© Manel Fallah, 2020
La construction d’une carte génétique consensus à
haute densité chez le soja basée sur des marqueurs
SNP dérivés du génotypage par séquençage (GBS)
Mémoire
Manel Fallah
Maîtrise en biologie végétale - avec mémoire
Maître ès sciences (M. Sc.)
II
Résumé
Les cartes génétiques dérivées de l’étude d’une seule population de cartographie souffrent typiquement de plusieurs lacunes dont une couverture incomplète du génome et une résolution limitée. Une carte génétique consensus est une carte issue de la fusion de multiples cartes génétiques individuelles et permet de remédier à ces lacunes. Cette étude avait pour objectif de construire une carte génétique consensus à haute densité pour le soja (Glycine max (L.) Merr.) canadien au moyen de marqueurs obtenus par génotypage par séquençage (GBS). Six populations biparentales, dont l’effectif variait entre 278 et 365 lignées, ont été génotypées par GBS. Dans un premier temps, nous avons généré une carte génétique pour chaque population. Les tailles de ces cartes variaient entre 1869,3 cM et 2286,7 cM. Au total, quatre-vingt-trois (83) intervalles non-couverts ayant une taille > 10 cM (le maximum étant de 34,8 cM) ont été recensés. Ensuite, les six (6) cartes génétiques individuelles ont été fusionnées pour produire une carte consensus couvrant 99,5 % du génome, totalisant 16 311 SNP, s’étendant sur 2075,2 cM et comptant seulement deux intervalles dépourvus de marqueurs dont la taille excédait 10 cM. Cette carte consensus a ainsi pu remédier aux carences des cartes individuelles. Elle est considérée de meilleure qualité comparée aux cartes consensus précédentes, y compris la plus récente issue de 40 populations NAM (Nested Association Mapping ). En effet, cette dernière recèle encore 36 intervalles non-couverts de > 10 cM et couvre une moins grande portion du génome. Finalement, la carte consensus GBS présente une meilleure cohérence entre la position physique et la position génétique des marqueurs. Grâce à cette carte consensus, nous avons pour chaque marqueur SNP dérivé du GBS une position à la fois sur la carte physique et sur la carte génétique, une information utile pour de nombreuses analyses génétiques et génomiques.
III
Abstract
Genetic linkage maps using only one mapping population are described as maps with low resolution. These maps typically retain gaps, i.e. regions that are not covered by any markers. Consensus genetic maps were developed to overcome such limitations. They are generated by merging individual maps and using the common markers as reference points. The aim of this study was to generate a high-density consensus map for Canadian soybean using markers generated by genotyping by sequencing (GBS). Six mapping populations of varying size (n = 278 to 365) were genotyped using GBS. In a first step, we generated individual genetic maps. The size of the resulting maps varied between 1869.3 cM and 2286.7 cM and a total of 83 gaps with a size > 10 cM (the largest gap size was 34.8 cM) were observed across the six maps. In a second stage, we merged the six individual genetic maps to generate a single consensus map. On this map, 16,311 SNPs were assigned a position and these markers covered 99.5% of the genome. The map extends over 2075.2 cM and the number of gaps > 10 cM was reduced to only two. This map therefore overcame the limitations of the individual genetic maps and is superior to the previous consensus genetic maps such as the consensus genetic map generated from 40 nested association mapping (NAM) populations. The NAM map contains 36 gaps with a size > 10 cM and covers a smaller portion of the genome. In addition, the order of markers was much more concordant in the GBS map, when comparing the genetic and the physical positions. Thanks to this consensus map, we were able to assign both a physical and a genetic position for every SNP generated using GBS. These two types of information are important for many genetic and genomic studies.
IV
Table des matières
Résumé ... II Abstract ... III Table des matières ... IV Liste des tableaux ... VI Liste des figures ... VI Liste des abréviations ... VIII Remerciements ... IX Avant-propos ... X
Introduction générale ... 1
I Revue bibliographique ... 3
I.1 Le soja ... 4
I.1.1 Description de sa morphologie, biologie et botanique ... 4
I.1.2 La production de soja ... 5
I.2 Les marqueurs moléculaires ... 9
I.2.1 Les types de marqueurs moléculaires ... 9
I.2.2 Les technologies de génotypage à haut débit ... 10
I.3 La cartographie génétique ... 12
I.4 Les étapes clés de la construction d’une carte génétique ... 16
I.5 Les cartes individuelles ... 17
I.6 Carte consensus ... 19
II The construction of a high-density consensus genetic map for soybean based on SNP markers derived from genotyping by sequencing (GBS) ... 24
Résumé ... 26
Abstract ... 27
II.1 Introduction ... 28
II.2 Materials and Methods ... 30
V
II.2.2 DNA extraction, preparation of genotyping-by-sequencing (GBS) libraries and
sequencing ... 30
II.2.3 SNP calling and data filtering ... 30
II.2.4 Construction of individual genetic linkage maps ... 31
II.2.5 Construction of the consensus map ... 31
II.2.6 Estimation of the number of recombination events ... 32
II.3 Results ... 33
II.3.1 SNP genotyping of six F5 mapping populations ... 33
II.3.2 Number of recombination events (REs) in the mapping populations ... 34
II.3.3 Individual genetic maps ... 34
II.4 Consensus linkage map ... 36
II.4.1 Initial consensus map ... 36
II.4.2 Correction of distances ... 36
II.4.3 Final consensus map ... 37
II.5 Discussion ... 40
II.5.1 Comparison of the physical coverage for the GBS map and the NAM map .... 40
II.5.2 Comparison of the genetic coverage of the GBS map and the NAM map ... 40
II.5.3 Relationship between physical and genetic positions: ... 43
II.5.4 Recombination events ... 44
II.5.5 Conclusion ... 44
References ... 45
Supplementary data ... 47
Conclusions ... 52
VI
Liste des tableaux
Tableau I-1 La classification taxonomique du soja (Glycine max) (Canadensys explorer, 2018) ... 5 Tableau I-2 Évolution de la production du soja (en tonnes) dans les principaux pays producteurs entre 2007 et 2017(Tiré de FAOSTAT, 2019) ... 6 Tableau I-3 L’évolution de la production du soja (en tonnes) dans les provinces productrices entre 2007 et 2017 (Tiré de SOY Canada, 2019) ... 7 Tableau I-4 Quelques cartes consensus précédemment publiées chez le soja ... 22 Table II-1 Number and properties of the SNPs segregating in the six mapping populations of soybean…..………33 Table II-2 Number of recombination events (REs) captured in the six mapping populations of soybean ... 34 Table II-3 Distribution of SNPs for the six individual genetic maps of soybean. ... 35 Table II-4 Distribution of SNPs in the consensus genetic map of soybean ... 39 Supplementary Table 1. The six mapping populations of soybean……….……….47 Supplementary Table 2. Comparison of the physical coverage between the GBS map and the NAM map of soybean. ... 50 Supplementary Table 3. Comparison of the genetic length between the GBS map and the NAM map of soyeban. ... 51
VII
Liste des figures
Figure I-1 La plante du soja (Glycine max (L.) Merr.) (Ainsworth et al., 2012)………4 Figure I-2 Évolution de la production d'huile de soja entre 2007 et 2014 aux les États-Unis, au Brésil et en Chine (FAOSTAT, 2019) ... 6 Figure I-3 Les régions de culture de soja au Canada (SOY Canada, 2019) ... 8 Figure I-4 Les étapes de la technique du génotypage par séquençage (GBS)(Elshire et al., 2011 ... 11 Figure I-5 Représentation schématique des populations biparentales de cartographie (Singh and Singh, 2015). ... 14 Figure I-6 Schéma représentatif d’un évènement de recombinaison lors de la méiose (Singh et Singh, 2015) ... 15 Figure I-7 Schéma des étapes pour de construction d’une carte génétique issue d’une population biparentale de type RIL (Singh et Singh, 2015) ... 18 Figure I-8 Schéma de de construction d’une carte génétique consensus ... 21 Figure II-1 Final consensus genetic map for soybean from six F5 mapping populations and GBS-derived markers. The 20 linkage groups (chromosomes) are extensively covered by SNPs (black lines) and any remaining gaps appear as white spaces. ... 38 Figure II-2 Distribution of SNP markers across the 20 chromosomes of soybean for the GBS map and the NAM map. The position of each SNP marker is indicated with a blue line. Chromosomes are shown in pairs, with the ones on the left representing the NAM map and the ones to the right ones showing GBS ones. ... 42 Figure II-3 Relationship between genetic and physical positions of SNP markers of soybean on the two maps for Gm05, 11 and 13. Each dot provides the coordinates for each marker on the genetic (y axis, in cM) and physical (x axis, in bp) maps. Markers derived from the NAM map are in blue and those from the GBS in red. ... 43 Supplementary Figure 1. Relationship between the physical and genetic positions of GBS-derived SNP markers on the 20 soybean linkage groups that form the GBS consensus map...………...47 Supplementary Figure 2. Correlation between the genetic distance on the NAM map and the GBS map. The mean ratio between genetic distances on the NAM map vs the GBS map was 0.67 . ... 48 Supplementary Figure 3. Correlation between the genetic and physical positions for SNP markers on the GBS map (red line) and the NAM map (blue line). On the y axis is the genetic distance (cM) and the x axis is the physical position in base pairs (bp). Each dot is the location of a SNP. ... 49
VIII
Liste des abréviations
ADN : Acide désoxyribonucléiqueAFLP: Amplified Fragment Length Polymorphism
BC: Backcross
HD: Haploïdes doublés
FAO: Food and Agriculture Organisation of the United Nations
LG: Linkage Group
LOD: Logarithm of the odds
NIL: Near-isogenic Lines
GBS: Genotyping by sequencing
QTL: Quantitative trait loci
RAPD: Random Amplified Polymorphic DNA
RFLP: Restriction Fragment Length Polymorphism
RIL: Recombinant Inbred Lines
SNP: Single Nucleotide Polymorphism
IX
Remerciements
Je tiens à remercier du fond du cœur le professeur François Belzile, qui m’a accueilli au sein de son équipe et m’a permis d’avoir cette expérience aussi importante et enrichissante pour mon parcours académique. Comme étant un directeur de recherche, je tiens à le remercier pour le temps qu’il m’a accordé, sa disponibilité, ses conseils, les directives au plan scientifique ainsi que son côté humain.
