© Dazhi Li, 2019
Synthesis and Applications of Novel Chiral NHC
Precursors. Synthesis of Urea Derivatives through
Decomposition of Cu-NHC under Air. Iron-mediated
Synthesis of Dihydroquinoxalinones
Thèse
Dazhi Li
Doctorat en chimie
Philosophiæ doctor (Ph. D.)
Québec, Canada
Synthesis and Applications of Novel Chiral NHC
Precursors. Synthesis of Urea Derivatives through
Decomposition of Cu-NHC under Air.
Iron-mediated Synthesis of Dihydroquinoxalinones
Thèse
Dazhi Li
Sous la direction de :
iii
Résumé
Depuis sa première isolation, les ligands carbènes N-hétérocycliques (NHC) s’avèrent très utiles pour la coordination avec les métaux de transition ainsi que pour la catalyse. Étant abondant et moins onéreux, le fer en tant que catalyseur a connu un essor considérable au cours de ces dernières décennies. De nombreux Fe-NHCs ont été synthétisés, mais le Fe-NHC chiral utilisé pour la catalyse asymétrique en est encore à ses débuts. En comparaison avec les métaux rares, le cuivre en tant que métal de transition polyvalent et moins coûteux, a également suscité beaucoup d’attention. Cependant, le développement du Cu-NHC chiral en tant que catalyseur efficace reste difficile. Ainsi, plusieurs types de nouveaux précurseurs de ligand NHC chiral ont été synthétisés. Les synthèses de Fe-NHCs et de Cu-NHCs chiraux ont été initiées à partir des précurseurs chiraux. Il a été constaté que les Fe-NHCs et les Cu-NHCs se décomposent au contact de l’air. Les Fe-NHCs et Cu-NHCs chiraux générés in situ sont utilisés dans les réactions d'hydrosilylation, les réactions de Mukaiyama aldol, l'insertion de carbène métallique dans la liaison SiH et les réactions de type Heck. Les Fe-NHCs in situ se sont avérés non utilisables dans la réaction d'hydrosilylation de l'acétophénone. Pour les réactions de Mukaiyama aldol, les conditions d'utilisation de Fe-NHCs in situ ont permis d'obtenir les produits souhaités avec un rendement allant jusqu'à 88%. Cependant, aucune énantiosélectivité n'a été observée, probablement pour des raisons de désactivation du ligand NHC. La réaction d'insertion du métal-carbène dans la liaison SiH catalysée in situ par des Cu-NHCs a donné un rendement pouvant atteindre 84% et 24% ee de produit. En outre, les réactions de type Heck ont été testées avec un catalyseur chiral Pd-NHC, qui a aboutit à un rendement supérieur à 91% sans avoir fournir d’énantiosélectivité. De plus, les décompositions de différents types de Cu-NHCs et Ag-NHCs dans des solutions sous air humide ont été étudiées. L’hydrolyse et l’oxydation de Cu-NHCs ont généré, sous air, des imidazoliums et des dérivés d'urée. Les Ag-Cu-NHCs ont été hydrolysés pour donner des formamides ou des imidazoliums en solution sous air humide. Par la suite, une nouvelle méthode de synthèse du dérivé d'urée utilisant du cuivre et de l'air en tant qu'oxydant a été developpée. Elle a permis d'obtenir des rendements modérés voire même très bons pour des substrats sans encombrement stérique. Les conditions d'oxydation douces conviennent à la synthèse de dérivés d'urée possédant des groupes alkyle, benzyle, aryle, hydroxy primaire, un groupe tert-butyloxycarbonyle sensible aux acides et des groupes amine tertiaire. Dans le dernier projet, une synthèse générale et efficace des dihydroquinoxalinones énantiopures a été développée. La cyclisation réductrice de N-(o-nitroaryl)amino esters a été réalisée en utilisant du fer et du zinc métallique dans des conditions douces pour donner des dihydroquinoxalinones avec des rendements modérés à élevés et une pureté énantiomérique élevée.
iv
Abstract
Since its first isolation, N-heterocyclic carbenes (NHC) have been found very useful to coordinate with metals and serve as ligand in catalysis. With the advantages of environmental friendliness, abundance and being less expensive, iron as a metal catalyst has received growing attention in recent decades. Despite that many Fe-NHCs have been synthesized, chiral Fe-NHC for asymmetric catalysis is still in its infancy. In comparison to precious metals, copper as a versatile and less expensive transition metal also has recieved much attention. However, the development of chiral Cu-NHC as efficient catalyst is still challenging. Thus, several types of novel chiral NHC ligand precursors have been synthesized. The synthesis of chiral Fe-NHCs and Cu-NHCs were attempted using those chiral precursors. It was found that the Fe-NHCs and Cu-NHCs would decompose under air. On the other hand, the applications of in situ generated generated chiral Fe-NHCs and Cu-NHCs were carried out for hydrosilylation reactions, Mukaiyama aldol reactions, insertion of metal-carbene into SiH bond and Heck-type reactions. The in situ generated Fe-NHCs were found not applicable in the hydrosilylation of acetophenone. For the Mukaiyama aldol reactions, the conditions using in situ generated Fe-NHCs led to the desired products in up to 88% yield. However, no enantioselectivity was observed for all attempts, probably due to the deactivation of NHC ligand. The insertion reaction of metal-carbene into SiH bond catalyzed by in situ generated Cu-NHCs afforded up to 84% yield and 24% ee of product. Besides, the Heck-type reactions were tested using a chiral Pd-NHC as catalyst. The reactions afforded up to 91% yield, but no enantioselectivity was observed. Furthermore, the decompositions of different types of Cu-NHCs and Ag-NHCs in solutions under humid air were studied. The Cu-NHCs underwent hydrolysis and oxidation to generate imidazoliums and urea derivatives under air. The Ag-NHCs were hydrolyzed to yield formamides or imidazoliums in solution under humid air. Subsequently, a new synthetic method of urea derivative using copper and air as oxidant was developed, which provided moderate to very good yields for sterically unhindered substrates. The mild oxidation conditions are suitable for the synthesis of urea derivatives possessing alkyl, benzyl, aryl, primary hydroxy, acid-sensitive tert-butyloxycarbonyl group, and tertiary amine groups. In the last project, a general and efficient synthesis of enantiopure dihydroquinoxalinones has been developed. The reductive cyclization of N-(o-nitroaryl)amino esters was performed by using iron and zinc metal under mild conditions to afford dihydroquinoxalinones in moderate to high yields and high enantiomeric purity.
