevertex.Attheendofthestatus,left hemispheric slowwaves, predominating incentral region, increasedandwereintermixedwithrarespikesandwaves (Fig.3).Jerk-lockedbackaveragingwasperformedto con-firm thecorticalorigin ofmyoclonus (Fig.3).The patient wastreatedwithclonazepamandvitamins(B6,B1,biotin, andpyridoxalphosphate).Phenytoinwasaddedattheageof 4months,providingpartialcontrolofseizures.Sincethen, hehas been having an average of 4 myoclonic status per year.
At3.5yearsoflife,hehadfrequentspontaneousfacial myoclonia.Weightwas13.5kg(mean),heightwas91.5cm (mean), and head circumference was 51cm (+1DS). He had moderate developmental delay. He could walk with help and say 10 words. He had a bulbous nose with flat nasalroot. Hehad nohandor foot anomalyand no deaf-ness.Globalorganizationoftheinterictalwake andsleep EEGwasnormal,exceptforrareposteriorslowspikesand increasedamplitudeoftheposteriorregions,during drowsi-ness. Brain MRIs, performed at 3 and 23 months, were normal.Metabolicscreeningincludingbiotinidase,pipecolic acid,lactate/pyruvate,bloodandurinaryaminoacid, uri-naryorganicacid,andneurotransmittersdosageinCSFwere normal.
Mutationanalysis
Informed consent for genetic analyses wasobtained from theparents,accordingtotheFrenchbioethicslaw.Sanger sequencing of the eight exons and intronic junctions of theTBC1D24genewasperformedinpatientII-2(Genbank
reference sequence: NM001199107). In silico predictions ofpathogenicitywasperformedusingtheAlamutSoftware (Interactive biosoftware, Mont Saint-Aignan, France). The segregationof themutation withthe phenotype was performedinDNAfromtheparentsandsiblings.
Results
The c.457G>A missense substitution, located in exon 2, wasfound at the homozygous state in patient II-2 and in patientII-3 (Fig.1B).This substitutionledtothe replace-mentof aglutamate, highly-conserved throughevolution, to a lysine, p.Glu153Lys (Fig. 1C). It was predicted to be diseases causing by SIFT (score: 0) and Polyphen-2 (score:1)softwares.Theparentsandthetwosisterswere heterozygouscarriersofthemutation(Fig.1A).This muta-tion has been reported in a single heterozygous carrier (1/6437 genotypes)in the ExomeVariant server database (http://evs.gs.washington.edu/EVS/) and in 10 individ-uals from the Exome Aggregation Consortium database (http://exac.broadinstitute.org/),whichgivesafrequency around10−5inthecontrolpopulation.
Discussion
The two brothers reported here had an unusual epilep-tic disease characterized by very early onset of focal myoclonic fits, lasting a few minutes to several hours, withoutlossofconsciousnessthatregularlyevolvedto gen-eralized myoclonus or myoclonic status. In patient II-3, facial myoclonia were triggered by feeding. In contrast, neurologicaloutcomewasmarkedbymoderateintellectual impairment withno regression, and only few paroxysmal abnormalitiesonstandardinterictalEEGs.Themoresevere neurologicalimpairmentof patientII-2wasrelatedtothe
anoxic brain injury. We could confirm the cortical origin ofmyoclonus by jerk-lockedback averaging.The electro-clinical constellation observed in these patients wasvery similartothepeculiarphenotype,describedasFIME,except formoderatecognitiveimpairment(Bergetal.,2010).
Tenyearsafter havingmappedthe FIMElocusto chro-mosome 16p13.3, mutations in the TBC1D24 gene have beenfoundbytargetednext-generationsequencing(Falace etal.,2010;deFalcoetal.,2001).Despitetherecognition ofFIMEasaspecificelectro-clinicalentityamongthe classi-ficationoftheInternationalLeagueAgainstEpilepsy(ILAE), other families witha similar phenotypehave been rarely reported.
We performed direct sequencing of TBC1D24 in our patientsbecausetheysharedasignificantnumberof pheno-menologicalaspectswiththosefromtheformerFIMEfamily, suchas early onset myoclonic epilepsy, withhigh seizure frequency andvariabletopography of myoclonicseizures, affecting hands, legs, eyelids, and eyeballs, which could evolve to diffuse myoclonia or myoclonus status. In our patients,areflexcomponentwaspresentandinterictalEEG showedonlymildparoxysmalabnormalities,asinthe origi-nalFIMEfamily(Falaceetal.,2010).