Je tiens à remercier madame Martine Jean, pour ses conseils, surtout sa disponibilité, les discussions intéressantes au niveau scientifique.
Je tiens à remercier mes collègues Chiheb, Sidiki, Marc-André et Thomas pour avoir rendu mon séjour agréable au sein de l’équipe.
Je tiens à remercier tous les membres de l’équipe du professeur Belzile, Amina, Everton, Livia, Walidiodio, Sébastien, Martin, Suzanne et tous ceux qui ont contribué à la réalisation de ce projet.
Enfin, je tiens à remercier ma famille qui était toujours derrière moi et m’a toujours encouragé et soutenu sur tous les plans.
X
Avant-propos
Ce mémoire comporte deux chapitres qui sont précédés d’une brève introduction générale et suivis de conclusions. L’introduction générale décrit le contexte général dans lequel s’inscrivait cette étude. Le premier chapitre est une revue bibliographique qui comprend la présentation de la biologie du soja, l’importance économique du soja au Canada, les marqueurs moléculaires ainsi que les technologies de séquençage à haut débit, la cartographie génétique les études précédentes en cartographie génétique chez le soja. Le deuxième chapitre correspond à un manuscrit préparé en vue de sa soumission dans un périodique scientifique. Celui-ci s’intitule: The construction of a high-density consensus genetic map based on GBS-derived SNP markers. Finalement, je présente les différentes utilités de la carte consensus produite.
1
Introduction générale
Le soja (Glycine max (L.) Merr.) est une légumineuse (2n = 40) originaire de l’Asie. Le centre de domestication du soja reste inconnu mais il existe plusieurs pays candidats tels la Chine, le Japon et la Corée (Sedivy et al., 2017). C’est une culture importante grâce à la richesse de ses graines en protéines (35-40 %), en huiles (15-25%) et en hydrates de carbone (33 %) (Sharma et al., 2014). Elle est exploitée pour la production d’huile et elle représente 90 % de la production totale des huiles aux États-Unis (USDA, 2019). Sur un autre plan, elle substitue souvent la viande comme source de protéines non carnées.
La production mondiale de soja a augmenté de 2,4 fois entre 1997 et 2017 pour atteindre une production totale de 352 MT (million de tonnes) (FAOSTAT, 2019). Au Canada, le soja occupe la troisième place du point de vue des revenus monétaires chez les grandes cultures qui étaient estimés à 3,06 milliards de dollars canadiens ($CDN) en 2018 (SOY Canada, 2019). C’est la quatrième espèce au Canada pour les superficies cultivées avec un total de 2,31 million d’hectares en 2019 ( SOY Canada, 2019). Sur le plan historique, le soja a d’abord été cultivé en Ontario, vu que les conditions climatiques le permettaient. Par la suite, le Québec a commencé à cultiver cette espèce qui représente aujourd’hui la seconde culture de grains, après le maïs, en étant cultivée sur une superficie 367 000 ha en 2019 (SOY Canada, 2019). Au Canada et au Québec, toute augmentation de la superficie cultivée en soja repose sur le développement de variétés à maturité encore plus hâtive.
La cartographie génétique représente un outil clé pour l’amélioration des plantes. Elle permet de localiser les marqueurs/gènes sur le génome. Elle est utilisée pour les études de cartographie des caractères quantitatifs (QTL) d’intérêt tels que le rendement, la maturité (Kong et al., 2018), la résistance aux maladies (da Silva et al., 2019), la teneur en huiles et protéines (Wang et al., 2015). Les cartes utilisées dans ces études sont des cartes issues d’une population biparentale. Ces dernières présentent une faible résolution à cause de la taille limitée des population employées (le plus souvent <300), ce qui limite le nombre d’évènements de recombinaison capturés. De plus, les cartes génétiques individuelles présentent une couverture du génome incomplète en raison des régions monomorphes, qui
2
sont définies comme étant des régions identiques chez les deux parents. Par conséquence, les cartes génétiques consensus ont été développées pour surmonter les limites des cartes individuelles. Ces cartes consensus sont basées sur l’analyse de plusieurs populations biparentales. Elles présentent une meilleure résolution et une meilleure précision des positions génétiques. Elles sont construites à partir d’un grand effectif ce qui maximise le nombre d’évènements de recombinaison capturés. En outre, les cartes consensus sont un outil pour la sélection génomique. La sélection génomique présente une méthode importante pour la sélection de nouvelles variétés, elle consiste à la prédiction du phénotype à partir des données du génotype. Cette méthode combine les données génotypiques et phénotypiques d’une population nommée population d’entrainement, afin obtenir les valeurs génomiques des individus dans une population test ayant les données génotypiques sans avoir les données phénotypiques.
Le projet SoyaGen s’intéresse au soja sur divers plans, dans le but de développer des variétés adaptées aux conditions climatiques locales, résistantes aux maladies et dotées d’un haut rendement. Mon projet de maitrise fait partie de ce projet, dont l’objectif est de générer une carte génétique consensus à haute densité chez le soja canadien. Dans le cadre de cette étude, nous avons caractérisé six populations de cartographie F4 :5, avec des effectifs variant de 278
à 365 individus. La taille des populations utilisées est considérée grande, ce qui maximise la capture des événements de recombinaison d’où la précision de l’estimation des distances génétiques. Les données génotypiques des lignées ont été obtenues par le biais du génotypage par séquençage (GBS) pour générer le catalogue de SNP utilisé dans l’étude. Dans une première étape, on a procédé à la génération d’une carte individuelle par population. Les six (6) cartes présentaient toujours des limitations, principalement sous la forme de régions non-couvertes par des marqueurs. Par la suite, on a fusionné les cartes individuelles pour produire une carte consensus. Les marqueurs en commun entre ces dernières ont été utilisés comme point de référence pour assigner des positions génétiques aux marqueurs non-communs. La carte consensus obtenue présente de nombreux avantages : haute résolution, grande densité de marqueurs, une distribution uniforme des marqueurs et une couverture quasi-complète du génome.