v
Table of Contents
Résumé ... iii
Abstract ... iv
Table of Contents ... v
List of Figures ... viii
List of Schemes ... xi
List of Tables ... xvi
List of Abbreviations ... xvii
Acknowledgments ... xx Foreword ... xxi General Introduction ... 1 N-Heterocyclic Carbene ... 1 Chemical Properties of NHC... 4 Dimerization of NHC ... 4
Basicity and Nucleophilicity of NHC ... 6
Chemical Reactions of NHC ... 8
Chiral NHC as Organocatalyst ... 14
Metal-NHC Complexes ... 25
Bonding between Metal and NHC ... 25
Drawing of M-NHC Bond ... 26
Metal-NHC Complexes ... 27
Synthesis of Metal-NHC Complexes ... 28
Fe-NHC Complexes and Catalysis ... 29
Structural Diversities of Fe-NHCs ... 29
Fe-NHCs as Catalysts... 30
Challenges of Fe-NHCs in Asymmetric Catalysis ... 40
Cu-NHC Complexes and Catalysis ... 43
Cu-NHC Complexes ... 43
Chiral Cu-NHCs in Asymmetric Catalysis ... 44
Challenges of Cu-NHCs in Asymmetric Catalysis ... 46
vi
Chapter 1 Synthesis and Applications of Chiral NHC Precursors ... 49
1.1 Synthesis of Chiral NHC Precursors ... 49
1.1.1 Synthesis of Pyridyl- and Bipyridyl-based Chiral NHC Precursors... 49
1.1.2 Synthesis of Chiral NHC Precursors Using Natural Amino Acids ... 53
1.2 Synthesis of Chiral M-NHCs ... 59
1.2.1 Synthesis of Chiral Fe-NHCs ... 59
1.2.2 Synthesis of Chiral Cu-NHCs ... 61
1.2.3 Synthesis of Chiral Pd-NHCs ... 64
1.3 Applications of Chiral NHC precursors ... 66
1.3.1 Hydrosilylation of Ketone ... 66
1.3.2 Mukaiyama Aldol Reaction ... 68
1.3.3 Insertion of Metal-Carbene into SiH bond ... 71
1.3.4 Heck-type reaction ... 74
Chapter 2 Decomposition of Metal-NHC Complexes under Air ... 76
2.1 Introduction ... 76
2.2 Results and Discussion ... 80
2.2.1 Unsuccessful Isolation of Fe-NHCs and Cu-NHCs under Air ... 80
2.2.2 Decomposition of Cu-NHC in Solution under Air ... 83
2.2.3 Steric Effect on Decomposition of Cu-NHC ... 96
2.2.4 Effect of Other Properties on Decomposition of Cu-NHC ... 102
2.2.5 Postulated Mechanism on Decomposition of Cu-NHC... 107
2.2.6 Decomposition of Ag-NHC in Solution under Air ... 113
Chapter 3 Synthesis of Urea Derivatives through Oxidation Using Copper and Air ... 120
3.1 Résumé français ... 122
3.2 Abstract ... 123
3.3 Introduction ... 124
3.4 Results and Discussion ... 126
3.5 References ... 132
Chapter 4 Iron- or Zinc-mediated Synthetic Approach to Enantiopure Dihydroquinoxalinones .... 134
4.1 Résumé français ... 136
4.2 Abstract ... 137
4.3 Introduction ... 138
vii
4.5 References and Notes ... 148
Chapter 5 Experimental Section ... 152
5.1 General Information ... 152
5.2 Synthesis of NHC Precursors ... 153
5.3 Synthesis of Metal-NHC complexes ... 195
5.4 Typical Procedures for Catalytic Reactions ... 203
5.5 Experimental Procedures for Cu-NHC Decomposition Study ... 206
5.6 Computational Details ... 207
5.7 Procedures for Synthesis of Ureas and Characterization Data ... 218
5.8 Procedure for Synthesis of Dihydroquinoxalinones and Characteriz-ation Data ... 229
Conclusions ... 248
References and Notes ... 253
Appendix ... 269
1 H and 13C NMR Spectra of NHC Precursors ... 269
1 H and 13C NMR of M-NHCs ... 289 1 H and 13C NMR of Ureas ... 308 1 H and 13C NMR of Dihydroquinoxalinones ... 328
viii
List of Figures
Figure 0.1 Orbital diagram of sp-hybridized and sp2-hybridized carbene... 1
Figure 0.2 Four possible electronic configurations of a bent carbene ... 1
Figure 0.3 Eletronic configuration of NHC ... 2
Figure 0.4 Three types of bondings between NHC and metal... 25
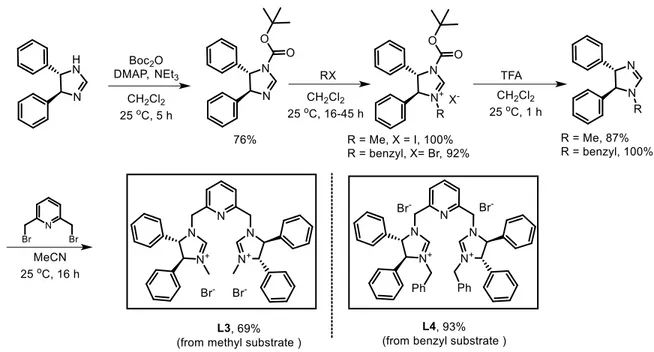
Figure 1.1 Crystal structure of L2 (PF6- and solvent are omitted for clarity) ... 51
Figure 1.2 Crystal structure of L4 (PF6 - counterions are omitted for clarity) ... 52
Figure 1.3 HRMS of a recovered Cu-catalyst ... 73
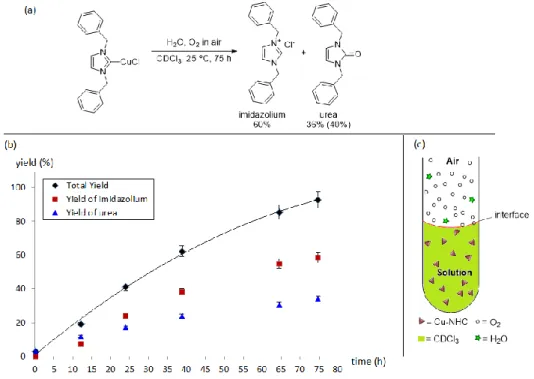
Figure 2.1 1H NMR of Cu-NHC 3a (initial 0.1 mol/L in CDCl3, 400MHz) under air at different times, and 1H NMR of isolated 1,3-dibenzyl-1H-imidazol-3-ium chloride and 1,3-dibenzyl-1,3-dihydro-2H-imidazol-2-one. ... 84
Figure 2.2 Appearance of Cu-NHC solutions (initial 0.1 mol/L in CDCl3) under air: before (a) and after (b) decomposition of 3a; before (c) and after (d) decomposition of 3c; before (e) and after (f) decomposition of 3d; before (g) and after (h) decomposition of 3g. ... 84
Figure 2.3 (a) Decomposition of Cu-NHC 3a under air; Yields determined by 1H NMR (isolated yield in parenthesis). (b) Time-dependent yields of imidazolium and urea during the decomposition of 3a (initial 0.1 mol/L in CDCl3). (c) The diagram of the Cu-NHC solution under air. ... 85
Figure 2.4 Plot of the natural logarithm of the Cu-NHC concentration (ln[Cu-NHC]) of 3a against time (initial 0.1 mol/L in CDCl3; concentration determined by 1 H NMR). ... 86
Figure 2.5 (a) Decomposition of Cu-NHC 3b under air; Yields determined by 1H NMR. (b) Time-dependent yields of imidazolium and urea during the decomposition of Cu-NHC 3b (initial 0.1 mol/L in CDCl3) under air. ... 87
Figure 2.6 1H NMR of Cu-NHC 3c (initial 0.1 mol/L in CDCl3, 400 MHz) under air. ... 88
Figure 2.7 (a) Decomposition of Cu-NHC 3c under air; Yields determined by 1H NMR (isolated yield in parenthesis). (b) Time-dependent yields of urea during the decomposition of Cu-NHC 3c (initial 0.1 mol/L in CDCl3) under air. ... 89
Figure 2.8 (a) Decomposition of Cu-NHC 3d under air; Yields determined by 1H NMR (isolated yield in parenthesis). (b) Time-dependent yields of benzimidazolium and urea during the decom-position of Cu-NHC 3d (initial 0.1 mol/L in CDCl3) under air. ... 90
ix
Figure 2.9 (a) Decomposition of Cu-NHC 3e under air; Yields determined by 1H NMR (isolated yield in parenthesis). (b) Time-dependent yields of benzimidazolium and urea during the decom-position of Cu-NHC 3e (initial 0.1 mol/L in CDCl3) under air. ... 91
Figure 2.10 (a) Decomposition of Cu-NHC 3f under air; Yields determined by 1H NMR. (b) Time-dependent yields of urea and imidazolinium during the decomposition of Cu-NHC 3f (initial 0.1 mol/L in CDCl3) under air. ... 92
Figure 2.11 (a) Decomposition of Cu-NHC 3g under air; Yields determined by 1H NMR. (b) Time-dependent yields of urea and imidazolinium during the decomposition of Cu-NHC 3g (initial 0.1 mol/L in CDCl3) under air. ... 93
Figure 2.12 (a) Decomposition of Cu-NHC 3h under air at 100 oC; Yields determined by 1H NMR. (b) Time-dependent yields of urea and imidazolinium during the decomposition of Cu-NHC 3h (initial 0.1 mol/L in (CD3)2SO) under air at 100
o
C. ... 94
Figure 2.13 (a) Decomposition of Cu-NHC 3i under air at 150 oC; Yields determined by 1H NMR. (b) Time-dependent yields of urea and imidazolinium during the decomposition of Cu-NHC 3i (initial 0.05 mol/L in (CD3)2SO) under air at 150
o
C. ... 95
Figure 2.14 (a) Decomposition of Cu-NHC 3j under air at 150 oC; Yields determined by 1H NMR. (b) Time-dependent yields of urea and imidazolinium during the decomposition of Cu-NHC 3j (initial 0.05 mol/L in (CD3)2SO) under air at 150
o
C. ... 95
Figure 2.15 (a) 3D diagram of NHC ligand of 3a buried in a sphere of radius 3.5 Å (centering at
