appliquer Heading 7 au texte que vous souhaitez faire apparaître ici.
Département de
Génie de Procédés
Rapport de soutenance
En vue de l’obtention du diplôme
de Licence professionnalisant en :
Génie Chimique
Thème :
Réalisé par :
Melle BELHAOUA nour elhouda
Devant le jury composé de :
Pr LATRACHE Président
Mme BENHOURIA Assia Examinatrice Mme DAIRI Nassima EncadreurTuteur de l’entreprise :
M R HELLAL sofiane Chef de service production
A laboratoire merinal
Année Universitaire 2017/2018
Fabrication et contrôle de qualité d’un
médicament : « Ranitidine MABO »
Avant tout, je remercie ALLAH le tout puissant qui m’a donnée le
courage, la volonté et la patience pour réaliser ce travail.
Nos vifs remerciements s’adressent à notre promotrice Mme. N.
DAIRI pour la prise en charge de la réalisation de ce travail, pour sa
disponibilité, pour son aide et ses précieux conseils.
Un grand merci à M
r.
HELLAL, mon tuteur au niveau des
laboratoires MERINAL pour sa disponibilité, son aide précieuse et
son soutien tout au long de ce travail.
Je voudrais adresser mes vifs remerciements aux membres du jury qui
ont accepté de juger ce modeste travail.
Tous les enseignants de l’Institut de Technologie, et les personnes de
l’administration
Je remercier profondément tous les étudiants de la promotion ainsi le
staff administratif
C’est avec toute l’ardeur de mes sentiments que je dédie ce
modeste travail à :
Mes très chers parents, qui m’ont soutenu, encouragé pour
que je puisse mener à bien mes études, et qui ont attendu ce
jour avec impatience.
Mes chères sœurs et mes belles sœurs
Familles : BELHAOUA et CHAMI
Mes enseignants et mes amis
Sommaire
Introduction………..…....1
Chapitre I Partie théorique I.1. Généralités sur les médicamentes …………... ...2
I.1.1.Définition d’un médicament………... ..2
I.1.2. Composition des médicaments………..……...2
I.1.2.1. Définition d’un principe actif ……….……...2
I.1.2.2. Excipient ……….….2
I.1.2.3. Pelliculage………..……...3
I.2. Contrôle de qualité des médicaments……….….3
I.2.1. Définition de la qualité ………...…….3
I.2.2.Définition du contrôle ………...…..3
I.2.3. Objectif du contrôle de qualité ………...…3
I.2.4. Différents types de contrôle de qualité ……….…..…4
I.2.4.1 Contrôle Physico-chimique ………...……...4
I.2.4.1.1..Friabilité ……….……....4
I.2.4.1.2. Dureté ……….…...4
I.2.4.2. Contrôle Microbiologique ………..…...…4
I.2.4 .3. Contrôle Toxicologique………...…….4
Chapitre II Fabrication et contrôle de qualité de Ranitidine MABO II.1.Présentation des laboratoires MERINAL ……….………..….5
II.1.1. Historique………..………..….5
II.1.2. Laboratoire Contrôle de qualité du médicament ………...…...6
II.2. Identification du médicament……….………..………….….6
II.2.1. Mode et voie d’administration………..………....6
II.2.3.1.Définition ………..7
II.2.3.2. Structure chimiques………...7
II.2.4. Les excipients de Ranitidine MABO ………..7
II.2.4.1. Cellulose microcristalline………7
II.2.4.2. Stéarate magnésium ………..…..…..7
II.3. Procèdes de fabrication du « RANITIDINE MABO » ……….………8
II.3.1. Peser et mélanger ……….8
II.3.2. Compression ………...9
II.3.3. Pelliculage………10
II.3.3.1. Premier pelliculage avec Sepifilm LP030. ………...11
II.3.3.2. Deuxième pelliculage avec Sepifilm LP770……….…………..13
II.3.4. Conditionnement ………14
II.3.4.1. Conditionnement primaire ……….……….14
II.3.4.2. Conditionnement secondaire ……….………….15
II.4. Contrôle de qualité………...16
II.4.1. Contrôle physico-chimique……….….16
II.4.1.1 Matières premières……….………..16
II.4.1.2. Produit au cours de fabrication ………..…….16
II.4.1.3. Produits finis……….…..….19
Chapitre III Résultats et discussion III.1.Résultats des tests en cours de fabrication ………..…….24
III.2.Résultats des tests (produit fini)……….……..27
Conclusion ………..… 30 Références
Figure II.1 : Historique des laboratoires MERINAL Figure II.2 : Etui de la Ranitidine MABO
Figure II.3 : Formule chimique du principe actif
Figure II.4 : Etapes de fabrication de la Ranitidine MABO Figure II.5 : BIN de 300L
Figure II.6 : Mélangeur
Figure II.7: Salle de compression Figure II.8 : Tourelle de la compression Figure II.9 : Salle de pelliculage
Figure II.10 : Pistoles de pulvérisation
Figure II.11 : Conditionnement primaire (blistéreuse) Figure II.12 : Conditionnement secondaire (encartonneuse)
Figure II.13 : Tapie roulon de sortir les boites de RANITIDINE MABO Figure II.14: Balance de précision avec imprimante
Figure II.15: Désagrégateur Figure II.16 : Friabilimètre Figure II.17: Duromètre
Figure II.18 : Karl Fisher (appareil de mesure teneur en eau ) Figure II.19 : Dissolutest
Figure II.20 : Plaque CCM
Figure II.21 : Plaque CCM après la révélation Figure II.22: Lampe UV
Tableau II.1: Matière premières utilisée selon l’ordre Tableau II.2 : Conditions de chauffage
Tableau II.3 Conditions de la 1ére étape de pulvérisation Tableau II.4 : Conditions de la 2éme étape de pulvérisation