3
4
I.1 Le soja
I.1.1 Description de sa morphologie, biologie et botanique
Le soja (Tableau I-1) est une espèce diploïde à 2n = 40 chromosomes. Il s’agit d’une plante annuelle qui appartient à la famille des Fabacées (Kumar et al., 2018). C’est une plante grimpante (Figure I-1) qui peut atteindre deux (2) mètres selon les variétés, lignées ou cultivars et les conditions de culture. Les feuilles se présentent sous forme trifoliée de 5 à 10 cm et les fleurs sont généralement de couleur violette ou blanche. Le fruit est une gousse de longueur entre 3 et 8 cm, laquelle forme la partie comestible de la plante, et contient 2 à 3 graines. C’est une espèce autogame, c’est-à-dire qu’elle se reproduit par autofécondation, ce qui permet d’avoir un taux d’homozygotie élevé à travers les générations. Elle a été domestiquée à partir de son ancêtre sauvage (G. soja)(Kim et al., 2012a), dont le centre de domestication reste inconnu : avec la Chine, il y a d’autres régions qui sont considérées comme régions candidates pour la domestication du soja telles que le Japon, la Corée…
5
Tableau I-1 La classification taxonomique du soja (Glycine max) (Canadensys explorer, 2018)
I.1.2 La production de soja
• Le soja dans le monde
Cette légumineuse est utilisée dans l’alimentation humaine et animale en raison de ses qualités oléagineuses et sa teneur élevée en protéines, ce qui permet sa consommation sous diverses formes (huile, lait, tofu…). En moyenne, le soja cultivé contient 40 % de protéines de haute qualité qui permettent d’assurer des quantités importantes d’acides aminés. La graine contient aussi 20 % d’huile de haute qualité caractérisée par une haute teneur en acides gras polyinsaturés (85 %) (Kumar et al., 2018). Grâce à ses valeurs nutritionnelles, le soja est devenu une espèce d’importance économique. La demande ne cesse d’augmenter pour le soja, la production mondiale a augmenté de 1,6 fois entre 2007 et 2017 pour passer de 219 MT à 352 MT. Comme le tableau I-2 le montre, actuellement les principaux pays producteurs sont les États-Unis, le Brésil et l’Argentine avec une production estimée, respectivement, à 119 millions de tonnes, 114 millions de tonnes et 54 millions de tonnes en 2017. En dix ans, le Brésil a presque doublé sa production pour être le deuxième producteur mondial après les États-Unis (taleau I-2; FAOSTAT, 2019).
Classe Equisetopsida
Sous-classe Magnoliidae
Ordre Fabales
Famille Fabaceae
Genre Glycine
6
Tableau I-2 Évolution de la production du soja (en tonnes) dans les principaux pays producteurs entre 2007 et 2017(Tiré de FAOSTAT, 2019)
Pays Production en 2007 (tonnes) Production en 2017 (tonnes)
Les États-Unis 72 859 180 119 518 490
Brésil 57 857 172 114 599 168
Argentine 47 482 784 54 971 626
Chine 12 725 147 13 152 688
Le soja se consomme selon différentes formes de produits dérivés tels que le tofu, l’edamame, le miso…mais le produit principal de soja est l’huile. Le soja est la deuxième culture pour la production d’huile après le palmier à l’huile avec une production, respectivement, de 45MT et 57MT en 2017 (FAOSTAT, 2019). La Chine est le principal producteur d’huile de soja (Figure I-2), presque la totalité de la quantité produite va à la production d’huile (12 MT) suivie des États-Unis (9MT) puis le Brésil (7MT) en 2014. La Chine s’intéresse à cette production de façon à presque doubler la quantité produite en seulement en sept ans, de 6,6 MT à 12,1 MT (Figure I-2).
Figure I-2 Évolution de la production d'huile de soja entre 2007 et 2014 aux les États-Unis, au Brésil et en Chine (FAOSTAT, 2019)
7
• Le soja au Canada
Le Canada est le 7e producteur mondial de soja avec 7 266 600 tonnes métriques. Cette production a presque triplé (2,7 fois) entre 2007 et 2017 (Tableau I-3). L’Ontario est classée première province en termes de production de soja avec 3 769 600 tonnes métriques suivie du Manitoba avec 2 245 300 tonnes métriques, le Québec occupe la troisième place avec 1 115 000 tonnes métriques. Le Manitoba a commencé à cultiver du soja en 2001, tandis que cette production est encore plus récente en Saskatchewan (2013) (Tableau I-3; SOY Canada, 2019)
Tableau I-3 L’évolution de la production du soja (en tonnes) dans les provinces productrices entre 2007 et 2017 (Tiré de SOY Canada, 2019)
Province Production en 2007 (Tonnes métriques) Production en 2017 (Tonnes métriques) Ontario 2 000 300 3 769 600 Manitoba 202 800 2 245 300 Québec 472 000 1 115 000 Saskatchewan --- 479 000 Maritimes 11 100 80 700 Total 2 686 200 7 266 600
Au début de la culture du soja au Canada (Figure I-2), cette dernière a été cultivée dans les régions les plus méridionales de l’Ontario, en raison des conditions climatiques qui y règnent, principalement la température et la photopériode. La photopériode est définie comme étant le rapport entre la durée du jour et la durée de la nuit. Ce rapport contrôle des activités physiologiques des végétaux tels que la reproduction et la maturité des graines. La plante est assignée à un groupe de maturité selon le nombre de jours qu’elle prend entre la plantation et la maturité physiologique des graines, sous une latitude définie et dans des conditions optimales pour la culture. Les groupes de maturité sont définis selon les conditions abiotiques principalement la photopériode, la température. On distingue 13 groupes de maturité (MG) en Amérique du Nord (Zhang et al., 2007), lesquels se distribuent selon un axe Nord-Sud. Les variétés les plus hâtives (MG0 ; 00 ; 000) étant cultivées dans les régions septentrionales et les variétés tardives (MGVII et plus) étant cultivées dans les régions les plus méridionales
8
du continent. Le choix des variétés à cultiver dans une région précise s’appuie principalement sur le groupe de maturité. Certaines provinces sont dotées d’un été plus court, par conséquent la maturité est un critère important pour la sélection d’où avec le développement des variétés à maturité précoce, il est maintenant rendu possible de produire du soja dans les autres provinces telles que le Manitoba, et la Saskatchewan (Figure I-3).
Figure I-3 Les régions de culture de soja au Canada (SOY Canada, 2019)
• Le soja au Québec
La culture de soja au Québec a débuté en 1986 et cette province est la troisième productrice au Canada avec ses 366 700 ha en culture en 2019, soit 17,3 % de la production totale au Canada (Statistique Canada, 2019). Cette culture est considérée une culture jeune au Québec, mais elle s’est rapidement déployée en gagnant de plus en plus de superficie. Durant les dernières années la superficie cultivée en soja a doublé, pour être presque équivalente à celle du maïs grain (382 500 ha en 2019). C’est une production rentable pour le Québec, et la demande ne cesse d’augmenter. Le soja produit au Québec peut être destiné à l’exportation dans des pays tels que la Chine, lequel constitue la première destination pour le soja canadien (60 % de ce qui est exporté). Pour qu’il survienne une poursuite de l’expansion du soja dans des régions encore plus nordiques, il faudra développer des variétés adaptées aux condition climatiques et résistantes aux ravageurs tels que les nématodes (Gendron St-Marseille et al., 2019) et cela nécessitera principalement des nouvelles variétés à maturité encore plus hâtive
9
ce qui est possible via l’amélioration des plantes. La cartographie génétique représente un outil clé dans les programmes de sélection. Cette méthode fait appel à des plateformes et des outils, principalement les marqueurs moléculaires et les technologies de séquençage et de génotypage à haut débit pour permettre de générer des cartes génétiques.