copper atom); (b) The steric map (contour) of NHC ligand of 3a. ... 96
Figure 2.16 The steric maps (contours from optimized structures) of NHC ligands 3a-j. ... 99 Figure 2.17 Plot of the yields of urea and imidazolium products against the NHC binding energy
gaps between H2O and O2 for different Cu-NHCs. Note that 3h reacted at 100 o
C, 3i and 3j reacted at 150 oC. ... 104
Figure 2.18 Plot of the C-Cu bond order against the %VBur. ... 107
Figure 2.19 The HOMO and LUMO orbitals of Cu-NHCs at PBE/LanL2DZ level ... 108 Figure 2.20 (a) The HRMS (ESI-TOF) spectrum of a Cu(I)-NHC after exposure to air. (b) The
experimental and (c) the theoretical isotopic distribution of the Cu(II)-NHC in HRMS. ... 110
Figure 2.21 Energy profile for the Cu-NHC hydrolysis calculated at PBE/LanL2DZ level. ... 111 Figure 2.22 Energy profile for the Cu-NHC oxidation calculated at PBE/LanL2DZ level ... 111 Figure 2.23 Decomposition of Ag-NHC 3k (initial 0.05 mol/L in CDCl3 as a mixture with
x
Figure 2.24 (a) Time-dependent concentration of Ag-NHC 3k and formamide 3l (as mixture) in
CDCl3 under air determined by 1
H NMR. (b) Plot of the natural logarithm of the Ag-NHC con-centration (ln[Ag-NHC]) of 3k against time. ... 116
Figure 2.25 The decomposition of Ag-NHC 3m (initial 0.38 mol/L in CH3CN/H2O, (2/1, v/v))
under air at 70 oC at different time ... 118
Figure 3.1 (a) HRMS (ESI-TOF) spectrum of the reaction mixture of 4c. (b) Experimental and (c)
theoretical isotope distribution of the Cu-monoNHC found in the mixture. ... 130
xi
List of Schemes
Scheme 0.1 Synthesis of 1,3-diadamantylimidazol-2-ylidene (C = 13C chemical shift of carbene) .. 2
Scheme 0.2 Structures of several persistent carbenes (C = 13 C chemical shift of carbene) ... 3
Scheme 0.3 The Wanzlick’s equilibrium bewteen free NHC and dimer ... 4
Scheme 0.4 Dimerization pathway of a noncyclic diaminocarbene ... 5
Scheme 0.5 Proton-catalyzed dissociation of NHC dimer ... 5
Scheme 0.6 Dimerization of a bridged NHC ... 6
Scheme 0.7 Preparation of free NHC through deprotonation ... 6
Scheme 0.8 pKa values of some NHC conjugate acids ... 7
Scheme 0.9 Reactions using NHC as nucleophile ... 8
Scheme 0.10 Increasing order of the nuleophilicity and eletrophilicity of NHC ... 8
Scheme 0.11 Chemicals reactions of 1,3-diphenyl-imidazolidinylide ... 9
Scheme 0.12 Chemicals reaction of 1,3-diphenyl-imidazolidinylide with benzaldehyde ... 9
Scheme 0.13 Reaction of 1,3-dimesityl-imidazolidinylide with benzaldehyde ... 10
Scheme 0.14 Reactions of a triazole-based free NHC ... 10
Scheme 0.15 Reactions of a triazole-based free NHC ... 11
Scheme 0.16 Reactions of NHC with some main group element compounds ... 12
Scheme 0.17 Insertion of carbene into C−H bond ... 13
Scheme 0.18 Air-stable free NHCs ... 13
Scheme 0.19 Formation of the nucleophilic Breslow intermediate ... 14
Scheme 0.20 Benzoin condensation catalyzed by in situ generated NHC ... 14
Scheme 0.21 Benzoin condensation catalyzed by different in situ generated NHCs ... 15
Scheme 0.22 Cross-benzoin condensation catalyzed by in situ generated NHC ... 16
xii
Scheme 0.24 Cross-benzoin condensation catalyzed by in situ generated NHC ... 17
Scheme 0.25 Intramolecular Stetter reaction catalyzed by in situ generated NHC ... 18
Scheme 0.26 Intramolecular Stetter reaction catalyzed by in situ generated NHC ... 18
Scheme 0.27 Racemization of 1,4-dicarbonyl compound in the presence of NHC ... 18
Scheme 0.28 Formation of quaternary stereocenters through Stetter reaction ... 19
Scheme 0.29 Enantio- and diastereoselective Stetter reaction catalyzed by NHC ... 19
Scheme 0.30 Intermolecular Stetter reaction catalyzed by in situ generated NHC ... 20
Scheme 0.31 Intermolecular Stetter reaction catalyzed by polymer-supported catalyst ... 21
Scheme 0.32 Kinetic resolution of chiral alcohol through redox esterification by NHC ... 22
Scheme 0.33 Homoenolate formation of conjugate aldehyde with NHC (conjugate umpolung)... 22
Scheme 0.34 Conjugate umpolung addition for the synthesis of -butyrolactone ... 23
Scheme 0.35 Enantioselective hetero-Diels−Alder reaction catalyzed by NHC ... 23
Scheme 0.36 1,2-Addition of trimethylsilylcyanide to aromatic aldehydes by NHC ... 24
Scheme 0.37 Different drawings of M-NHC bonds found in the literature ... 26
Scheme 0.38 Some M-NHCs from 1A to 5A main group ... 27
Scheme 0.39 Some M-NHCs from 1B to 8B transitional group ... 28
Scheme 0.40 Three general synthetic methods of M-NHCs ... 29
Scheme 0.41 Some Fe-NHCs with different Fe oxidation states ... 30
Scheme 0.42 Cross-coupling catalyzed by Fe(II)-NHC ... 32
Scheme 0.43 Cross-coupling via C-O bond activation catalyzed by in situ generated Fe(II)-NHC 33 Scheme 0.44 Cross-coupling catalyzed by in situ generated Fe(III)-NHC ... 33
Scheme 0.45 Cross-coupling catalyzed by Fe(II)-NHC ... 34
Scheme 0.46 Aldehyde olefination catalyzed by Fe(II)-NHC ... 35
xiii
Scheme 0.48 Hydrosilylation of aldehydes and ketones catalyzed by Fe(II)-NHC ... 36
Scheme 0.49 Hydrosilylation of aldehydes and ketones catalyzed by Fe(II)-NHC ... 36
Scheme 0.50 Transfer hydrogenation for aldehydes and ketones catalyzed by Fe(II)-NHC ... 37
Scheme 0.51 Selective reduction of esters to aldehydes catalyzed by Fe(0)-NHC ... 37
Scheme 0.52 Reduction of imines to amines catalyzed by Fe(0)-NHC ... 38
Scheme 0.53 Reduction of sulfoxides to sulfides catalyzed by Fe(II)-NHC ... 38
Scheme 0.54 Oxidative aromatic esterification of aldehydes using boronic acids ... 39
Scheme 0.55 Oxidation of p-xylene catalyzed by Fe(II)-NHC ... 40
Scheme 0.56 Asymmetric diethylzinc addition to aldehyde catalyzed by in situ generated Fe-NHCs ... 41
Scheme 0.57 Transfer hydrogenation catalyzed by Fe(II)-NHC ... 41
Scheme 0.58 Eantioselective C−H alkylation catalyzed by in situ generated Fe(III)-NHC ... 42
Scheme 0.59 Asymmetric Michael addition catalyzed by Fe(II)-NHC ... 42
Scheme 0.60 Structures of some Cu-NHCs ... 43
Scheme 0.61 Enantioselective allylic alkylations catalyzed by in situ generated Cu(I)-NHC ... 44
Scheme 0.62 Enantioselective allylic arylation catalyzed by chiral Cu(I)-NHC ... 45
Scheme 0.63 Enantioselective conjugate addition catalyzed by in situ generated Cu(I)-NHC ... 45
Scheme 0.64 Enantioselective conjugate -borylation catalyzed by Cu(I)-NHC ... 46
Scheme 0.65 Diastereoselective silylation of 3-acylindoles catalyzed by Cu(I)-NHC ... 46
Scheme 0.66 Design of novel NHC precursors ... 48
Scheme 0.67 Asymmetric reactions to be studied using chiral M-NHCs ... 48
Scheme 1.1 NHC precursors bearing pyridyl or bipyridyl or deriving from amino acid ... 49
Scheme 1.2 Synthesis of NHC precursor L1 and L2... 50
Scheme 1.3 Synthesis of NHC precursor L3 and L4... 51
xiv
Scheme 1.5 Synthesis of NHC precursor L6 and L7... 53
Scheme 1.6 Synthesis of NHC precursor L8, L9 and L10 ... 53
Scheme 1.7 Synthesis of NHC precursor L11 ... 54
Scheme 1.8 Synthesis of NHC precursor L12 ... 55
Scheme 1.9 Synthesis of NHC precursor L13-L16 ... 55