Additional mutations of TBC1D24 have been reported recently,thankstoexomesequencinginpatientswitha pri-oriunrelateddisorders.Severalpatientshadamoresevere neurologicalpresentation(Corbettetal.,2010;Milhetal., 2013;Duruetal.,2010;Straˇziˇsaretal.,2014).Corbettetal. reportedalargeconsanguineousfamilywithsevenaffected individuals, who were described as having focal seizures with prominent eye blinking, as well as facial and limb jerking,whichstartedattwomonthsofage andpersisted throughoutlife (Corbettet al.,2010;Afawi etal.,2013). Thosepatientshadmild-to-moderateintellectualdisability. EEGdata werenotreported.Other mutationsinTBC1D24 havebeen found in threefamilieswithdevastating infan-tileepilepticencephalopathy(Milhetal.,2013;Duruetal., 2010;Straˇziˇsaretal.,2014).Inthefirstone,thepatients hadlong-lastingmyoclonicseizuresthatwereexacerbated by commoninfections and that did notrespond to medi-cation,withneurologicaldeterioration(Duruetal.,2010). EEGrecordingsin theinitialstages werecharacterizedby multiplespikesthatlaterevolvedtoprogressiveslowingof backgroundactivity.Theelectro-clinicalpresentationofthe twoaffectedsistersfromthesecondfamilywasconsistent withthediagnosisofmalignantmigratingpartialseizuresof infancy(Milhetal.,2013).Inthethirdfamily,theepileptic phenotypewasverysimilartothe patients reportedhere despiteverysevereneurologicaloutcome,withearly letha-lityinonepatient(Straˇziˇsaretal.,2014).
Mutations of TBC1D24 were recently shown to be the majorcause of DOORSsyndrome (Campeau etal., 2014). Althoughourtwopatientssharesomedysmorphicfeatures withpatientsaffectedwithDOORSsyndrome,theylackthe other featuresconstitutive of thissyndrome. Avariety of seizuretypes have been observedin patients withDOORS syndrome,includinggeneralizedtonic—clonic,complex par-tial focal clonic seizures, and infantile spasm (Campeau etal., 2014).Although precise description of the epilep-ticfeatures wasnot available in that series, a myoclonic presentation was mentioned in more than 20% of the patients.
In myoclonic epilepsy related to TBC1D24 mutations, myoclonia seem to involve more frequently the face, andespeciallytheperi-orbicularandperi-oralmuscles,as well asthe upper limbs, suggesting an increased cortical excitability affecting more selectively the Rolandic area. TBC1D24 belongs to the TBC domain containing proteins, highly conserved throughevolution, and is a regulator of neuritelengthandbranching(Falaceetal.,2010;Corbett et al., 2010; Campeau et al., 2014). Highest expression levels of Tbc1d24 in mice brain tissue were seen in the somatomotor and auditory areas, and in the hippocam-palformation(Allen BrainAtlas:http://mouse.brain-map. org/experiment/show?id=69531049).MutationsinTBC1D24 mayleadtohyperexcitabilityoftheRolandicregion,which generatescorticalmyoclonus.Thep.Glu153Lysmutationis locatedwithintheTBCdomainoftheprotein,closetothe p.Asn147Hismutationthatwasfound(compound heterozy-gous with the p.Ala515Val mutation) in the former FIME family,anddistinctfromthosefoundinpatientswithmore severe phenotypes(Falaceetal.,2010).Thismaysuggest that missense mutations of TBC1D24 may have different functionalconsequencesaccordingtheirlocationonthe pro-teinsequence.Itisalsopossiblethattruncatingmutations maycauseamoreseverecourseofdisease,withhigh mor-talityrate(GuvenandTolun,2010;Straˇziˇsaretal.,2014).
Thetwopatientsfromthepresentfamilyhadan electro-clinicalpresentationconsistentwiththediagnosisofFIME. Ourpatients, however,had moresevere cognitive impair-ment and myoclonia were resistant to treatment. This observation, combinedtorecent data fromthe literature suggest that mutations in TBCD24 cause a pathological continuum,withFIMEatthe‘‘benign’’endandsevere drug-refractory encephalopathyonthe severeend. Early-onset myoclonic epilepsy with focal and generalized myoclonic seizuresisthemostcommonfeatureofthiscontinuum.
Acknowledgements
Wethankthepatientsandparentsfortheirparticipatedto thestudy.
References
Afawi, Z., Mandelstam, S., Korczyn, A.D., Kivity, S., Walid, S., Shalata, A., Oliver, K.L., Corbett, M., Gecz, J., Berkovic, S.F., Jackson,G.D.,2013. TBC1D24 mutationassociatedwith focalepilepsy,cognitiveimpairmentandadistinctive cerebro-cerebellarmalformation.EpilepsyRes.105,240—244.