I.2 Les marqueurs moléculaires
I.2.1 Les types de marqueurs moléculaires
Les premiers marqueurs utilisés pour la sélection sont les marqueurs morphologiques. Le choix des individus se base sur le phénotype. On peut penser à des caractères comme la taille des feuilles, la forme des feuilles, la couleur de la fleur… Cette méthode présente des limites, tels que le phénotype n’est pas souvent associé à des caractères agronomiques importants comme le rendement (Jiang, 2013), ce qui fait qu’ils sont moins efficaces. Par la suite, il y a eu le passage aux allozymes qui sont définis comme étant des enzymes codées par différents allèles du même gène et sont considérés comme étant les premiers marqueurs moléculaires. Ces marqueurs peuvent être différenciés par électrophorèse selon le poids et la charge. L’obstacle majeur de ces marqueurs est qu’ils sont incapables de détecter d’une façon directe les variations sur le génome (Schlötterer, 2004). Avec l’avancement de la biotechnologie, il y a eu la mise en place des marqueurs moléculaires qui sont définis comme étant une séquence d’ADN, qui est utilisée pour détecter le polymorphisme (délétion, insertion, ou substitution) entre deux ou plusieurs génotypes. Les marqueurs moléculaires jouent un rôle primordial en amélioration des plantes tel que la sélection assistée par marqueurs, la cartographie qui peut servir à localiser des locus contrôlant des caractères importants (QTL) sur le génome, la génétique d’association et la construction des cartes génétiques. Au fil des années, plusieurs types de marqueurs ont été utilisés tels que les RFLP “restriction fragment
length polymorphism ”, les AFLP “ amplified fragment length polymorphism ”, les RAPD “ random amplified polymorphic DNA ”, les SSR “simple sequence repeat ” et les SNP “ single nucleotide polymorphism ”. Les SNP sont devenus les marqueurs les plus utilisés ces
dernières années. Par définition, ils marquent la différence d’une paire de bases sur le génome et sont typiquement bi-alléliques (Jehan et Lakhanpaul, 2006). Leur faible coût et leur distribution uniforme et dense au niveau du génome leur permettent d’être de plus en plus
10
utilisés. Chez le soja plusieurs études ont fait appel à ces marqueurs à diverses fins comme la cartographie des QTL (da Silva et al., 2019; Liu et al., 2019; Wang et al., 2015, 2019), les études d’association pour divers caractères (Bastien et al., 2014) ainsi que pour la construction d’une carte génétique consensus chez le soja (Song et al., 2017).
I.2.2 Les technologies de génotypage à haut débit
Les technologies de génotypage à haut débit génèrent des milliers et même des millions de marqueurs SNP. Ces technologies sont considérées moins chères et plus rapides que les méthodes précédentes utilisées telles que le séquençage Sanger. Ces plateformes sont différentes au niveau des protocoles utilisés ce qui affecte directement la qualité des données produites et le coût (Metzker, 2010). Parmi les technologies les plus communes sont le génotypage par séquençage (GBS) « genotyping by sequencing » et la puce à ADN. Les deux technologies diffèrent quant à la façon dont les marqueurs sont découverts et génotypés, mais toutes deux permettent de générer un grand nombre de SNP.
• GBS
La méthode de génotypage par séquençage (GBS) a été développée comme étant une méthode simple mais robuste pour la réduction de complexité du génome (Poland et al., 2012), laquelle est particulièrement utile chez les espèces dont le génome est de grande taille. Elle permet de générer un grand nombre de SNP avec une répartition uniforme et une couverture complète du génome. Le GBS décrit bien le polymorphisme existant chez les échantillons analysés. Le principe de la méthode (Figure I-4) se base, dans un premier temps, sur la fragmentation de l’ADN des échantillons à l’aide des enzymes de restriction. En un second temps, les barcodes moléculaires (adaptateurs) spécifiques de chaque individu, vont se liguer aux fragments d’ADN. Les barcodes non liés seront éliminés, et les adaptateurs “primers ” appropriés interviennent pour permettre l’amplification
(PCR) “ polymerase chain reaction ” Finalement, la librairie produite sera séquencée et les
séquences sont alignées par rapport au génome de référence de l’espèce en question (Elshire et al., 2011). Vu son faible coût et son efficacité, cette technique (GBS) a été utilisée dans le cadre de plusieurs études et chez différentes espèces telles que les études de la génétique d’association chez le soja (Bastien et al., 2014; Sonah et al., 2015), chez le raisin (Guo et al.,
11
2019), la cartographie génétique des QTL chez le blé (Hussain et al., 2017), chez le melon (Pereira et al., 2018), le riz (Yadav et al., 2019) et le pois chiche (Verma et al., 2015). Le GBS a permis aussi la génération d’une carte consensus à haute densité chez des espèces telles que le canneberge (Covarrubias-Pazaran et al., 2016), le blé (Edae et al., 2017), l’hévéa (Pootakham et al., 2015) mais chezle soja, il n’existe pas une carte consensus dont les marqueurs SNP utilisés sont issus du GBS.
Figure I-4 Les étapes de la technique du génotypage par séquençage (GBS) (Elshire et al., 2011)
• Puce de génotypage
La puce de génotypage détermine l’allèle présent chez les SNP puis celui chez les génotypes analysés en se basant sur le dosage des deux allèles. C’est une technologie qui est utilisée dans plusieurs études et chez différentes espèces mais considérée chère par rapport au GBS. La première puce chez le soja comptait seulement 1536 SNP « 1536 Universal Soy Linkage Panel » et elle a servi pour la construction de la carte consensus V4 (Hyten et al., 2010). Par la suite, en 2013, il y a eu le développement de la puce SoySNP50K iSelect BeadChip par
12
Song et al. (2013). La validation de la puce a été faite sur 96 cultivars locaux, 96 cultivars élites et 96 accessions sauvages. Au sein de cette collection de lignées variées, 47 337 SNP ont été trouvés polymorphes. La même équipe a aussi généré la puce SoyNAM6K BeadChip qui a servi pour la génération d’une carte consensus chez le soja (Song et al., 2017).
I.3 La cartographie génétique
Depuis les contributions d’Emerson et al. en 1935 (cités par Rhoades, 1984) sur les concepts de génétique quantitative, les cartes de caractères morphologiques et ceux des isozymes ont été publiées mais elles n’ont souvent qu’un nombre limité de marqueurs par groupe de liaison. Pour cela, il fallait procéder parfois à des multiples tests de croisement pour développer plus de marqueurs (Schwarzacher, 1994). Par la suite le développement des marqueurs moléculaires a fait la révolution pour la cartographie génétique. Les cartes ont été construites en utilisant les RFLP, RAPD, SSR et de nos jours les marqueurs SNP.
Pour assurer la construction d’une carte génétique, il faudra une population de cartographie, un catalogue de marqueurs et un logiciel de cartographie (Singh and Singh, 2015). On distingue deux types de cartes génétiques en se basant sur le nombre de populations utilisées. Le premier type de carte est la carte individuelle issue d’une seule population biparentale tandis que la carte consensus est issue de multiples populations. En cartographie, on peut employer plusieurs types de populations de cartographie selon l’objectif de l’étude.