Scheme 1.10 Synthesis of NHC precursor L17 ... 56
Scheme 1.11 Synthesis of NHC precursor L18 ... 57
Scheme 1.12 Synthesis of NHC precursor L19 ... 57
Scheme 1.13 Synthesis of NHC precursor L20 ... 58
Scheme 1.14 Synthesis of NHC precursor L21 ... 58
Scheme 1.15 Formation of NHC-tBuOH adduct ... 62
Scheme 1.16 Synthesis of Cu-NHC bearing aromatic backbone through transmetalation ... 63
Scheme 1.17 Structures of Ag-NHCs and Cu-NHCs bearing aromatic backbones ... 63
Scheme 1.18 Synthesis of chiral Pd-NHCs... 64
Scheme 1.19 Enantioselective hydrosilylation of acetophenone catalyzed by Fe(OAc)2... 66
Scheme 1.20 Enantioselective Mukaiyama aldol reaction catalyzed by Fe(DS)2... 69
Scheme 1.21 Insertion of metal-carbene into SiH bond catalyzed by Fe(II) and Cu(I) salts ... 71
Scheme 2.1 Synthesis of urea derivative from imidazolinium ... 77
Scheme 2.2 Urea product obtained after catalytic reaction ... 77
Scheme 2.3 Decomposition of (a) free NHC and (b) Cu(II)-NHC ... 78
Scheme 2.4 Hydrolysis of three types of Ni-NHC ... 78
Scheme 2.5 The decomposition pathway of Pd-NHC in Mizoroki−Heck reaction ... 79
Scheme 2.6 Arylation and decomposition of Cu-NHC ... 79
xv
Scheme 2.8 By-products from Cu-NHCs after work-up under air ... 81
Scheme 2.9 By-products from Cu-NHCs after work-up under air ... 82
Scheme 2.10 Structures of the investigated Cu-NHCs ... 83
Scheme 2.11 Hypothesis of Cu-NHC oxidation with 3O2 and 1 O2 ... 90
Scheme 2.12 Postulated mechanisms of the decomposition of Cu-NHC under humid air... 109
Scheme 2.13 Stability comparison of Cu(II)-NHCs bearing different backbones... 113
Scheme 2.14 Hydrolysis of NHC precursors through Ag2O to generate formamides ... 114
Scheme 2.15 Decomposition pathway of Ag-NHC with saturated backbone ... 117
Scheme 2.16 Structures of Ag-NHCs isolable under air without decomposition ... 117
Scheme 2.17 The decomposition of Ag-NHC 3m (0.38 mol/L in CH3CN/H2O) ... 119
Scheme 3.1 Structures of selected biologically active urea derivatives ... 124
Scheme 3.2 Synthesis of urea derivatives by oxidation ... 125
Scheme 3.3 Synthesis of chiral imidazolidinones and benzimidazolones ... 126
Scheme 3.4 Synthesis of imidazolidinones, imidazolones, and benzimidazolones ... 128
Scheme 3.5 Proposed reaction pathway of the urea formation through oxidation of Cu-NHCs .... 130
Scheme 4.1 Unexpected synthesis of enantiopure dihydroquinoxalinone ... 138
Scheme 4.2 Synthetic routes of chiral dihydroquinoxalinones... 139
Scheme 4.3 Synthesis of N-(o-nitroaryl) amino acid esters ... 141
Scheme 4.4 Reductive cyclization reaction into of 3,4-dihydroquinoxalin-2(1H)-ones–Scope ... 144
Scheme 4.5 Synthesis of 3,4-dihydroquinoxalin-2(1H)-ones 5w, 5x, and 5y ... 145
Scheme 4.6 Ammonolysis of substrates 5l and 5m ... 146
Scheme 6.1 Synthesis of potential chiral ligands and Cu- and Fe-complexes ... 251
xvi
List of Tables
Table 1.1 Synthesis of Fe-NHCs under different conditions ... 60
Table 1.2 Synthesis of Cu-NHCs under different conditions ... 61
Table 1.3 Comparison of Fe, Cu, Ag, and Pd elements ... 65
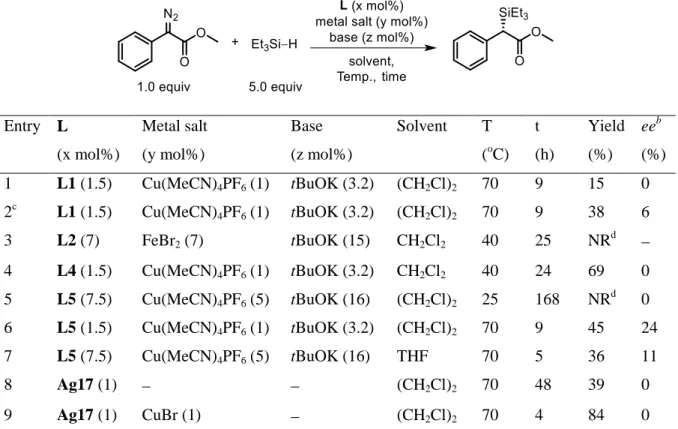
Table 1.4 Hydrosilylation of ketone catalyzed by chiral in situ generated M-NHCs ... 67
Table 1.5 Mukaiyama aldol reaction of silyl enol ether with benzaldehyde ... 69
Table 1.6 Insertion of metal-carbene into SiH bond ... 72
Table 1.7 Heck-type reaction catalyzed by chiral Pd-NHC... 74
Table 2.1 %VBur of NHC and C-Cu bond length of 3j using optimized structures computed by different DFT methods. ... 97
Table 2.2 Buried volume, rate constants (k) and half-reaction times (t50) of Cu-NHCs ... 98
Table 2.3 %VBur of chiral NHCs in the corresponding Cu-NHCs calculated by different methods 101 Table 2.4 Dipole moments, log P, HOMO/LUMO energies and binding energies of Cu-NHC .... 102
Table 2.5 13C NMR of NHC (NHC), Mulliken charge of NHC carbon, and the yields of urea and imidazolium for different Cu-NHCs ... 105
Table 3.1 Oxidation of dibenzylimidazolium bromide–optimization of the reaction conditions ... 127
Table 4.1 Optimization of the reaction conditions of dihydroquinoxalinones ... 142
xvii
List of Abbreviations
Å Angstrom (10-10 m) Ac Acetyl Bn Benzyl Boc tert-Butyloxycarbonyl o C Celsius degree Cp CyclopentadienylC=O Carbonyl group
d Doublet
DABCO 1,4-diazabicyclo[2.2.2]octane DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DCM Dichloromethame
dd Double doublet
DFT Density functional theory
DMAP 4-Dimethylaminopyridine DMF Dimethylformamide DMSO Dimethylsulfoxide dq Double quartet DS Dodecyl sulfate dt Double triplet ee Enantiomeric excess Et Ethyl g Gram
GGA Generalized gradient approximation
xviii HMPA Hexamethylphosphoramide HOMO Highest occupied molecular orbital HPLC High performance liquid chromatography
Hz Hertz
IR Infrared
J Coupling constant
KHMDS Potassium hexamethyldisilazide
L Liter
LUMO lowest unoccupied molecular orbital
m Multiplet
m-CPBA meta-Chloroperoxybenzoic acid
Me Methyl
Mg Milligram
mmol Millimole
mol Mole
MS Mass spectrometry or molecular sieves
nBu Normal butyl
NHC N-Heterocyclic carbene NMR Nuclear magnetic resonance OAc (or AcO) Acetate
o- ortho- Ph Phenyl PhI(OAc)2 (Diacetoxyiodo)benzene q Quartet quin Quintet R Radius
xix
s Singlet
t Time (or Triplet)
T Temperature
TBME tert-Butyl methyl ether TFA Trifluoroacetic acid
THF Tetrahydrofuran
TMEDA Tetramethylethane-1,2-diamine
tBu tert-Butyl
xx
Acknowledgments
This thesis has been accomplished under the supervision of Professor Thierry Ollevier. I appreciate the guidance and support provided by our supervisor, especially in the aspect of choosing projects, experiment design, and the writing of scientific articles and my dissertation. During my PhD study, Prof. Ollevier has been showing us a conscientious attitude towards learning, dynamic academic thinking and efficient working style, which will definitely have a positive influence on my future life and work. Here, I would like to express my sincere gratitute and best regard to my supervisor.
I would like to thank my dear colleagues, Dr. Hoda Keipour, Dr. Angela Jalba, Dr. Di Meng, Dr. Mao Li, Wan Xu, Virginie Carreras, Nour Tanbouza and Samuel Lauzon whom are very responsible for laboratory duties and are always ready to help each other. Special thanks must go to Dr. Mao Li and Dr. Di Meng for sharing living and study tips during my early period in Québec. Sincere gratitute must go to my intern Rongyu Sun for helping me to carry out many catalytic reactions. I would like to thank my friends, Dandan Miao, Tiezhu Liu, Fangru Meng, Liya Zhang, Tingfeng Han, Dan Wang, Ying Lu and Daniela Ron for the helps.
I would also like to thank Mr. Pierre Audet for his useful guidance on instrument operations. Sincere thanks also go to the department faculties: Prof. Jean-François Morin, Prof. Denis Giguére, Mme Denyse Michaud, Mme Mélanie Tremblay, Mr. Christian Côté and Mr. Jean Laferrière. I really appreciate Prof. Jean-François Morin, Prof. John Boukouvalas and Prof. Denis Giguère’s groups for the generous sharing of chemicals.