Tableau II.5 : Conditions du premier séchage Tableau II.6 : Conditions du 2éme séchage Tableau II.7: Conditions de refroidissement
Tableau III.1 : Résultats des tests au cours de fabrication Tableau III.2 : Résultats des tests des produits finis
UICPA : Union international de Chimie Pure et Appliquée
PE : Pharmacopée EuropéenneCCM : Chromatographie sur Couche Mince OMS : Organisation Mondiale de la Santé PA : Principe Actif
Introduction
2
Introduction
L’industrie pharmaceutique est de nos jours une industrie florissante et importante tant du point de vue de l’innovation que du business. L'enjeu au niveau de la santé publique que représente la production de médicaments nécessite de nombreuses réglementations strictes et contraignantes qui ont pour préoccupations premières d’assurer la qualité, la sureté, l'efficacité des produits et la satisfaction des clients et des consommateurs. C’est pourquoi les industriels n’ont cessé d’améliorer la qualité de leurs services au fil des temps [1].
La qualité a pris une importance considérable au cours de l’histoire dans l’industrie, à tel point que des outils spécifiques ont été créés pour permettre son management et son amélioration continue. Les entreprises pharmaceutiques notamment, doivent se conformer à un système d’assurance qualité strict pour être aux normes et répondre aux exigences en matière de qualité, de sécurité et d’efficacité [1].
Ce travail a été réalisé dans le cadre de la fabrication et du contrôle de qualité d’un médicament « RANITIDINE MABO ». Pour mener cette étude, le plan de travail suivant a été adopté :
Le premier chapitre contient deux parties, la première présente des généralités sur les médicaments. La seconde est consacrée aux contrôles pharmaceutiques.
Dans le deuxième chapitre, nous présentons la partie expérimentale qui contient quatre points :
Présentation des laboratoires MERINA ; Identification du médicament ;
Procèdes de fabrication ; Contrôle de qualité.
Le troisième chapitre est réservé aux « résultats et discussion » des tests effectués sur les différents produits.
2
I.1.
Généralités sur les médicamentes
L’industrie pharmaceutique algérienne est confortée à la nécessité de se mettre au diapason de l’évolution des exigences internationales en matière de recherche et de développement de leurs objectifs, la fabrication de médicament de dernière génération capable de prendre en cherche les pathologies les plus fréquentes, et ce à moindre cout, tout en respectant les critères d’efficacité et de qualité, de sécurité et de tolérance [2].
I.1.1 Définition d’un médicament
Un médicament c’est toute substance utilisée pour prévenir, atténuer, ou guérir une maladie ou ses symptômes [2].
Toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l’homme ou chez l’animal, ou pouvant leur être administrée en vue d’établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique [2].
I.1.2 Composition des médicaments
Un médicament se compose d’un ou de plusieurs principes actifs et d’excipients [2].
I.1.2.1 Définition d’un principe actif
Le principe actif d’un médicament est une substance d’origine chimique ou naturelle caractérisée par un mécanisme d’action curatif ou préventif précis dans l’organisme [2]. Est une substance active douée de propriétés pharmacologiques, et est donc à la base de l’effet thérapeutique [2].
I.1.2.2 Excipient
Tous les éléments entrant dans la composition d’un médicament, autres que le principe actif, sont généralement des excipients [3].
Chapitre I Partie théorique
3
L’excipient est une substance d’origine chimique ou naturelle qui facilitent l’utilisation du médicament mais ne présente pas d’effet curatif ou préventif.
Les excipients sont classés en plusieurs catégories à savoir : Les diluants
Ils jouent un rôle de remplissage lorsque la quantité du PA est insuffisante pour faire un Cp de taille convenable [3].
Les liants ou agglutinants
Leur rôle est de lier entre les. Leur présence permet de réduire la force de compression [3].
Les lubrifiants
Ils jouent un triple rôle dans la fabrication des Cp :
– Amélioration de la fluidité du grain, donc du remplissage de la chambre de compression, qui est important pour la régularité du poids (pouvoir glissant),
– Diminution de l’adhérence du grain aux poinçons et à la matrice (pouvoir anti adhèrent),
– Réduction des frictions entre les particules pendant la compression, ce qui assure une meilleure transmission de la force de compression dans la masse du grain (pouvoir anti-friction).
A ces trois rôles importants vient s’ajouter un intérêt supplémentaire des lubrifiants : ils donnent un bel aspect, brillant et non poussiéreux aux Cp [3].
I.1.2.2 Pelliculage
Cette étape consiste à enrober un matériau support (comprimé) par une fine couche d’agents filmogènes.