• La population de cartographie
La population qui sert à la construction d’une carte génétique est appelée population de cartographie. Généralement ces populations sont construites à partir du croisement de deux parents homozygotes qui appartiennent à la même espèce (Singh and Singh, 2015). Dans certains cas où la variation intra-spécifique est limitée, on a recours à des croisements entre espèces proches. La population de cartographie est utilisée pour déterminer la position génétique des locus et calculer les distances génétiques entre les paires de marqueurs en se basant sur les évènements de recombinaison. Les types de populations de cartographie les
13
plus fréquents (Figure I-5) sont F2, F2 :3, rétrocroisement (BC), haploïdes doublés (HD), ainsi
que les lignées recombinantes fixées ou “ recombinant inbred lines ” (RIL). La première étape
pour la construction de la population de cartographie est le choix du type de la population qui dépend de l’objectif de l’étude, la possibilité de la plante à l’autofécondation, le temps du développement. Le choix des parents est primordial pour la construction de la population ; les parents sont généralement homozygotes et contrastés pour au moins un ou plusieurs caractères. La différence génotypique et phénotypique des parents permettra de maximiser la capture du polymorphisme. Par la suite, ceci permet d’augmenter le nombre de marqueurs polymorphes couvrant plus de régions du génome afin de construire une carte avec une meilleure couverture. Suite au croisement des parents, on obtient la première génération F1
qui est 100 % hétérozygote pour les loci différents chez les parents au niveau génotypique. Les plantes de la F1 sont par la suite autofécondées pour produire la F2, la F3 et ainsi de suite
jusqu’à la F7 :8, un stadeoù le degré de fixation génétique permet de parler de RIL. Avec
l’avancement de chaque génération, l’hétérozygotie diminue de moitié (50 %) par rapport à la génération précédente et la population tend vers une plus grande homozygotie. Les F2 et
F2 :3 sont considérées comme des populations non fixées puisqu’elles sont composées de
plantes ayant des loci homozygotes et hétérozygotes. En revanche, les RIL sont composés uniquement d’homozygotes. On distingue aussi les populations NAM “ Nested association
mapping ” qui font partie des RIL. Le principe de la construction de la population NAM est
le choix d’un parent pivot qui sera croisé avec tous les autres parents pour générer les populations biparentales. Quarante familles NAM totalisant 5600 lignées ont été générées pour construire une carte consensus génétique chez le soja (Song et al., 2017). Par ailleurs, le rétrocroisement consiste à croiser la descendance de la F1 avec un des deux parents
originaux tout en effectuant une sélection au sein de chaque descendance pour conserver un caractère d'intérêt. Finalement, les haploïdes doublés sont produits à partir du doublement des chromosomes de plantes haploïdes dérivées par culture d’anthères de la F1 ce qui permet
de donner des plantes aux chromosomes identiques. Durant le processus de génération des populations de cartographie, pour tous les types de population, il n’existe aucune pression de sélection pour aucun caractère (à l'exception des rétrocroisements).
14
Figure I-5 Représentation schématique des populations biparentales de cartographie (Singh and Singh, 2015).
• Les évènements de recombinaison
La carte génétique montre l’ordre génétique des marqueurs tout au long du génome et les distances sont calculées en se basant sur la fréquence de recombinaison (Beyer et al., 2007; Paterson, 2009). Au plan du matériel génétique, les gènes sont codés par des allèles qui représentent la variation nucléotidique. Ces allèles peuvent être identiques (Figure I-6), donc pour l’allèle A (exemple : AA) sinon il y aura une hétérozygotie. Lors de la méiose, il se déroule un échange de portion des chromosomes de façon aléatoire ce qui génère de nouvelles combinaisons alléliques (Figure I-6). Cet évènement joue un rôle primordial dans la création de la diversité génotypique et phénotypique. Il existe une relation entre la recombinaison génétique et la distance entre deux locus : plus les locus sont distants sur le chromosome plus il y a de fortes possibilités de brassage génétique. En conséquence, l’information de la position génétique d’un locus et la distance entre deux locus est très importante pour les programmes de sélection car elle permet de savoir la fréquence à laquelle deux gènes seront transmis ensembles d’une génération à la suivanteAinsi, deux gènes
Parent A X Parent B
F1
Populations F2
Recombinant Inbred Lines (RIL)
Autofécondation de la F1 Autofécondation de la F2 Population F2:3 Autofécondation de la F2:3 jusqu’à la F7:8 Population rétrocroisement BC1 F1 Near-isogenic population (NIL)
La production des haploïdes Haploïdes
15
proches seront fréquemment transmis ensemble tandis que deux gènes distants seront moins fréquemment transmis ensemble.
Il existe une relation de proportionnalité entre la précision de l’estimation de la distance génétique et l’effectif de la population de cartographie : plus la taille de la population augmente, plus on capte des évènements de recombinaison et plus la précision augmente. L’unité de mesure des distances génétiques est le centimorgan (cM). La distance génétique est directement liée à la fréquence de recombinaison par la formule suivante (1% FR = 1 cM).
Figure I-6 Schéma représentatif d’un évènement de recombinaison lors de la méiose (Singh et Singh, 2015)
En cartographie génétique, on distingue la carte génétique et la carte physique. La carte physique montre l’ordre physique entre les marqueurs en mesurant la distance en paires de bases. Les distances physiques sont souvent déterminées par différentes techniques telles que la cartographie par radiation hybride “ radiation hybrid mapping ”, l’hybridation in situ en
fluorescence “ fluorescence in situ hybridization ” ou le séquençage de l’ADN (Beyer et al.,
Chromosomes Parentaux
La recombinaison génétique
16
2007). Il existe une relation de complémentarité entre la carte physique et la carte génétique puisque chacune fournit une information différente mais complémentaire pour l’autre (Beyer et al., 2007). La carte génétique et la carte physique ne présentent aucune conformité ou proportionnalité entre la distance physique et génétique, seul l’ordre des marqueurs est maintenu : si deux marqueurs sont localisés proches sur la carte physique ils peuvent être localisés à une grande distance sur la carte génétique et vice versa. La probabilité qu’un évènement de recombinaison génétique a lieu au niveau du chromosome diffère selon sa localisation : la région centromérique est souvent pauvre en évènements de recombinaison tandis que les régions télomeriques sont riches.
I.4 Les étapes clés de la construction d’une carte génétique
• La formation des groupes de liaison
Lors de la construction d’une carte génétique, l’étape clé est la formation des groupes de liaison qui représentent les chromosomes. Dans un cas idéal, le nombre de groupes de liaison attendu doit être égal au nombre de chromosomes de l’espèce en question. Par exemple, le soja possède 20 chromosomes, donc il devrait y avoir 20 groupes de liaison. La méthode la plus fréquente dans la construction des cartes génétiques est la méthode de vraisemblance (LOD : “Logarithm of the odds ”), laquelle permet de mesurer l’intensité des liens (en matière
de co-ségrégation) entre les paires de marqueurs. Le LOD est un paramètre qu’on calcule entre les paires de marqueurs (Kocherina et al., 2011). S’il est supérieur à un seuil critique, les marqueurs seront déclarés comme appartenant au même groupe de liaison. Sinon, on conclura qu’ils n’appartiennent pas au même groupe de liaison. Cette méthode est la plus répandue car elle se base seulement sur l’information génétique (la recombinaison observée) et pour la facilité de l’accès à cette information. Une méthode utilisée pour déterminer l’ordre des marqueurs est RECORD “REcombination Counting and ORDering”, laquelle consiste à
minimiser le nombre total d’évènements de recombinaison requis pour produire les données observées. Cette méthode est considérée comme étant la plus performante pour les cartes à haute densité (Van Os et al., 2005). Avec l’avènement des génomes de référence qui nous
17
permettent de connaître avec exactitude la position physique des SNP, on peut former des groupes de liaison en se basant sur la position physique connue des marqueurs.
• Le calcul des distances génétiques
Ces fonctions de calcul de la distance génétique ont pour objectif de transformer un phénomène biologique complexe qu'est la recombinaison génétique en distance génétique. Pour de grandes distances entre deux locus, les évènements de recombinaison restent une mesure non-additive. Par conséquent, la construction des cartes génétiques nécessite la transformation des paramètres non-additifs en des distances génétiques additives. Ces transformations se font via ce que l’on appelle des fonctions de cartographie (Huehn, 2011). Les fonctions les plus utilisées dans les études de cartographie sont celles de Haldane et celle de Kosambi (1944), cités par Huehn (2011). A ce stade, on distingue trois types de fonctions : celles qui considèrent l’interférence complète, interférence incomplète et pas d’interférence. L’interférence peut être définie comme étant l’effet d’un évènement de recombinaison sur l'occurrence d'un autre événement de recombinaison dans la même région, ce qui fait que les évènements de recombinaison n’ont pas lieu d’une façon tout à fait aléatoire (Lin et al., 2001). L’interférence complète ne permet pas les doubles recombinants, l’interférence incomplète permet quelques doubles recombinants (tel que prévu par la fonction de Kosambi) et la non-interférence est le cas prévu par la fonction de Haldane. La fonction de Kosambi reste la plus utilisée pour ses avantages et sa meilleure estimation des distances génétiques (Vinod, 2011).