I also must thank my family members, and in particular my parents for giving their full love and support to my study and life. I would like to thank China Scholarship Council for the financial support for living during my PhD study.
xxi
Foreword
This thesis consists of a general introduction, five chapters: synthesis and applications of chiral NHC precursors (Chapter 1), decomposition of metal-NHC complexes under air (Chapter 2), synthesis of urea derivatives through oxidation using copper and air (Chapter 3), iron- or zinc-mediated synthetic approach to enantiopure dihydroquinoxalinones (Chapter 4) and experimental section (Chapter 5), and a general conclusion, followed by the references and appendix.
Chapter 3 and Chapter 4 are manuscripts of articles written in English without modification, which have been published in international scientific journals. The first article in Chapter 3, entitled Synthesis of Imidazolidinone, Imidazolone, and
Benzimidazolone Derivatives through Oxidation Using Copper and Air (Li, D.; Ollevier,
T.), was submitted to the international journal Organic Letters on 19 March 2019, and have been published on 6 May 2019. The second article in Chapter 4, entitled Iron- or
Zinc-Mediated Synthetic Approach to Enantiopure Dihydroquinoxalinones (Li, D.; Ollevier, T.),
was submitted to the international journal European Journal of Organic Chemistry on 3 November 2018, and have been published on 2 December 2018.
The contribution of each co-author in the two articles is described as follows:
1. Dazhi Li, Ph.D. candidate in chemistry, Département de chimie, Université Laval, first author on the two articles: experimentals, data processing, interpreting the results and writing of the drafts of the two articles in all sections.
2. Thierry Ollevier, Ph.D., professor and research director, Département de chimie, Université Laval, second author on the two articles: checking of data, revision of the manuscripts and submission of articles to publishers.
The written authorization of each co-author has been obtained so that the articles may be inserted in this thesis respecting the copyright clauses issued by Université Laval.
1
General Introduction
N-Heterocyclic Carbene
In chemistry, a carbene is a neutral, divalent carbon atom with six electrons in valence shell.1 The geometry of a carbene could be linear or bent, depending on the orbital hybridization of the carbon atom.2 A linear geometry of carbene indicates an sp-hybridization of carbon, possessing two nonbonding degenerated Px and Py orbitals (Figure 0.1(a)). A bent geometry of carbene adopts an sp2-hybridization of carbon. Thus, the degeneracy of Px and Py orbitals is interrupted. The Px orbital in a bent carbene acquires some s-orbital character, so it is also called σ-orbital. The Py orbital remains almost unchanged and it is usually called P orbital (Figure 0.1(b)).
Figure 0.1 Orbital diagram of sp-hybridized and sp2-hybridized carbene2
Due to the loss of degeneracy of Px and Py orbitals in a bent geometry, four electronic configurations are possible for an sp2-hybridized carbene (Figure 0.2). Three electronic configurations (Figure 0.2(a-c)) possess singlet multiplicity, and the last one (Figure 0.2(d)) has triplet multiplicity. Due to some of s-orbital character, the σ-orbital is lower in energy
2
than P orbital. The multiplicity of these bent carbene systems is closely related to the carbene -substituents (R1 and R2).1-2
As an sp2-hybridized carbene, N-Heterocyclic carbene (NHC) has received a great deal of attention in the past few decades, since the first isolable NHC at room temperature in 1991 (Scheme 0.1).3 In this unique and stable species, the two -amino substituents donate the electron density into the empty p orbital from the lone pairs of heteroatoms, while the
σ-inductive electron-withdrawing effect of the nitrogen atoms reduces the electron density of the non-bonding lone pair of the carbene center.1, 4 For this reason, the σ-orbital and the p
orbital has a large energy difference ( = 52.1 kcal/mol5 or 58.56 kcal/mol estimated by the quantum chemical calculations). Thus, NHCs exist as singlet state (Figure 0.2(a) and Figure 0.3).7-8 In addition, because of this special interaction, a four-electron-three-center π-system and a partial multiple bond character of C-N bond were considered (The bond length is in between those of C-N single and C=N double bond).9
Scheme 0.1 Synthesis of 1,3-diadamantylimidazol-2-ylidene3 (C = 13
C chemical shift of carbene)
Figure 0.3 Eletronic configuration of NHC
The stability of the first isolable NHC was considered attributable to the large steric hindrance of the N-admantyl groups, which prevents the dimerization of carbene. However, this factor was found not neccessary after the isolation of the NHC bearing small N-methyl groups in 1992, and no dimerization of this compund was found (Scheme 0.2(a)).10Thus,
3
Scheme 0.2 Structures of several persistent carbenes (C = 13
C chemical shift of carbene)
imidazole-2-ylidenes are thermodynamically stable under inert atmosphere. In addition to the steric property, the aromatic character of the five-membered ring system was also considered as an important factor in the early of 1990’s. The C=C double bond in the ring system can conjugate with the lone pairs of the nitrogens, thus the unsaturated NHC backbone was thought to have a delocalized system as a stabilizing factor. In 1995, the isolation of a free carbene lacking the C=C double bond (Scheme 0.2(b)) showed that the aromatic character of the backbone was not a dominant feature for the stabilization of NHC.11 Later in 1996, a noncylic NHC crystal (Scheme 0.2(c)) was isolated under nitrogen and studied by X-ray diffraction.12 This case proved that the five-membered heterocycle of NHC was not necessary. The NCN bond angle of the noncyclic NHC was determined as 121.05o, indicative of sp2-hybridization of the carbene (the usual NCN bond angle of a cyclic NHC was in the range of 100-110o due to the ring constraint).2 The carbenes introduced above are all stabilized by two adjacent amino groups, which was thought as an important character. However, in 1997 and 1998, the isolation of carbenes adjacent to sulfur (Scheme 0.2(d))13 and oxygen (Scheme 0.2(e))14 precluded the necessity of two amino substituents within the structure. Later, the presence of two heteroatoms adjacent to carbene center was proven unnecessary by Bertrand (Scheme 0.2 (f)).15 In addition to the carbene of five-membered ring and acyclic carbene, the isolation of the carbene of six-membered ring (Scheme 0.2 (g))16 and seven-membered ring (Scheme 0.2 (h))17 further
4
expend the scopes of NHC. The stabilizing factors of NHC are still disputable and it seems to be a combination of both steric and electronic properties of NHC. So far, there have been many types of NHCs found in the literature.18-21 In this thesis, the focus is the diamino-substituted N-heterocyclic carbene with five-membered ring, and the other types of NHCs will not be discussed in detail.
As shown in Scheme 0.1 and 0.2, the 13C chemical shift of usual NHC carbene is in the range of 210260 ppm, representative of a very down-field carbon. When adjacent to only one nitrogen, the 13C chemical shift is even larger than 300 ppm. The typical 13C signal of NHC is an important and usful method for its charaterization. Because of the unique eletron properties, the family of NHC has special chemical behaviors, which will be discused next.
Chemical Properties of NHC
Dimerization of NHC
In 1962, Wanzlick reported an equilibrium between 1,3-diphenyl-imidazolidinylide and its dimer during the study of the NHC (Scheme 0.3).22 The isolation of the NHC was unsuccessful, and the dimer which was claimed to dissociate at 170 oC was obtained. The dissociation of the dimer and the equilibrium were turned out to be impossible by Lemal in 196423 and Winberg in 1965.24 The report by Wanzlick led to the notion that free NHCs dimerize easily at room temperature. However, many NHCs actually did not dimerize or decompose either in solution or in solid state at room temperature as long as they were kept in an inert environment, including the sterically unhindered NHC shown in Scheme 0.2(a).
5
Moreover, the evidence for this equilibrium has been shown by some specific benzimidazolylidenes.25 Thus, the Wanzlick’s equilibrium has raised an intense debate.26-27 In spite of the debate about Wanzlick’s equilibrium, the dimerized product of NHC was indeed observed and obtained. However, for the most of cases, the dimerization probably did not occurr directly through two molecules of carbenes, but through proton27 or Lewis acid-catalyzed pathway,26 or through the nucleophilic attack of carbene to the amidinium salt (Scheme 0.4(a), which also could be considered as proton-catalyzed pathway).28-29 In fact, in the presence of enough base (then no amidinium salt or proton was left), the free diaminocarbene shown in Scheme 0.4 remained essentially unchanged in a THF solution under nitrogen atmosphere at room temperature for at least a week (Scheme 0.4(b)).28 Also, the trace proton or Lewis acid in the solution could catalyze the dissociation of the NHC dimer as well, which could be an explanation for the observation of the Wanzlick’s equilibrium (Scheme 0.5).27, 30
Scheme 0.4 Dimerization pathway of a noncyclic diaminocarbene
6
According to Chen, only in a double-bridged and constrained configuration, were the free NHCs forced to dimerize (Scheme 0.6).31 For double-bridged NHC of larger bridge size, mono-bridged NHC and sterically non-hindered NHC, no dimerization was observed in solution (Scheme 0.6).