Berg,A.T.,Berkovic,S.F.,Brodie,M.J.,Buchhalter,J.,Cross,J.H., vanEmdeBoas,W.,Engel,J.,French,J.,Glauser,T.A.,Mathern, G.W.,Moshé, S.L.,Nordli,D.,Plouin, P.,Scheffer,I.E.,2010.
Revisedterminologyandconceptsfororganizationofseizures andepilepsies:reportoftheILAECommissiononClassification andTerminology,2005—2009.Epilepsia51,676—685.
Campeau,P.M., Kasperaviciute,D.,Lu, J.T.,Burrage,L.C., Kim, C., Hori, M., Powell, B.R., Stewart, F., Félix, T.M.,van den Ende,J.,Wisniewska,M.,Kayserili,H.,Rump,P.,Nampoothiri, S.,Aftimos,S.,Mey,A.,Nair,L.D.,Begleiter,M.L.,DeBie,I., Meenakshi,G.,Murray,M.L.,Repetto,G.M.,Golabi,M.,Blair,E., Male,A.,Giuliano,F.,Kariminejad,A.,Newman,W.G.,Bhaskar, S.S.,Dickerson,J.E.,Kerr,B.,Banka,S.,Giltay,J.C.,Wieczorek, D., Tostevin, A., Wiszniewska, J., Cheung, S.W., Hennekam, R.C.,Gibbs,R.A.,Lee,B.H.,Sisodiya,S.M.,2014.Thegenetic
basisofDOORSsyndrome:anexome-sequencingstudy.Lancet Neurol.13,44—58.
Corbett,M.A.,Bahlo,M.,Jolly,L.,Afawi,Z.,Gardner,A.E.,Oliver, K.L.,Tan,S.,Coffey,A.,Mulley,J.C.,Dibbens,L.M.,Simri,W., Shalata,A.,Kivity,S.,Jackson,G.D.,Berkovic,S.F.,Gecz,J., 2010.Afocalepilepsyandintellectualdisabilitysyndromeisdue toamutationinTBC1D24.Am.J.Hum.Genet.87,371—375.
deFalco,F.A.,Majello,L.,Santangelo,R.,Stabile,M.,Bricarelli, F.D., Zara, F., 2001. Familial infantile myoclonic epilepsy: clinicalfeatures ina large kindred with autosomal recessive inheritance.Epilepsia42,1541—1548.
Duru, N., Iseri, S.A.U., Selcuk, N., Tolun, A., 2010. Early-onset progressivemyoclonicepilepsywithdystoniamappingto 16pter-p13.3.J.Neurogenet.24,207—215.
Falace, A., Filipello,F., La Padula, V., Vanni, N., Madia, F., De PietriTonelli,D., deFalco,F.A.,Striano,P.,Dagna Bricarelli, F., Minetti, C., Benfenati, F., Fassio, A., Zara, F., 2010.
TBC1D24, an ARF6-interacting protein, is mutated in famil-ial infantile myoclonic epilepsy. Am. J. Hum. Genet. 87, 365—370.
Guven,A.,Tolun,A.,2010.TBC1D24truncatingmutationresulting insevereneurodegeneration.J.Med.Genet.50,199—202.
Milh, M., Falace, A., Villeneuve, N., Vanni, N., Cacciagli, P., Assereto,S.,Nabbout,R.,Benfenati,F.,Zara,F.,Chabrol,B., Villard, L., Fassio, A., 2013. Novel compound heterozy-gous mutationsinTBC1D24causefamilialmalignantmigratingpartial seizuresofinfancy.Hum.Mutat.34,869—872.
Rehman,A.U.,Santos-Cortez,R.L.,Morell,R.J.,Drummond,M.C., Ito,T.,Lee,K.,Khan,A.A.,Basra,M.A.,Wasif,N.,Ayub,M.,Ali, R.A.,Raza,S.I.,UniversityofWashingtonCenterforMendelian Genomics,Nickerson,D.A.,Shendure,J.,Bamshad,M., Riazud-din,S.,Billington,N.,Khan,S.N.,Friedman,P.L.,Griffith,A.J., Ahmad, W., Riazuddin, S.,Leal, S.M., Friedman, T.B., 2014.
Mutations in TBC1D24, a gene associated withepilepsy, also causenonsyndromicdeafnessDFNB86.Am.J.Hum.Genet.94, 144—152.
Straˇziˇsar,B.G., Neubauer,D., Paro Panjan, D., Writzl,K., 2014.
Early-onset epilepticencephalopathywithhearinglossintwo siblingswithTBC1D24recessivemutations.Eur.J.Paediatr. Neu-rol.S1090-3798,212—218.