I.2 Contrôle de qualité des médicaments
L’organisation mondiale de la santé (OMS) s’occupe non seulement des aspects pharmaceutiques de la qualité des médicaments mais encore de l’innocuité et de l’efficacité intrinsèque de leurs principes actifs [2].
4
I.2.1 Définition de la qualité
La qualité est : « l’ensemble des caractéristiques d’une entité qui lui confèrent l’aptitude à satisfaire des besoins exprimés et implicites [2].
I.2.2 Définition du contrôle
Le mot contrôle peut-être utilisé dans le sens de vérification ou dans celui de maitrise. Le contrôle à mesurer une ou plusieurs caractéristiques d’une entité et à comparer les résultats obtenus à des spécifications préétablies.
I.2.3 Objectif du contrôle de qualité
Le contrôle de qualité consiste à déceler les erreurs dépassants les limites jugées raisonnables, de manière à en corriger les causes ou à les prévenir [2].
I.2.4 Différents types du contrôle de qualité
Le contrôle de qualité des médicaments est divisé en : I.2.4.1 Contrôle Physico-chimique
Il aura pour rôle de vérifier la structure de la molécule et d’établir les propriétés physiques et chimiques. Il a pour but de vérifier la substance annoncée (analyses qualitatives, réaction d’identification les plus sélectives possibles) et s’assurer de son bon usage [2].
Dureté
Le test de dureté permet de s’assurer que les Cp présentent une résistance mécanique suffisante pour ne pas se briser lors de leurs manipulations ou d’étapes de production ultérieures [3].
Friabilité
Le test de friabilité permet de s’assurer que les Cp présentent une résistance mécanique suffisante, pour que leurs surfaces ne soient pas endommagées ou ne présentent pas des signes d’abrasion ou de rupture, sous l’effet de toutes les manipulations (chocs mécaniques, frottements, attrition) qu’ils vont subir jusqu’au moment de leur utilisation [3].
Chapitre I Partie théorique
5
Uniformité de masse
L’essai d’uniformité de masse des Cp permet de s’assurer qu’au cours de la fabrication, la répartition du mélange initial de poudre ou de granulés, en unités de prises (chaque Cp), a été suffisamment précise et uniforme pour garantir une même masse et donc une même teneur en PA pour l’ensemble des Cp d’un même lot [3].
Désagrégation
Le test de désagrégation des Cp permet de s’assurer, que leur vitesse de désagrégation ne constitue pas le facteur limitant de la dissolution du PA qu’ils contiennent [3].
Dissolution
Le test de dissolution in vitro appliqué aux Cp permet de s’assurer, qu’une fois administrés, ces derniers libèreront le PA qu’ils contiennent pour le mettre à la disposition de l’organisme, et ceci dans les limites de concentration et de vitesse déterminées, afin de garantir l’effet thérapeutique désiré [3].
Test d’identification du PA
L’identification d’un PA contenu dans un Cp, a pour but de s’assurer que ce dernier contient bel et bien le PA spécifié par le fabricant.
Les méthodes d’identification du PA les plus citées parles pharmacopées sont : → Spectrophotométrie d’absorption dans l’infrarouge ;
→ Spectrophotométrie d’absorption dans l’ultraviolet et le visible ; → Chromatographie liquide à haute performance (HPLC) ;
→ Chromatographie sur couche mince ;
→ Réactions chimiques caractéristiques du PA (réactions colorées en tube par exemple) [3]. I.2.4.2 Contrôle Microbiologique
Les contrôles microbiologiques doivent permettre de garantir une bonne qualité hygiénique et marchande du produit fabriqué, et minimisent les pertes dues aux mauvaises conditions de fabrication [2].
6
I.2.4.3 Contrôle Toxicologique
Les molécules destinées à la thérapeutique humaine doivent subir avant tout essai clinique des tests de toxicité aiguë et chronique sur les animaux [2].
Les études toxicologiques permettent d’éliminer de très nombreuses molécules dont les risques outrepassent les avantages [2].
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
7
Ce travail a été réalisé aux laboratoires MERINAL d’Oued SMAR, durant trois (03) mois (mars, avril, mai) de l’année 2018. L’objectif était de suivre les étapes de fabrication d’un médicament et également de contrôler sa qualité.
II.1. Présentation des laboratoires MERINAL
II.1.1. Historique
A l’instar de beaucoup de laboratoires pharmaceutiques dans le monde, c’est d’une pharmacie d’officine fondée en 1969, qui est née l’idée de créer les laboratoires MERINAL. Dès le démarrage de l’unité de production en 2002, MERINAL a opté pour une stratégie de développement axée sur le générique et sur la constitution d’une gamme de produits appartenant à MERINAL. En effet l'objectif souhaité est non seulement d'être en mesure de maîtriser ses activités et son développement, mais aussi de se projeter au-delà du marché algérien. Ainsi, MERINAL met sur le marché annuellement plus de 50 millions d'unités de vente, tout en élargissant sa présence à l’international [4].