I.5 Les cartes individuelles
Le processus de génération d’une carte individuelle commence par le choix du type de population (RIL, F2 :3, HD, BC), le choix des parents, puis la formation d’une population
issue de parents, généralement homozygotes. Les cartes issues d’une population biparentale sont souvent utilisées dans le cadre des études de cartographie des QTL ce qui permet de localiser des QTL responsables d’un caractère étudié (Figure I-7). Ces cartes ont été utilisées pour différents caractères agronomiques chez différentes espèces telles que la luzerne tropicale (Tang et al., 2019); pour le nombre et la taille des fruits chez la tomate (Brekke et
18
al., 2019). La première carte génétique chez le soja pour la cartographie des QTL pour différents caractères a été produite en 1990 par Keim et al. (1990). Depuis, il y a eu beaucoup d’études qui se sont intéressées à la cartographie des QTL chez le soja telles que pour des caractères agronomiques et la qualité des grains (Diers et al., 2018; Liu et al., 2017; Zhang
et al., 2004; Zhang et al., 2018b), ou pour la maturité et la floraison (Kong et al., 2018).
Figure I-7 Schéma des étapes de construction d’une carte génétique issue d’une population biparentale de type RIL
• Les limites des cartes individuelles
Les cartes individuelles se basent sur une population qui est souvent de petite taille (N <300), ce qui ne permet pas de capturer un grand nombre d’évènements de recombinaison. Un tel faible échantillonnage des évènements de recombinaison diminue la précision de l’estimation des positions génétiques des marqueurs. Plus l’effectif de la population de cartographie augmente, plus le nombre d’évènements de recombinaison capturés est maximisé. Ces cartes
X F1 RIL Parent A Carte individuelle Autofécondation Parent B F7:8
19
présentent aussi des limites en ce qui a trait à la couverture du génome. Celle-ci est souvent incomplète, ce qui est dû souvent à la présence de régions monomorphes qui sont identiques chez les deux parents de la population (Wen et al., 2017). Il est donc impossible d’avoir du polymorphisme dans ces régions du génome. Des régions non couvertes peuvent être aussi le résultat de la non-uniformité de la distribution des marqueurs tout au long du génome ou du faible nombre de marqueurs utilisés, un problème qui était souvent rencontré avant la découverte des SNP.
I.6 Carte consensus
Une carte consensus est construite sur la base de la fusion de plusieurs cartes individuelles (chacune issue d’une population biparentale). Par conséquent, l’effectif utilisé pour la carte consensus sera le total des populations individuelles. L’utilisation de multiples populations maximise la capture des évènements de recombinaison, et améliore la précision des positions génétiques des marqueurs sur la carte. Par la suite, les marqueurs en commun entre cartes individuelles sont utilisés comme points de référence pour calculer les distances génétiques. L’avantage aussi avec ces cartes est la détection de plus de polymorphisme puisqu’on utilise différents parents. Ainsi, une région qui était possiblement monomorphe au sein d’une population sera polymorphe (et donc informative) au sein d’une autre population (Figure I-8). Ces régions non-couvertes peuvent être situées au début ou à la fin du chromosome (Galeano et al., 2011). Comme le montre la Figure 7, la région entre les SNP1 et SNP4 n’est pas couverte sur la carte individuelle 1 par contre elle est polymorphe sur la carte 2, et suite à la fusion des deux cartes on a pu couvrir cette zone sur le chromosome consensus ce qui permet d’améliorer la couverture du génome.
La fusion de nombreuses cartes, en utilisant les marqueurs en commun comme référence, permet ainsi d’augmenter le nombre de marqueurs utilisés, ce qui offre donc une meilleure couverture et une distribution plus uniforme des marqueurs tout au long du génome. Le nombre de populations est un paramètre clé utilisé pour générer une carte consensus, ceci va capturer la diversité au sein de l’espèce et cherche plus de polymorphisme et pourra assurer une meilleure couverture et une meilleure résolution pour la carte consensus générée.
20
Grâce aux avantages amenés par les cartes consensus, elles ont été développées pour différentes espèces telles que l’avoine (Chaffin et al., 2016), la pastèque (Ren et al., 2014), le pommier (Di Pierro et al., 2016), la laitue (Truco et al., 2013), l’orge (Li et al., 2010; Muñoz-Amatriaín et al., 2011) , le blé (Edae et al., 2017, 2017; Wen et al., 2017) le lin (J. Zhang et al., 2018). Ces cartes peuvent être utilisées dans le cadre de différentes études telles que la sélection génomique, la génomique comparative et le clonage positionnel.
21
Figure I-8 Schéma de construction d’une carte génétique consensus
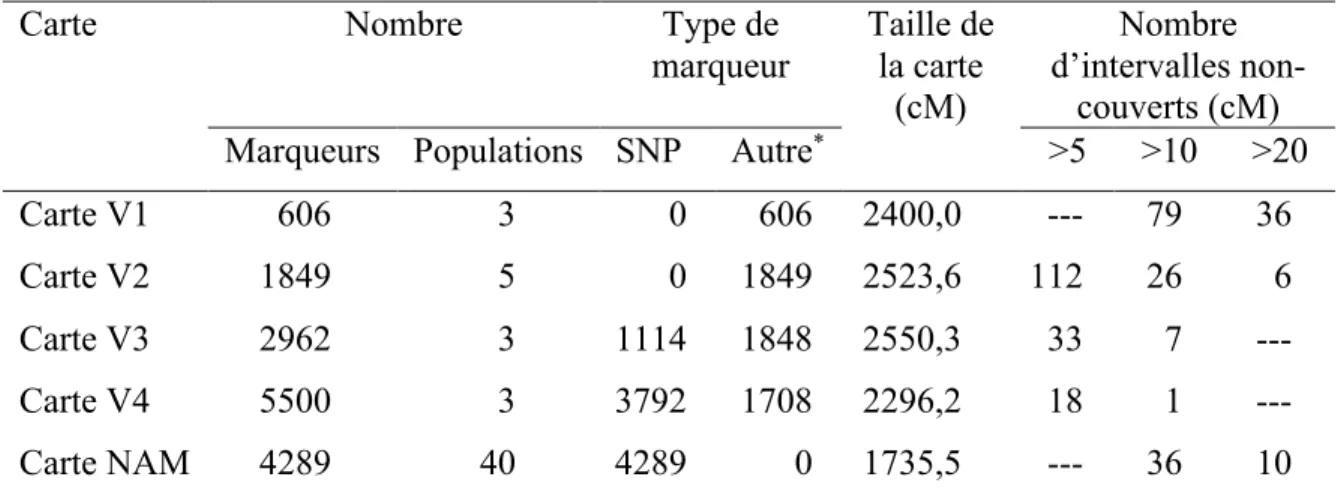
Au fil du temps, il y a eu le développement de cartes consensus chez le soja (Tableau 4) en utilisant différents types de marqueurs et de populations. La première carte consensus (carte V1) a été développée par Cregan et al. (1999) en se basant sur trois populations et 606
Population 1 Carte individuelle 1 X Population 2 Carte individuelle 2 X + Carte consensus + Chromosome 1 de la carte 1 Chromosome 1 de la carte 2 Chromosome de la carte consensus SNP1 SNP4 SNP2 SNP3 SNP2 SNP3 SNP1 SNP4
22
marqueurs SSR. Cette carte avait une taille totale de 2400 cM, mais comptait 79 intervalles non couverts dont la taille était > 10 cM. Pour améliorer la qualité de la carte, l’équipe de Song s’est intéressée à développer davantage de marqueurs SSR à produire une nouvelle carte consensus améliorée qui est considérée comme la deuxième version (carte V2) (Tableau I-4). La carte de Song et al. (2004) est d’une taille de 2523,6 cM et le nombre d’intervalles non-couverts (de plus de 10 cM) a été réduit à 26 (Tableau I-4). Avec l’avancement de la biotechnologie, Choi et al. (2007) se sont intéressés à générer une carte consensus V3, mais en ajoutant 1141 marqueurs SNP à la carte précédente. Cette dernière est d’une taille de 2550,3 cM et on y trouve 7 intervalles non-couverts (>10 cM). Par la suite, une carte consensus V4 a été développée par Hyten et al. (2010) en utilisant trois populations et en ajoutant 2500 SNP. Cette carte a une taille de 2296,2 cM et comporte un seul intervalle > 10 cM. Ces dernières années, Song et al. (2017) ont développé une nouvelle carte consensus, en utilisant sur 40 familles NAM et 4289 marqueurs SNP. La carte générée est d’une taille de 1735,5 cM et ne comporte que des marqueurs SNP. Toutes ces cartes sont considérées de faible résolution et le nombre élevé d'intervalles non-couverts (> 5cM; 10 cM et 20 cM) représente une carence majeure de ces cartes.