Scheme 0.6 Dimerization of a bridged NHC
Basicity and Nucleophilicity of NHC
The preparation of free NHC generally was through the deprotonation of the corresponding carbene precursors, e.g. imidazolium, imidazolinium, benzimidazolium, and triazolium (Scheme 0.7). For this reason, NHC could be also regarded as a base, and its basicity could be evaluated through the pKa of its conjugate acid (carbene precursor).
Scheme 0.7 Preparation of free NHC through deprotonation
The first measurement of the basicity of 1,3-diisopropyl-4,5-dimethyl-imidazol-2-ylidene was conducted by Williams and coworkers,32 and the pKa = 24.0 of its conjugated acid was determined by 1H NMR in (CD3)2SO. Subsequently, Toth’s group33 and O’Donoghue’s group34 determined the pKa of several imidazoliums and benzimidazoliums. In 2017, the acidity scale of a series of triazolium-based N-heterocyclic carbene precursors were determined by Cheng and coworkers.35 The effect of the N-substituents on the acidity of the carbene precursors was evaluated, and the results were compared with the prior reports.
7
Scheme 0.8 pKa values of some NHC conjugate acids32-35
It was found that the replacement of N-phenyl with electron donating or withdrawing groups caused appreciable change on the acidity. The pKa values of triazolium in DMSO are smaller than that in H2O. According to the previous work, the general and some specific pKa values of several types of NHC precursors are presented in Scheme 0.8. Among the structures shown in Scheme 0.8, the triazoliums are the most acidic (pKa = 1216; thus the NHC of triazolylidenes are the least basic), and the imidazoliums with N-aryl substituents are in the middle (pKa = 1922). The least acidic NHC precursors are the imidazoliums with N-alkyl substituents (pKa = 2325).
In addition to basicity, free NHC could react as a nucleophile with, for example, alkyl halide32 and ,-unsaturated aldehyde (Scheme 0.9)36 to offer 2-alkylated imidazolium and
8
enolate. The basicity and the nucleophilicity of NHC arise from the lone electron pair in the σ-orbital (Figure 0.3). Although the p orbital of NHC remains empty, due to the electron delocalization from the nitrogen atoms, the electrophilicity of NHC is diminished to a considerable extent.9 Incorporation of boron in the ring system of NHC is one of the methods to enhance its electrophilicity.37 Another strategy is to replace one of nitrogen atoms with carbon in an expanded ring size,38 as shown in Scheme 0.10, which results in higher HOMO (highest occupied molecular orbital) energy and lower LUMO (lowest unoccupied molecular orbital) energy (more -accepting) and consequently smaller HOMO-LUMO energy gap.
Scheme 0.9 Reactions using NHC as nucleophile
Scheme 0.10 Increasing order of the nuleophilicity and eletrophilicity of NHC
Chemical Reactions of NHC
As introduced before, the nuleophilic property enables NHC to react with electrophiles. In 1962, Wanzlick studied the chemical reactions of 1,3-diphenyl-imidazolidinylide which was generated in situ from pyrolysis (Scheme 0.11).22 The NHC was found to form addition product with O2 (Scheme 0.11(a)), alcohol adduct (insertion product into HOR
9
Scheme 0.11 Chemicals reactions of 1,3-diphenyl-imidazolidinylide
bond) (Scheme 0.11(b)), ring-opening product with H2O (Scheme 0.11(c)) and protonated product with HCl (Scheme 0.11(d)). The NHC was able to react with benzaldehyde to form a ketone (Scheme 0.11(e)), in which the nucleophilic attack of NHC to the aldehyde was considered as the first step (Scheme 0.12). In addition, the NHC could react with electron-deficient alkene through [2+1] cycloaddition to produce cyclopropane derivatives (Scheme 0.11(f)).
Scheme 0.12 Chemicals reaction of 1,3-diphenyl-imidazolidinylide with benzaldehyde
According to the studies by Berkessel,39-41 when sterically hindered carbene, such as 1,3-dimesityl-imidazolidinylide, was mixed with benzaldehyde, the product of enol was obtained instead of the ketone (Scheme 0.13). The data from theoretical computation also
10
supported that the enol from sterically hindered carbene was more thermodynamically stable than the ketone form. However, the ketones could be obtained through another synthetic method.40 For the less sterically hindered carbene, the ketone was more stable, such as the one shown in Scheme 0.11(e).
Scheme 0.13 Reaction of 1,3-dimesityl-imidazolidinylide with benzaldehyde
From 1995 to 1997, Enders investigated the chemical reactions of a triazole-based free NHC with different reagents (Scheme 0.14; only one substrate is shown for each entry).42-44 Instead of NHC generated in situ, in their study the NHC was indeed a free carbene which
11
was well characterized by 13C NMR and single crystal X-ray diffraction. The NHC was able to insert into HOR and HNR2 bonds with different alcohols (e.g. methanol, ethanol and pyridin-4-ylmethanol) and amines (e.g. morpholine, pyrrolidine and benzylamine) in 95-99% of yield (Scheme 0.14(a), (b)). The reaction of NHC with electron-deficient alkenes generated a [2+1] addition intermediate, followed by 1,2-H shift, and finally led to 5-methylene triazolines (Scheme 0.14(c)). In addition, the free NHC also reacted with oxygen, sulfur and selenium to afford the corresponding triazolinone, triazolinthione and triazolinselenone in good yields (Scheme 0.14 (d)). The triazole-based free NHC could be protonated by acid as expected, such as HClO4, leading to triazolium (not shown in Scheme).
The reaction of the NHC with trialkyloxonium salts (alkylating reagents) offered the corresponding alkylated triazolium in 93-95% of yield (Scheme 0.15(a)). The NHC was reactive with activated alkynes as well, such as methyl acetylenedicarboxylate, leading to a spirocyclic compound (Scheme 0.15(b)). This spirocyclic product was formed via addition
12
of the carbene to the triple bond, followed by 1,3-dipolar cycloaddition of the intermediate to a second molecule of alkyne.
Since the isolation of [2 + 1] cycloadducts turned out to be unsuccessful for this NHC, then Enders and coworkers attempted to obtain a [4 + 1] cycloadduct from an electron-deficient diene-type system, such as 3,6-diphenyl-l,2,4,5-tetrazine (Scheme 0.15(c)). However, the cycloadduct was not stable enough for the isolation. A spirocyclic product was isolated at the end. According to the study, the free carbene also reacted with heterocumulenes such as carbon disulfide, phenyl isothiocyanate and phenyl isocyanate to yield betaines. In the case of phenyl isocyanate, the corresponding betaine would continue to react with residual phenyl isocyanate to afford a spirocyclic compound (Scheme 0.15(d)). The reactions of triazolylidene could be regarded as several models for the chemical reactions of NHC.
With the progress of studies, NHC was found to form adducts or react with many compounds consisting of main group non-metal elements, such as boranes, halosilanes, nitrous oxide and diphosphenes (Scheme 0.16).18, 20, 45 It is noteworthy to mention that free NHCs were able to form adducts with CO2 reversibly (Scheme 0.16).46 The NHC-CO2 adducts have become very useful compounds (or intermediates) for the capture of CO2 and for the conversion of aromatic aldehydes with CO2 to form carboxylic acids.47
13
With only one nitrogen neighboring to the carbene in a six-membered ring, the NHC will be very reactive (both highly electrophilic and nucleophilic), and even allows for the intramolecular insertion into an unactivated C(sp3)−H bond (Scheme 0.17).38
Scheme 0.17 Insertion of carbene into C−H bond38
Although most of the reported free NHCs were sensitive to oxygen and moisture, some cases of air-stable free NHCs were found. In 1997, Arduengo and coworkers synthesized a 4,5-dichloroimidazol-2-ylidene (Scheme 0.18(a)).48 The free NHC as solid showed no reaction or decomposition after exposure to air in a laboratory hood for two days. A benzene solution of this NHC was also exposed to air overnight and no decomposition was detected. In 2015, Cole and coworkers published a 4,5-dibromoimidazol-2-ylidene (Scheme 0.18(b)).49 The dibromo-substituted free NHC was able to tolerate air for several weeks when stored in a desiccator. Storage in the absence of a desiccator resulted in decomposition over a week affording a complicated mixture. The reason for the special stability of 4,5-dihalogen-imidazol-2-ylidene NHC attributes to the strong -inductive effect of the chlorines or bromines, thus reducing the basicity and nucleophilicity of NHC to a considerable extent.
14
Chiral NHC as Organocatalyst
Since the first isolation of NHC in 1991, tremendous efforts have been made to develop various reactions as well as natural product synthesis using chiral NHC as organocatalyst. Many reviews have been published in this research area.50-55 One of the most important values of NHC is probably the NHC-catalyzed umpolung (German word umpolung for inversed polarity) of electrophilic aldehydes, through formation of the nucleophilic Breslow intermediate for many reactions (Scheme 0.19).52, 56 In this section, several types of asymmetric reactions using chiral NHCs will be discussed briefly.