8
II.1.2. Laboratoire Contrôle de qualité du médicament
Les Laboratoires MERINAL sont équipés d’un laboratoire de contrôle qualité (physico-chimiques et microbiologique), validé par le Laboratoire National de Contrôle des Produits Pharmaceutiques « LNCPP ». Des contrôles sont effectués en amont, au cours ainsi qu’en fin de procès, selon des procédures validées et approuvées [4].
II.2. Identification du médicament
Le produit choisi pour notre stage est un comprimé pelliculé : une boite de 30 ou 60 comprimés portant l’inscription Ranitidine MABO comme le montre la photo ci-dessous.
Figure 2 : Etui de la Ranitidine
II.2.1. Mode et voie d’administration
Voie orale, les comprimés doivent être avalés tels quels avec de l’eau. Il peut être pris au cours ou en dehors des repas [5].
II.2.2. Effets thérapeutiques de la Ranitidine MABO Ce médicament est indiqué dans :
L’ulcère gastrique ou duodénal évolutif ; L’œsophagite par reflux gastro-œsophagien ; Traitement d’entretien de l’ulcère duodénal ;
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
9
II.2.3. PA de la Ranitidine MABO II.2.3.1. Définition
Ranitidine chlorhydrate
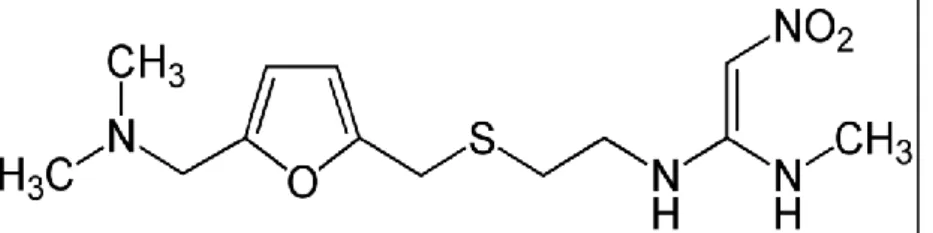
Le pourcentage du PA qui se trouve dans un comprimé est de 150 mg [5]. II.2.3.2. Structure chimique
Formule brute : C13H23CIN4O3S
Nom UICPA : Chlorhydrate de N-(2-(((5-((diméthylamino) méthyl) furan-2-yl) méthyl) sulfanyl) éthyl)-N-méthyl-2-nitroéth-1-éne-1,1-diamine [5].
Formule chimique :
Figure 3: Formule chimique du principe actif
II.2.4 Les excipients de la Ranitidine MABO
Cellulose microcristalline Stéarate magnésium [5].
II.3. Procèdes de fabrication de la Ranitidine MABO
La fabrication de la
Ranitidine MABO
est basée sur quatre étapes présentées sur la figure ci dessous:Figure 4: Etapes de fabrication de la Ranitidine MABO
peser et méllanger
compression
pelliculage
conditionnement
10
II.3.1 Peser et mélanger
Le but de cette étape est de préparer le mélange constitué du principe actif et des excipients. A/ La pesée des matières premières se fait dans un BIN de 300 L qui contient un sac de gel de silice pour chasser l’humidité.
L’introduction des matières premières est réalisée selon l’ordre présenté sur le tableau 01, afin d’avoir un lot de 112.5 Kg.
Tableau01 : Matières premières utilisées selon l’ordre
Désignation Quantité introduite (Kg)
Cellulose microcristalline 20.000
Chlorhydrate de RANITIDINE compactée 62 .775
Stéarate de magnésium 1.875
Cellulose microcristalline 20.000
Cellulose microcristalline 7.850
B/ Pour bien homogénéiser le mélange des matières premières, le BIN est combiné avec le mélangeur pendant 10 min à 12 tours par minute.
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
11
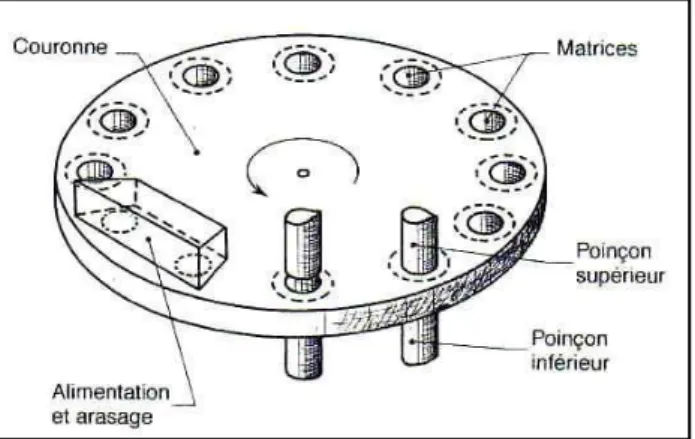
II.3.2. Compression
Dans cette étape de fabrication, le mélange préparé précédemment est transformé en comprimés.
Figure 7 : Salle de compression
Apres avoir mélangé les matières premières, le BIN est détaché du mélangeur et placé au dessus de la Compremeuse à l’aide d’un élévateur. Par la suite, le BIN sera ouvert pour que la poudre s’écoule dans le sabot d’alimentation à travers la trémie de la compresseuse, ce dernier distribue la poudre à l’aide d’une roue dans la tourelle qui contient les chambres qui seront remplis par la poudre. Par des mouvements de rotation de la tourelle, les poinçons supérieurs de diamètre de 9,5 mm pénètrent dans les chambres pour compresser la poudre, les comprimés seront conservés dans des futs de 25 Kg.