Tableau I-4 Quelques cartes consensus précédemment publiées chez le soja
Carte Nombre Type de
marqueur Taille de la carte (cM) Nombre d’intervalles non-couverts (cM) Marqueurs Populations SNP Autre* >5 >10 >20
Carte V1 606 3 0 606 2400,0 --- 79 36
Carte V2 1849 5 0 1849 2523,6 112 26 6
Carte V3 2962 3 1114 1848 2550,3 33 7 ---
Carte V4 5500 3 3792 1708 2296,2 18 1 ---
Carte NAM 4289 40 4289 0 1735,5 --- 36 10
* : tout autre type de de marqueurs à part les SNP tels que les RFLP, SSR, AFLP…
(Cartes V1 à 4 et carte NAM tirées de : V1, Cregan et al. 1999 ; V2, Song et al. 2004 ; V3, Choi et al. 2017 ; V4, Hyten et al. 2010 ; NAM, Song et al. 2017)
23
Hypothèses et objectifs
Les cartes génétiques denses représentent un outil clé pour la sélection. Les cartes issues des puces de génotypage sont dites de faible résolution principalement à cause de la couverture incomplète du génome et de la distribution non-uniforme des marqueurs. A l’aide du GBS, il y a eu génération d’un ensemble de marqueurs SNP dont la position physique est connue et dont on cherche à déterminer la position génétique sur le génome du soja. Dans cette étude on veut tester les hypothèses suivantes, à savoir que la carte consensus génétique générée offrira :
• Une couverture exhaustive du génome du soja
• Une répartition uniforme des marqueurs tout au long du génome • Une haute densité
Pour tester ces hypothèses, il y a un objectif principal qui consiste à générer une carte consensus à haute densité chez le soja qu’on réalise en trois étapes :
a. Optimiser les données génotypiques pour les fins de la cartographie génétique b. Produire une carte génétique individuelle par population
24
II The construction of a high-density consensus
genetic map for soybean based on SNP
markers derived from genotyping by
sequencing (GBS)
25
The construction of a high-density
consensus genetic map for soybean based
on SNP markers derived from genotyping
by sequencing (GBS)
Manel Fallah
1,2, Martine Jean
1,2and François Belzile
1,21
Département de phytologie Université Laval, Québec City, Québec, Canada
G1V0A6
2
Institut de Biologie Intégrative et des Systèmes (IBIS), Université Laval,
Québec City, Québec, Canada G1V0A6
Corresponding author
26
Résumé
L’objectif de ce projet est de générer une carte génétique consensus de haute densité pour le soja. Six populations de F4 :5 (n=278-365), totalisant 1857 individus qui ont été utilisées dans
le cadre de cette étude. Les marqueurs SNP utilisés sont issus de génotypage par séquençage (GBS). Par la suite, il y a eu la génération d’une carte génétique individuelle pour chaque population. Les six cartes individuelles ont été fusionnées pour produire la carte consensus. Cette dernière a pu localiser 16311 SNP qui couvre la presque totalité du génome. Finalement il persiste seulement deux régions non-couvertes avec une taille supérieure à 10 cM. En comparant cette carte aux cartes consensus précédentes pour le soja, on constate que celle-ci offre une meilleure couverture du génome et une distribution plus uniforme des marqueurs SNP.
27
Key words
Soybean, Genetic mapping, consensus genetic map, individual maps, genotyping by sequencing, recombination events,
Key message
A high-density consensus map was generated for soybean and it includes over 16,000 GBS-derived SNPs covering 99.5% of the reference genome.
Abstract
Genetic linkage maps are used to localise markers on the genome based on the recombination frequency. Most often, these maps are based on the segregation observed within a single biparental population. Usually, the limited size of these populations (n <300) entails that a relatively small number of recombination events are sampled, which limits the precision of the estimated distance separating markers. A second issue with genetic linkage maps is that any mapping population will present monomorphic regions that carry the same alleles in both parents, thus precluding any measure of recombination frequencies within these intervals. Together, these two limitations affect both the resolution of extent of genome coverage of such linkage maps. Consensus genetic maps were created to overcome the limitations of individual genetic maps. These are generated by merging the information from multiple segregating populations, thus increasing the number of recombination events and extending genome coverage as a greater diversity of parental combinations will reduce the number of monomorphic regions. The aim of this study was to construct a high-density consensus genetic map for SNP markers obtained through a genotyping-by-sequencing (GBS) approach. Six F4:5 mapping populations (n=278-365), totaling 1857 individuals were
genotyped, and individual genetic maps were generated for each. The six linkage maps were then merged to produce a consensus map comprising a total of 16,311 mapped SNPs that jointly cover 99.5% of the soybean genome and only two gaps larger than 10 cM. Compared to previous soybean consensus maps, it offers a more extensive and uniform coverage.
28
II.1 Introduction
Soybean (Glycine max (L.) Merr.) is a diploid species (2n = 40) that is believed to have been domesticated in eastern Asia 6,000-9,000 years ago (Sedivy et al., 2017). Offering a high level of protein and oil (Kim et al., 2012), it has become the world’s largest oilseed crop with its uses including human consumption, industrial products and animal feed (Hartman et al., 2011). In the Americas, it has established itself as the largest crop of all. In 2017, the three largest producers were the USA (119.5 MMT), Brazil (114.5 MMT) and Argentina (54.9 MMT) (FAOSTAT, 2019). In Canada, the seventh-largest producer, this crop has seen a large increase in the last decade with production more than doubling between 2008 and 2017 from 3.3 to 7.7 MMT (SOY Canada, 2019).
Various genetic improvement technologies, from marker-assisted breeding to genetic modification, have played major roles in the success of this crop. A large contributor to the spectacular expansion of soybeans has been the success in breeding varieties adapted to an increasingly wide area, from the tropical soybeans grown in the Cerrado region of Brazil to the Prairies of western Canada. Genetic mapping of key genes involved in controlling soybean adaptation (including maturity, determinate growth and tolerance to various stresses) have paved the way to the wide use of markers in soybean breeding.
Genetic linkage maps have been used for many purposes but are often used for quantitative trait locus (QTL) mapping. In QTL mapping, individual genetic maps, based on a single segregating biparental population, are needed to identify genomic regions responsible for important traits. In soybean, many studies have been conducted to map QTLs controlling traits such as flowering time (Yamanaka, 2001), oil and protein content (Wang et al., 2015) as well as resistance to various pests and diseases such as nematodes (Arriagada et al., 2012), and Sclerotinia sclerotiorum (Huynh et al., 2010; Zhao et al., 2015). Although highly useful, QTL mapping typically provides limited resolution because biparental populations (n < 300) capture a limited number of recombination events. Also, such mapping populations are limited in the number of alleles (i.e. 2) that can be discovered at a locus. Finally, there will inevitably be incomplete marker coverage across the genome as some regions will not be polymorphic between the two parents. Thus, genetic maps derived from the analysis of a single biparental cross are limited in resolution, allelic diversity and genome coverage.