Scheme 0.19 Formation of the nucleophilic Breslow intermediate
Benzoin Condensation
The asymmetric benzoin reactions catalyzed by thiazoliums have been known since 1966 by Sheehan.57 However, the early studies led to low yields and low enantioselectivity (22%
ee, enantiomeric excess). In 1996, the studies using a chiral triazolium by Enders and
coworker led to better yields and better enantioselectivities of benzoins (Scheme 0.20; other substrates are not shown in this scheme for simplicity).
15
Later, many efforts were invested to improve the enantioselectivities of the benzoin reactions. Three examples of NHC precatalyst by Leeper,58 Enders,59 and Connon,60 respectively, are present here (Scheme 0.21; other aldehyde substrates are not shown in this scheme for simplicity). The NHC precatalyst from Leeper’s group (Scheme 0.21(a)) contained a chiral center which is fixed by being incorporated into a bicyclic system. The fixed chiral center (i.e., the chiral center is not rotatable along CN bond) was expected to have a better chiral induction compare to the NHC shown in Scheme 0.20. Indeed, this NHC led to 80% ee of the benzoin product,58 which was 5% higher than the previous result shown in Scheme 0.20. In 2002, Enders and coworkers continued to optimize the condition. A bicyclic chiral NHC precatalyst with a bulky tert-butyl group was synthesized and tested in the reaction (Scheme 0.21(b)). The yield and enantioselectivity were further improved to 83% and 90%, respectively.59 In 2009, a chiral NHC precatalyst bearing a bulky hydroxy-diphenylmethyl and N-pentafluorophenyl group was synthesized by Connon’s group (Scheme 0.21(c)). The electron-withdrawing –C6F5 group led to an acidic NHC precatalyst, which is acidic enough to be deprotonated by a weak base (e.g. K2CO3 and iPr2NEt) to generate free NHC.60 This rigid and chiral NHC organocatalyst led to 90% yield and >99%
ee.
16
In addition to homo-benzoin, NHC precatalysts have been used for cross-benzoin reactions successfully. For example, in 2014, Johnson’s group reported a dynamic kinetic asymmetric cross-benzoin addition of aldehydes to ‑keto esters (Scheme 0.22).61 Different substrates were tested, and general good results were obtained. However, the conditions were not applicable to sterically hindered aromatic aldehydes (e.g. ortho-tolualdehyde, 0% yiled) and aliphatic aldehydes (e.g. isobutyraldehyde, 56:44 er (enantiomeric ratio), 51% yield). When the cross-benzoin was run on a 1 g scale, the same stereoselectivity was observed, but homo-benzoin (side reaction) increased and led to a by-product in 9% yield.
Scheme 0.22 Cross-benzoin condensation catalyzed by in situ generated NHC
In 2017, Soeta and coworkers studied the asymmetric cross-benzoin condensations between aliphatic and aromatic aldehydes, which were catalyzed by chiral triazolium NHCs bearing a pyridine moiety (Scheme 0.23).62 Twelve chiral NHC precatalysts were synthesized and tested. The NHC precatalyst leading to the best results are shown in Scheme 0.23. The studied found that the bulky substrates, such as iPrCHO (trace yield),
iBuCHO (24% yield, 17% ee) and ortho-tolualdehyde (0% yield), led to undesirable results.
Other linear aliphatic aldehydes offered moderate enantioselectivities.
17
In 2019, Ren and coworkers studied a series of cross-benzoin reactions between aldehydes and isatins catalyzed by chiral NHC (Scheme 0.24).63 The results showed that linear aliphatic aldehydes were well tolerated, as well as isatins bearing different electron-donating and electron-withdrawing groups. The studied also shown that the bulky aldehydes or sterically hindered isatins did not undergo any reactions. The aromatic aldehydes resulted in 32-63% yield and 28-43% ee (3 examples). In addition, the isatins with electron-withdrawing groups (F, Cl, Br and I) led to the desired products in approximate 50% yield as well as approximate 50% yield of undesired hydroacylation by-products. When the NO2-substituted isatin was used, only unknown mixture was obtained.
Scheme 0.24 Cross-benzoin condensation catalyzed by in situ generated NHC
The NHCs shown in Scheme 0.200.24 are all based on triazolium backbones probably due to the lower pKa values of this type of NHC precatalyst compared to other types of backbones (Scheme 0.8). The disadvantages of triazolium-based NHC organocatalysts in benzoin reactions include the intolerance of sterically hindered substrates, and varied enantioselectivities from a few percent to >99% depending on the substrates. In contrast with triazolium NHCs, imidazolium-based and benzimidazolium-based chiral NHC organocatalysts have not been explored for the asymetric benzoin condensations to date.
Stetter Reaction
In 1970’s, Stetter reported the reaction of the Breslow intermediate with ,-unsaturated carbonyl compounds (Michael acceptors), giving access to valuable 1,4dicarbonyl compounds, which is now called Stetter reaction.52, 64 In 1996, Enders reported the first example of the asymmetric intramolecular Stetter reaction, catalyzed by a chiral
triazolium-18
based NHC using K2CO3 as base (Scheme 0.25).65 Later, NHC-catalyzed asymmetric Stetter reactions continued to be studied by other research groups. In 2002, Rovis and coworkers synthesized and tested eight chiral NHC precatalysts in the intramolecular Stetter reactions.66 The NHC precatalyst in Scheme 0.26 showed the best results, affording up to 95% yield and 97% ee. When methyl (E)-3-(2-formylphenoxy)acrylate was used as
Scheme 0.25 Intramolecular Stetter reaction catalyzed by in situ generated NHC65
Scheme 0.26 Intramolecular Stetter reaction catalyzed by in situ generated NHC66
19
substrate, a racemic benzofuran-3(2H)-one derivative was obtained at the end (Scheme 0.27). At the beginning of the reaction (conversion = 10%), the ee of the product was determined as 80%. Due to the acidic nature of the benzofuran-3(2H)-one methine proton (pKa ~13), the compound rapidly reached at the equilibrium with its corresponding enolate, thus erosion of ee happened as the reaction progressed. This result indicated that strongly basic NHCs (pKa > 13) should be avoided when methyl (E)-3-(2-formylphenoxy)acrylate was used as substrate. In 2004, the same group reported the formation of quaternary stereocenters through Stetter reaction (Scheme 0.28).67 Due to the absence of -H next to the benzoyl, the product was not able to racemize through enolate, leading to high enantioselectivities. The established conditions were applicable to aliphatic aldehydes as well (Scheme 0.28(b)). In the subsequent year, the same group reported a series of highly
Scheme 0.28 Formation of quaternary stereocenters through Stetter reaction67
20
enantio- and diastereoselective Stetter reactions (Scheme 0.29).68 The studied found that the products would undergo epimerization in the presence of strong bases, such as KHMDS and DBU. Thus, the conditions using in situ generated NHC by deprotonation should be avoided. On the other hand, the use of isolated NHC was found not to cause epimerization, and the results were reproducible with high enantio- and diastereoselectivities.
In recent years, new triazolium-based NHC precatalysts have been synthesized and applied to various substrates, and excellent results were obtained.69-71 The immobilizations of the chiral NHCs for Stetter reaction were achieved as well, and the reusable catalysts showed promising results.72-73 In addition to intramolecular Stetter reaction, the intermolecular reactions have been also explored.71, 74-76 For example, Glorius and coworkers published the Stetter reactions of aldehydes with N-acylamido acrylates in 2011 (Scheme 0.30).77 The base was crucial for the reaction, and KOtBu was found as the best one. According to the computational study by Sunoj, KOtBu lowered the transition state energy and played an important role in the intermolecular proton transfer, thus providing the best results.75 It was also important to put less molar percent of base (8 mol%) than the NHC precatalyst (10 mol%), because the residue of strong base might lead to racemization of products.
Scheme 0.30 Intermolecular Stetter reaction catalyzed by in situ generated NHC77
In 2018, Ma and coworkers published the intermolecular Stetter reactions of salicyl-aldehydes with dimethyl acetylene dicarboxylate using organic polymeric nanospheres-supported triazolium carbene (Scheme 0.31).72 The polymer-supported catalyst led to the same enantioselectivity as homogeneous triazolium carbene did, and showed good
21
reusability for repeated experiments. The immobilized catalyst could possibly be used in other Stetter reactions, and further study could be carried out.
Scheme 0.31 Intermolecular Stetter reaction catalyzed by polymer-supported catalyst72
The Stetter reaction has become a very useful reaction to obtain 1,4-dicarbonyl compounds. The asymmetric Stetter reactions have been realized by different triazolium NHCs, providing good yields and good enantioselectivities for different substrates, either through an intramolecular way or intermolecular way. Due to the potential racemization of the products through enolate form, attention should be given to the choice and the amount of the base used in the Stetter reaction.