CHEKMASTER
COMPRIMEUSE PUPITRE
12
Au cours de la compression, et chaque 20 min, les comprimés seront déplacés au chekmaster qui va contrôler chaque 10 comprimé à la fois. Le chekmaster permet de déterminer la masse, l’épaisseur et la dureté de chaque comprimé.
Figure 8 : Tourelle de la compression
II.3.3. Pelliculage
Figure 9 : Salle de pelliculage
II.3.3.1. Premier pelliculage avec Sepifilm LP030
Solution de pelliculage
La solution de Sepifilm LP030 est un mélange de 130L d’eau purifiée et de 17,750 Kg de sepifilm LP030. Cette solution a pour but de protéger les comprimés contre l’humidité.
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
13
Chargement
Charger les comprimés nus de Ranitidine MABO dans la turbine de pelliculage. Chauffage
Les comprimés ont été chauffés sous les conditions données dans le tableau ci après :
Tableau 2 : Conditions de chauffage
Paramètre Valeur
Température d’entrée d’air 65°C Débit d’entrée d’air 3000 m3/h
Dépression tambour -150 Pa
L’arrêt du chauffage est déterminé par la température de sortie d’air T= 44°C Pulvérisation
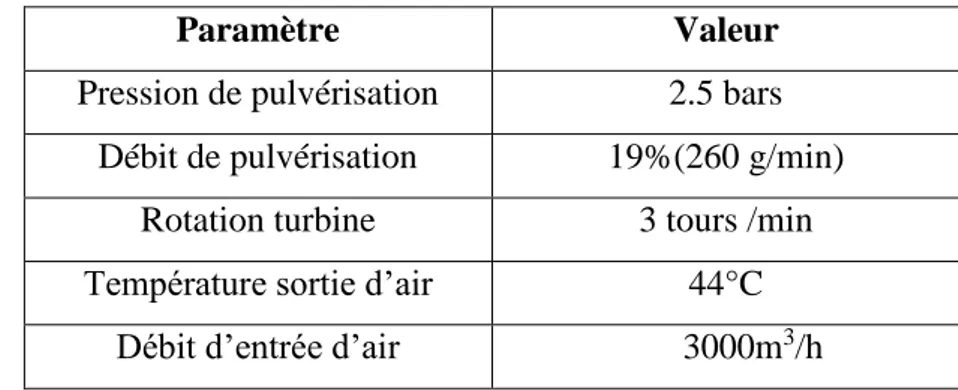
La pulvérisation est effectuée en deux étapes : 1èreétape :
Tableau 3 : Conditions de la 1ère étape de Pulvérisation
Paramètre Valeur
Pression de pulvérisation 2.5 bars Débit de pulvérisation 19%(260 g/min)
Rotation turbine 3 tours /min Température sortie d’air 44°C
Débit d’entrée d’air 3000m3/h
2 éme étape
Tableau 4 : Conditions de la 2 éme étape de Pulvérisation
paramètre Valeur
Pression de pulvérisation 3.5 bars Débit de pulvérisation 16%(180g/min)
Rotation turbine 3.5 tour/min Température sortie d’air 44°C
14
L’arrêt de la pulvérisation est déterminé par la masse du comprimé pelliculé qui est de 320 mg (300mg + 20 mg), la masse initiale du comprimé est de 300 mg.
Séchage
Le séchage s’effectue en deux étapes : 1ére étape :
Tableau5 : Conditions du 1er séchage
2ème étape : s’effectue selon les paramètres suivants :
Tableau 6 : Conditions du 2ème séchage
Refroidissement
S’effectue selon les paramètres suivants :
Tableau 7 : Conditions de refroidissement
Paramètre Valeur à introduite Température entrée d’air 10°C
Débit d’entrée d’air 3000 m3/h
L’arrêt du refroidissement est déterminé par la température de sortie d’air T=25°C.
Paramètre Valeur
Durée 01min
Température de sortie d’air 44°C Vitesse rotation tambour 5 tour /min
Débit d’air 3000 m3/h
Paramètre Valeur
Durée 5 min
Température 44°C
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
15
II.3.3.2. Deuxième pelliculage avec Sepifilm LP770
Après le premier pelliculage, les comprimés pelliculés de masse de 320 mg ont été récupérés après refroidissement pour un deuxième pelliculage.
Solution de pelliculage
La solution Sepifilm LP770 composée de 43 L d’eau purifiée et de 5 ,850 Kg de Sepifilm LP770. L’objectif de cette solution est de donner une couleur, un gout et une odeur au comprimé, et encore pour différencier le médicament des autres.
La deuxième étape de pelliculage a été faite par la solution Sepifilm LP770. Les mêmes étapes suivies dans le premier pelliculage ont été repris dans ce deuxième pelliculage.
La pulvérisation de la deuxième étape de pelliculage est déterminée par la masse du comprimé pelliculé qui du 326 mg (320 mg + 6 mg).
Remarques :
La préparation des deux solutions de pulvérisation a été faite dans une cuve en acier, sous une agitation vigoureuse durant 45min, puis à faible vitesse pendant 60 min.