29
Consensus genetic maps were developed to overcome the limitations of individual genetic maps especially the regions of low marker density (Pootakham et al., 2015). The idea is to merge several individual maps based on the common markers between the maps, which are used as reference points to connect the maps. Thanks to the combination of data from multiple sets of progenies, more recombination events are captured, and this will increase the resolution of the map. Similarly, the use of multiple populations improves genome coverage as a region that is monomorphic within a population may be polymorphic in another. As a result, consensus genetic maps typically offer improved coverage and higher resolution. Genomic selection (GS) is another tool used by geneticists and breeders. It aims to predict the phenotype of an individual based only on its genotype. To do this, it is necessary to build a statistical model that accurately captures the relationship between the genotype and the phenotype. Phenotypic and genotypic data for a training population are used to build such a GS model, which can then be used to produce a genomic estimated breeding value (GEBV), i.e. to predict trait values for a line based only on its genotype (Crossa et al., 2017). Recently, the use of GS models for the prediction of the most promising crosses has been explored and it relies on the production of a simulated set of progeny (Mohammadi et al., 2015). To do this, the genetic position of the SNP markers that are part of the genotypic dataset must be known. Unfortunately, for genotyping-by-sequencing (GBS)-derived markers in soybean, there is limited information on the exact genetic position of these markers. The objective of this study was to generate a high-density consensus genetic linkage map for soybean using the GBS-derived SNP markers identified in six biparental populations.
30
II.2 Materials and Methods
II.2.1 Mapping populations
In this study, we used a total of six F4:5 mapping populations derived from crossing selected
Canadian soybean cultivars (Supplementary Table 1) developed at CÉROM “Le centre de recherche sur les grains”. Generations were initially advanced in the form of a population until the F4 generation when individual plants were randomly selected to harvest F5 seed to
establish F4:5 lines. The resulting populations were composed of between 278 and 365 lines.
Some parental lines were used in more than one cross: Costaud and OT96-23 were each a parent in three different populations, while Misty was a parent in two populations.
II.2.2 DNA extraction, preparation of genotyping-by-sequencing (GBS)
libraries and sequencing
Genomic DNA was extracted using a CTAB-based protocol on four dried leaf punches collected off a single plant. The DNA concentration was measured using fluorometric quantification and it was adjusted to 10 ng/μl per sample. These DNAs were used to prepare
GBS libraries using the ApeKI enzyme as per Elshire et al. (2011) and 3 to 4 libraries were
prepared per population. Each library was sequenced separately on a single Ion PIÔ chip on
an Ion Torrent Proton sequencer at the «Plateforme d’analyses génomiques» (IBIS,
Université Laval).
II.2.3 SNP calling and data filtering
The resulting GBS-derived reads were processed using the Fast-GBS pipeline (Torkamaneh et al., 2017) and the soybean reference genome for cv. Williams 82 (Wm82.a2.v1). Briefly, SNP calls were made at loci where at least 5 reads were captured for an individual. Additionally, only calls with a genotype quality (GQ) score > 30 were retained. In the resulting catalogue, SNPs with missing data (MD) > 80% and a minor allele count (MAC < 8) were excluded as were lines with MD > 90 %. Finally, missing data were imputed using Beagle v 4.1 (Browning and Browning, 2007). By using TASSEL V5.0 (Bradbury et al., 2007), the imputed data set was further filtered to remove lines and SNPs with excess
31
heterozygosity, the exact threshold being determined in each population using the extreme outlier method (see Supplementary data). Finally, only SNPs with a minor allele frequency (MAF) > 0.3 were kept. Using a python script developed in-house, each resulting SNP dataset was converted (homozygotes being coded as “A” or “B”, heterozygotes as “H”) based on the origin of the allele. The converted data sets were corrected for double recombinants (DRs) using the Genotype Corrector (GC) script (Miao et al., 2018). A final conversion (homozygotes = 0 or 2, heterozygous = 1) was performed in view of the mapping step.
II.2.4 Construction of individual genetic linkage maps
The six individual linkage maps were generated using QTL IcIMapping V4.1 (Meng et al., 2015). As this software offers a limited choice in terms of the type of segregating population (F3 or RILs), we chose the F3 option to reflect the fact that our lines were not fixed. Genetic
distances were estimated using the Kosambi mapping function (Huehn, 2011). To produce the maps, we first performed binning to retain a single marker exhibiting a given segregation pattern within a population. In a first analysis, this reduced set of SNPs were grouped based on “the recombination events”, this parameter allows the grouping of markers based on the segregation of these markers. Then, marker ordering was also performed on the basis of marker segregation using the “nnTwoOpt” and a rippling step. In a second and independent analysis, the same set of SNP markers was used to form linkage groups on the basis of the known physical position of each marker (“anchor info” option). Markers were then ordered and assigned a genetic position, again with the help of the knowledge of their physical positions (“input order”). In a final step, rippling of markers was performed to optimize their location.
II.2.5 Construction of the consensus map
The consensus genetic linkage map was constructed using the six individual linkage maps. An R package, “LPmerge” (https://cran.r-project.org/web/packages/LPmerge/index.html) (Endelman and Plomion, 2014) was used with its default options to integrate the data from the different mapping populations. LPmerge resolves conflicts in marker order by minimizing the error between the consensus map and the individual linkage maps.
32
II.2.6 Estimation of the number of recombination events
A recombination event (RE) was observed when an allele switch occurred along a chromosome, i.e. a long succession of alleles from one parent being replaced by another suite inherited from the other parent. The number of such switches was counted along each chromosome for each individual line.
33
II.3 Results
II.3.1 SNP genotyping of six F
5mapping populations
In view of producing genetic maps, a total of 1,857 F5 individuals from six mapping
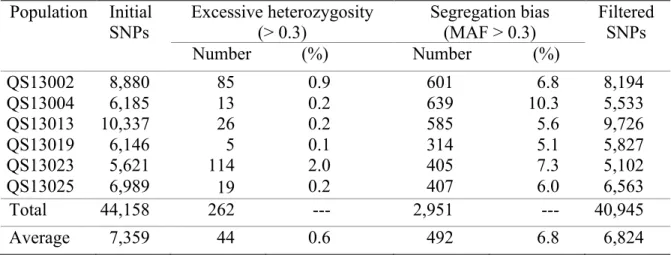
populations (n=278 to 365) were genotyped via a GBS approach. On average, 806K reads (mean length = 149 bp) were obtained per line and used to call SNPs. As shown in Table II-1, the number of SNPs called initially within a mapping population ranged between 5,621 and 10,337 (mean = 7,359). These initial SNP data were then filtered to eliminate loci presenting either an excessive proportion of heterozygosity (> 0.3, typically due to paralogues) or a segregation bias (MAF < 0.3). The proportion of SNPs eliminated due to excess heterozygosity averaged only 0.6% and was no more than 2%. The fraction eliminated on the basis of segregation bias averaged 6.8% and ranged between 5.1 and 10.3%. The final set of SNPs, after filtration, averaged 6,824 and varied between 5,102 and 9,726 markers.
Table II-1 Number and properties of the SNPs segregating in the six mapping populations of soybean.
Using these filtered SNPs, we could verify the composition of the segregating populations. In the six populations, all progenies were segregating for the markers found to be polymorphic between the parental lines and thus truly progeny of the same cross. None of the progenies were found to be the result of selfing and a total of only four lines (2 in QS13002 and 2 in QS13004) were eliminated because they presented an excessive degree of heterozygosity (> 0.36), possibly due to cross contamination of these DNA samples.
Population Initial SNPs Excessive heterozygosity (> 0.3) Segregation bias (MAF > 0.3) Filtered SNPs Number (%) Number (%) QS13002 8,880 85 0.9 601 6.8 8,194 QS13004 6,185 13 0.2 639 10.3 5,533 QS13013 10,337 26 0.2 585 5.6 9,726 QS13019 6,146 5 0.1 314 5.1 5,827 QS13023 5,621 114 2.0 405 7.3 5,102 QS13025 6,989 19 0.2 407 6.0 6,563 Total 44,158 262 --- 2,951 --- 40,945 Average 7,359 44 0.6 492 6.8 6,824