Other Reactions
In addition to the reactions mentioned before, NHC organocatalysts were found applicable to many other reactions, such as redox esterification, conjugate umpolung addition (through homoenolate equivalent), hetero-Diels-Alder reaction, 1,2-addition of silylcyanide to aldehydes, etc. Some examples have been highlighted below.
In 2005, Scheidt and coworker reported a redox esterification of ,-unsaturated aldehyde with racemic 1-phenylethanol in the presence of chiral benzimidazolium as precatalyst and PhOH as additive (Scheme 0.32).78 The kinetic resolution of secondary alcohol led to 24% ee of enantioenriched alcohol and 55% ee of ,-saturated ester when conversion reached 40% (selectivity factor, s factor = 4.8). The conditions were applicable to different primary and secondary alcohols with different ,-unsaturated aldehydes to obtain racemic products in up to 90% yield (15 examples), using
1,3-dimethyl-1H-22
benzo[d]imidazol-3-ium as achiral precatalyst. In spite of good yields, the enantioselectivity still needs to be improved.
Scheme 0.32 Kinetic resolution of chiral alcohol through redox esterification by NHC78
Scheme 0.33 Homoenolate formation of conjugate aldehyde with NHC (conjugate umpolung)
The addition of NHC to ,-unsaturated aldehyde leads to the formation of homoenolate (Scheme 0.33),52 which is quite useful in organic synthesis. For example, in 2008, Bode and coworkers published several different asymmetric reactions catalyzed by chiral imidazolium-based NHC, including the synthesis of -butyrolactone (Scheme 0.34).79 55% total yield and 32% ee for cis-product (major) were obtained. For some reactions including the synthesis of -butyrolactone, the results using this NHC precatalyst were better than that using triazolium-based precatalyst. In 2013, Yu and coworkers reported the synthesis of -butyrolactones using chiral imidazolium precatalysts, which involved the conjugate umpolung addition to aryl aldehydes (Scheme 0.34, other precatalysts and substrates from Yu’s group are not shown).80
23
The study of chiral NHC-catalyzed annulations of cinnamaldehyde with aldehydes is still limited, and the results need to be improved, especially for the enantioselectivity.
Scheme 0.34 Conjugate umpolung addition for the synthesis of -butyrolactone79-80
The NHC organocatalysts were found useful in hetero-Diels−Alder reactions as well. For example, in 2015, Smith and coworkers published the asymmetric redox hetero-Diels−Alder reactions catalyzed by chiral NHC (Scheme 0.35).81
High diastereo- and
24
enantioselectivity were obtained. The authors also proposed the mechanism for the reaction, and a simplified version is presented in Scheme 0.35. The deprotonation of triazolium generated a free NHC, which subsequently formed the Breslow intermediate. The leaving of arylcarboxylate group led to an enol, followed by deprotonation to form an enolate. The enolate reacted with ,-unsaturated ketone through [2+4] cycloaddition to form a cyclic intermediate, which collapsed to afford the final product and regenerate the free NHC.
In 2006, Suzuki and coworkers reported the chiral NHC-catalyzed 1,2-addition of trimethylsilylcyanide to aromatic aldehydes, as well as the proposed mechanism (Scheme 0.36).82 The authors also reported the results using different achiral NHC precatalysts (8 examples) and different substrates to obtained racemic cyanohydrins (9 examples; non-enantioselective examples are not shown in the scheme). The products were obtained in 65-98% yields. However, the two asymmetric examples showed low enantioselectivities. The enantioselective 1,2-addition to aldehydes are still underdeveloped.
Scheme 0.36 1,2-Addition of trimethylsilylcyanide to aromatic aldehydes by NHC82
As discussed before, NHC has become a very useful organocatalyst in many reactions, such as benzoin condensation, Stetter reaction, redox esterification, conjugate umpolung addition, hetero-Diels-Alder reaction and 1,2-addition of silylcyanide to aldehydes, etc. The formation of Breslow intermediate through NHC nucleophilic attack to carbonyl group has been proven as the key factor for the NHC-catalyzed umpolung. NHC as organocatalyst
25
undoubtedly will continue to offer more other useful and practical synthetic breakthroughs in the near future.52
Metal-NHC Complexes
Bonding between Metal and NHC
As shown in Figure 0.4, NHC possesses a lone electron pair in its -orbital, which makes NHC a good ligand for main group metals20, 83 and transitional metals83-84 through a dative coordination bond.85 The bondings between NHC and metals are classified as three types:
-bonding from the lone pair of NHC into the orbital of metal (Figure 0.4(a)), π-donation (Figure 0.4(b)), and π-backπ-donation (Figure 0.4(c)) between the π system of NHC and the dxz (or dyz) orbital of metal.4 The -donation (-bond) is the main contribution to
the bonding between NHC and metals (Figure 0.4(a)). The P orbital of NHC was considered not fully empty because the delocalization of electron from the neighboring nitrogen atoms. Thus, the π-donation from NHC to metal could be envisioned as well (Figure 0.4(b)). On the other hand, due to the fact that the P orbital could still accommodate electrons, π-backdonation from the filled orbital of metals to NHC is also possible (Figure 0.4(c)). The π-donation and π-backdonation are usually weak,86 because the four-electron-three-center π-system of N-C-N is already well stabilized.85 According to the discussion above, the bonding between NHC and transitional metal is quite complex. In some cases, the bonding between NHC and metal was mainly considered as single bond. In other cases, the bonding was considered to possess 15-30% of double bond character.4, 85 The Metal–NHC (M-NHC) interaction depends not only on the structure and properties of the NHC, but also on the electronic properties of the metal center.4 Hence, it would be better to carry out a specific analysis for each case of M-NHC for the bonding details.
26
Drawing of M-NHC Bond
There are different ways to draw the bond of metal-NHC (M-NHC) in the literature, and there seems to be no standard for the drawings (Scheme 0.37).
Scheme 0.37 Different drawings of M-NHC bonds found in the literature
Scheme 0.37(a)-(c) shows the drawing as an arrow from NHC to metal, which is the traditional drawing for a dative bond.20, 87 The NHCs in Scheme 0.37(a)-(c) are drawn clearly as a carbene with two dots beside the carbon, representing the lone electron pair. This kind of drawing are often applied to main group elements, such as M = Li, Na, K, Mg, B and Si,20-21 and sometimes applied to transition metals.19 Thus, the complexes containing main group elements are often called as the corresponding element-NHC adducts. In most cases, the drawing is often shown as a solid single bond (Scheme 0.37 (d)(h)). The drawing in Scheme 0.37(d) marks the carbene carbon using the capital letter “C” to emphasize that no hydrogen atom is attached to that carbon.83 Thus, the ambiguity that whether or not there is a hydrogen atom at C2 position like Scheme 0.37(e) is avoided. The drawing in Scheme 0.37(d) is also found for main group metals.83 Despite the ambiguous drawing in Scheme 0.37(e), many papers used this drawing for simplicity.84, 88-91 Scheme 0.37 (f),92-94 0.37(g)95-96 and 0.37(h)86, 97-98 show the popular drawings of the M-NHCs, emphasizing the delocalization of electrons and showing clearly no hydrogen attached to the C2 carbon. In rare cases, the bond of NHC with metal, such as Ag99 and Rh100 was drawn as double bond to show the significant π-backdonation (however, -donation is still
27
the main contribution to the bonding). In this thesis, the drawing of M-NHC in Scheme 0.37(h) is used.
Metal-NHC Complexes
Like in a phosphine, the lone pair of NHC is able to form a dative coordination bond towards metals. Because of the strong -donation ability, metal-NHC bond is usually stronger than metal-phosphine bond.101-102 In terms of organometallic chemistry, NHC is often considered as a close neighbor to phosphines.4 Now it should be clear that, in most cases of M-NHCs, the main bonding between NHC and metal is a dative covalent bond, just like the bond between phosphine and metal, or the bond between water and metal cations in metal aquo-complexes [M(H2O)x]n+, thus the formal oxidation state of the carbene and metals remain unchanged after complexation.
Due to easy modification of NHC structures and strong coordination of NHC to different metals, a great variety of M-NHCs have been synthesized in the recent decades. Some examples of M-NHCs with main group metals, such as Li,103 K,104 Be,105 Mg,106 Al,107 Ga,108 Ge,109 and Bi,110 are shown in Scheme 0.38.





![Figure 2.4 Plot of the natural logarithm of the Cu-NHC concentration (ln[Cu-NHC]) of 3a against time (initial 0.1 mol/L in CDCl 3 ; concentration determined by 1 H NMR)](https://thumb-eu.123doks.com/thumbv2/123doknet/3201454.91483/107.918.223.714.108.373/figure-plot-natural-logarithm-concentration-initial-concentration-determined.webp)