La pulvérisation a été réalisée par un des pistolets déposés sur un support, la distance entre chaque pistolet est de 17 cm et entre le pistolet et le lit des comprimés est de l’ordre de 23 cm.
Figure 10 : Pistoles de pulvérisation
Vidange de la turbine et stockage des comprimés
Après le refroidissement, les comprimés pelliculés sont récupérés et stockés dans des futs tarés, étiquetés, dans des sacs en plastique à usage unique, chaque fut pèse 22 kg.
16
II.3.4.Conditionnement
Le conditionnement a été fait en étapes
II.3.4.1 Conditionnement primaire
L’objectif du Conditionnement primaire est de conserver les comprimés pelliculés dans des blistères. Cette étape est réalisée par une blistéreuse.
Figure 11 : Conditionnement primaire (Blistéreuse)
Les conditions de fonctionnement de la blistéreuse sont les suivants :
Paramètre généraux (vitesse de la machine =20-50s /m2, vitesse alimentation
produit=50-70 s/m2).
Matériau de formage (film utilisé : Alu-tropicalisé (inférieur), Alu imprimé (supérieur)
Préchauffage: T=23C°
Formage (matrice de formage)
Chargement (plaque de distribué le comprimé dans les alvéoles dans les blisters) Scellage (film supérieur est fixé au film inférieur)
Découpe (copie les blisters chacune à 10 comprimés) Sortie (rassemblement des blisters dans le magasin). II.3.4.2. Conditionnement secondaire
L’objectif du Conditionnement secondaire est de conserver les blistères dans des boites. Cette étape est réalisée par une Encartonneuse.
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
17
Figure 12: Conditionnement secondaire (Encartonneuse)
Les étapes de conditionnement secondaires sont : Magasin des blisters ;
Palpeur (nombre des blisters +l’ordre des notices et les étuis) ; Des poussoirs (poser le blister et la notice dans la boite) ; Des fermetures (fermer la boite) ;
Numérateur (N° de lot) ;
Vignetteuse (placer la vignette à l’extérieure de la boite) ; Capteur +caméra (vérification de la boite) ;
Sortie des boites et remplissage des cartons.
18
II.4. Contrôle de qualité
II.4.1 Contrôle physico-chimique
II.4.1.1 Matières premières
Les analyses sont été faites bien avant la fabrication des comprimés par les laboratoires MERINAL.
II.4.1.2 Produit au cours de fabrication A / Au cours de la compression
Au cours de la compression, les comprimés ont subi des contrôles avant l’éjection finale, ces contrôles ont été faits par le Chekmaster relié à la machine de compression.
1. Masse individuelle, masse moyenne
La masse individuelle et la masse moyenne de 10 comprimés sont déterminées chaque 20 min.
2. Epaisseur et dureté
L’épaisseur et la dureté des 10 comprimés sont déterminées chaque 30 min.
Au niveau du laboratoire de contrôle, les comprimés seront contrôlés une deuxième fois. 1. Uniformité de masse et la masse moyenne
Chaque une heure, la masse et la masse moyenne de 20 comprimés ont été déterminées en utilisant une balance connectée à une imprimante présentée sur la figure ci-dessous.
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
19
Ces analyses ont été faites chaque une heure, au début, au milieu et en fin du lot. 2. Aspect : Observation visuelle
3. Désagrégation
Dans chacun des 6 tubes, introduire un comprimé, et placer l’assemblage dans le vase cylindrique contenant de l’eau à une température de 37°C, faire fonctionner l’appareil, l’essai est satisfait si tous les échantillons sont désagrèges au temps prescrit.
Figure 15:Désagrégateur 4. Friabilité
Entrer20 comprimés de masse initial mi dans un friabilimètre et régler l’appareil à 100 tours
pondant 4 min, la masse finale mf des comprimés a été calculée.
Figure16 : Friabilimètre
Le pourcentage de friabilité est calculé par la formule suivante :
% (friabilité) = m f
20
5. Dureté
Le comprimé est placé à plat. Deux mâchoires se faisant face et se déplaçant l'une vers L’autre va exercer une pression constante sur le comprimé jusqu'à sa cassure. C'est cette force qui va être mesurée Kilo Pound et qui va exprimer la dureté.
Figure 17 : Duromètre
B/ Au cours de pelliculage
Chaque 20 min, contrôler la masse moyenne des 100 comprimes tout au long du procédé de pelliculage.
C/ Au cours de conditionnement
1. Contrôle du conditionnement primaire
Contrôler l’état général de blister : centrage des alvéoles, n° de lot, date de péremption, centrage aluminium, nombre de comprimé par blister.
Teste d’étanchéité
Ce test a pour objectif de contrôler la pénétration de l’air et de l’eau à l’intérieur des blistères.
Ce test consiste à mettre 6 blisters dans la solution bleue de méthylène et régler la pression p = 0.2 b et le temps à une minute.
2. Contrôle du conditionnement secondaire
Contrôler l’état général de la boite : 3 blisters (30 comprimés), ou bien 6 blisters (60 comprimés), la notice, N° de lot, la date de préparation et péremption, la vignette.
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
21
II.4.1.3. Produits finis 1. Aspect
Observation visuelle
2. Masse moyenne et uniformité de masse
Peser individuellement 20 comprimés et déterminer la masse moyenne (Mm) par la formule
ci-dessous :
Mm=∑i=20 mi / 20
L’uniformité de masse (UM) par la loi suivante : UM=MM ± 5% (18/20), UM=MM ± 10% (20/20) 3. Désagrégation
Le mode opératoire est cité préalablement. 4. Teneur en eau
Réactifs : hydranal titrant, hydranal solvant
La teneur en eau est déterminée sur une prise d’essai de 200 mg.
22
5. Dissolution Solutions à préparer Solution standard
- Peser et transférer une prise d’essai de 18 ,6mg de Ranitidine HCL (substance de référence) standard dans une fiole de 100 ml d’eau. (concentration de la Ranitidine = 0,186 mg/ml).
- Diluer 1ml de cette solution dans 10 ml d’eau. Solution de l’échantillon
- Remplir les 6 cloches par 900 ml d’eau dans chacune. - La température de cette dernière doit atteindre 37°C. - Régler la vitesse de rotation des palettes à 50 tr/ min.
- Placer 1 comprimé dans chacune des 6 cloches, les comprimés doivent être au fond des cloches.
- Au bout de 45 mn de dissolution, prélever 15ml dans une zone située à mi-distance de la surface du milieu de dissolution et le haut de la palette, et à moins 1 cm de la paroi du récipient en utilisant des seringues individuelles.
- Filtrer les solutions avec un filtre PVDF 0,45µm, laisser refroidir le filtrat. - Diluer 1 ml du filtrat dans 10 ml d’eau.
- Utiliser une cuve de 1cm en quartz.
- Mesurer l’absorbance de la solution standard et des solutions échantillons à 314 nm en utilisant l’eau purifiée comme blanc.
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
23
Le pourcentage du principe actif dans le comprimé est calculé par l’opérateur des laboratoires MERINAL.
6. Identification et dosage des substances apparentes Substance de référence : Ranitidine HCL, Imp A - Plaque CCM de silicagel F254 (20 × 20)
- Saturation de la cuve avec la phase mobile - Volume de dépôt : 10 µl
- Parcours : 15 cm Solutions à préparer
Phase mobile
- Mélanger 50 ml d’acétate d’éthyle, 30ml d’Isopropanol, 10 ml d’hydroxyde d’ammonium et 2ml d’eau.
Solution standard de résolution
- Dans une fiole de 10 ml, dissoudre une prise d’essai de 12.7 mg de la Ranitidine impureté A dans du méthanol.
- Agiter pendant 5 min (concentration en Ranitidine impureté A = 1.27 mg/ml). Solution essai
- Préparer une solution de la Ranitidine en broyant 20 comprimés traités de la même façon que pour le dosage moyen (eau purifiée).
- Peser une quantité de poudre équivalente à 200 mg de la Ranitidine base dans une fiole de 10 ml.
- Dissoudre avec 8 ml de méthanol ;
- Agiter pendant 10 min, compléter au volume avec du méthanol ; - Filtrer cette solution et utiliser le filtrat comme solution essai.
Solution standard
- Peser et transférer 22 mg de la Ranitidine HCL standard dans une fiole de 100 ml.
- Dissoudre et compléter au volume avec du méthanol (concentration en Ranitidine = 0.20 mg/ml).
24
Solution standard diluée A
- Diluer 5ml de la solution standard dans 10 ml de méthanol (concentration en Ranitidine = 0.1 mg/ml) ;
Solution standard diluée B
- Diluer 6 ml de la solution standard dans 20 ml de méthanol (concentration en Ranitidine =0.06 mg /ml)
Solution standard diluée C
- Diluer 1ml de la solution standard dans 10 ml de méthanol (concentration en Ranitidine =0.02mg/ml).
Solution standard diluée D
- Diluer 1ml de la solution standard diluée A dans 10 ml de méthanol (concentration en Ranitidine =0.01 mg/ml)
- Déposer séparément sur une plaque CCM 10 µl de chacune des solutions suivantes : solution essai, solution standard et les solutions diluées A, B, C, D.
- Déposer également sur un même point (différent des points précédemment cités), 10 µl de la solution essai et 10µl de la solution de résolution (tache combinée)
- Laisser sécher les taches puis mettre la plaque dans la cuve à CCM.
- Après une migration de 15 cm environ, retirer la plaque et laisser la sécher à l’aire à siccité.
Les limites d’acceptation : l’analyse est considérée correcte :
S’il existe une séparation complète entre les taches principales du chromatogramme combiné c’est –à-dire entre la solution essai et la solution de résolution
Si dans le chromatogramme de la solution standard diluée d’une tache est observée. Pour déterminer la pureté de l’échantillon, comparer toutes les taches secondaires
possibles obtenues avec la solution à évaluer avec les taches principales obtenues avec la solution standard diluée A, B, C, D.
Aucune tache secondaire n’est plus intense que la tâche principale obtenue avec la solution témoin diluée A (0.5%)
Aucune tache secondaire éventuelle n’est plus intense que la tâche principale obtenue avec la solution diluée B (0.3%).
Chapitre II Fabrication et contrôle de qualité de la Ranitidine MABO
25
Figure 21 : Cuve à élution Figure22 : Plaque CCM après révélation
26
III.1. Résultats des tests au cours de fabrication Au cours de la compression
La masse individuelle et moyenne des comprimés
Tableau10 : Résultats des tests au cours de fabrication
Test Résultat Norme Conformité
Masse individuelle (mg) 292 297 301 291 306 302 305 298 307 299 300 ±5% [285-312] conforme Masse moyenne (mg) 299 ,8 300 ± 3% [295-309] conforme Epaisseur (mm) 4,5 4,8 4,4 4,7 4 ,5 4,8 4,7 4,6 4,4 4,7 [4,4 –4,8] conforme Dureté (kp) 9 5 6 7 8 10 7 5 8 9 [5kp -10 kp] Conforme Aspect
comprimé rond, biconvexe de couleur blanche crémeuse. Comprimé rond, biconvexe de couleur blanche crémeuse. conforme
Désagrégation (min) 28 ≤ 30 conforme
Friabilité m f=5995, m i=6000 % (friabilité)=𝟓𝟗𝟗𝟓 𝟔𝟎𝟎𝟎
=0,99
≤ 1% conformeDiscussion
III.1.1.Masse individuelleLes résultats de la masse individuelle sont conformes à la norme. III.1.2. Masse moyenne
Pour la masse moyenne, le résultat trouvé est de 299,8 mg, il est conforme à la norme [295-309] mg.
Chapitre III Résultats et discussion
27
III.1.3.Epaisseur
Nous notons que les valeurs obtenus sont tous concluantes ; l’épaisseur des 20 comprimés se situe dans l’intervalle recommandé et ne dépasse pas la norme [4,4 - 4,8] mm
III.1.4. Dureté
D’après le résultat obtenu, on observe que les valeurs de la dureté des 10 comprimés se situent dans l’intervalle correspondant aux normes [5 - 10] KP. Donc la dureté est conforme aux normes de la Pharmacopée Européenne.
III.1.5. Aspect
Les comprimés nus (non pelliculés) sont de couleur blanche. Un résultat qui conforme aux normes.
III.1.6. Désagrégation
Nous avons constaté qu’au bout de 28 min aucun résidu n’est présent dans le bécher, donc le résultat est concluant et répond aux normes de la PE.
III.1.7. Friabilité
D’après le résultat obtenu, on remarque que la friabilité est inférieure 1% (≤ 1%), le résultat est conforme.
28
III.2. Résultats des tests (produit fini)
Tableau 11 : Résultats des tests produit fini
Test Résultat Norme Conformité
Aspect Comprimé pelliculé rond, biconvexe de
couleur blanche crémeuse.
Comprimé pelliculé rond, biconvexe de couleur blanche crémeuse. conforme Masse moyenne (mg) 310 315 ± 5% [229 – 331] conforme
Teneur en eau (%) 4 ≤ 5% conforme
Désagrégation (min) 28 ≤ 30 conforme
Discussion
III.2.1.AspectOn n’observe que les comprimés pelliculés de couleur blanche crémeuse. Un résultat qui conforme aux normes
III.2.2.Masse moyenne
Pour la masse moyenne Le résultat calculé est 310 mg, donc est conforme à la norme [229 – 331] mg.
III.2.3.Teneur en eau
Le pourcentage d’eau est de 4%, une valeur inférieure à 5%, donc le résultat est confirme aux normes de la PE.
III.2.4.Désagrégation
On a constaté qu’au bout de 28 min aucun résidu n’est présent dans le bécher, donc le résultat est concluant et répond aux normes de la PE.
Chapitre III Résultats et discussion
29
III.2.5. CCM
Le résultat de la chromatographie sur couche mince montre que toutes les solutions analysées sont pures, car on observe une seul tache sur la même ligne. Ceci traduit la pureté des solutions du principe actif de la Ranitidine MABO.
30
Ce rapport de stage a pour objectif de répondre à la question d’étude, ’comment fabriquer un médicament comprimé pelliculé et quel est le contrôle effectué pour l’assurance de sa qualité ?’.
Le suivi de la fabrication nous a permis de mettre le point sur toutes les étapes de fabrication de la Ranitidine MABO, d’acquérir une bonne connaissance sur les bonnes pratiques de fabrication et d’enrichir nos connaissances dans le domaine pharmaceutique.
Les résultats du contrôle physico-chimique sont conformes aux normes décrites par la Pharmacopée Européenne 2014. Ceci confirme la bonne qualité du médicament.
[1] N.HAMADOU, A.MOKHNACHE, « Evaluation microbiologique (bioévalution) et physicochimique dans l’industrie pharmaceutique, (2017)
[2] S.MERSELLAB, H.ANGOUD, « Contrôle physico-chimique, microbiologique, et toxicologique d’une solution injectable clofenal 75mg /3 ml, (2015)
[3] Koissi Joël Franck, « contrôle de qualité des comprimes non enrobés cas d’un générique et d’un principe de doxycycline, (2008)
[4 ] sitweb : WWW .MERINAL.COM. [ 5] Notice de la « Ranitidine MABO »