Rabat
N° d’ordre 2322
THÈSE DE DOCTORAT D’ETAT
Présentée par
IDRISSI Laila
Discipline : Chimie
Spécialité : Chimie Analytique
ETUDE ET DEVELOPPEMENT DE NOUVELLES METHODES
ELECTROCHIMIQUES POUR LA DETERMINATION DES IONS
ORTHOPHOSPHATE, NITRITE, NITRATE ET AMMONIUM
Soutenue le 27 Décembre 2006
Devant le jury
Président :
A. Amine Professeur - Faculté des Sciences et Techniques de
Mohammedia
Examinateurs :
F. Cherkaoui El Moursli
Professeur - Faculté des Sciences - Rabat
A. Elyahyaoui Professeur - Faculté des Sciences - Rabat
A. Bouklouze Professeur - Faculté de Médecine et de Pharmacie
de Rabat
K. Digua Professeur - Faculté des Sciences et Techniques de
Mohammedia
Rabat
THÈSE DE DOCTORAT D’ETAT
Présentée par
IDRISSI Laila
Discipline : Chimie
Spécialité : Chimie Analytique
ETUDE ET DEVELOPPEMENT DE NOUVELLES METHODES
ELECTROCHIMIQUES POUR LA DETERMINATION DES IONS
ORTHOPHOSPHATE, NITRITE, NITRATE ET AMMONIUM
Soutenue le 27 Décembre 2006
Devant le jury
Président :
A. Amine Professeur - Faculté des Sciences et Techniques de
Mohammedia
Examinateurs :
F. Cherkaoui El Moursli
Professeur - Faculté des Sciences - Rabat
A. Elyahyaoui Professeur - Faculté des Sciences - Rabat
A. Bouklouze Professeur - Faculté de Médecine et de Pharmacie
de Rabat
K. Digua Professeur - Faculté des Sciences et Techniques
Mohammedia
Ce travail a été effectué sous la direction du Professeur Fouzia CHERKAOUI EL MOURSLI du Laboratoire d’Electrochimie et de Chimie Analytique de la Faculté des Sciences de Rabat.
Il est également le résultat d’une collaboration avec le Laboratoire d’Analyses Chimiques et Biocapteurs de la Faculté des Sciences et Techniques de Mohammedia.
Ma première pensée va tout naturellement à mon encadrante, le Professeur Fouzia CHERKAOUI EL MOURSLI dont j’ai apprécié sa grande chaleur humaine et sa disponibilité. Je la remercie pour la confiance qu’elle m’a témoignée et pour m’avoir permis de mener ce projet à son terme.
J’exprime ma vive reconnaissance au Professeur Aziz AMINE, co-directeur de thèse, pour m’avoir apporté un appui constant au cours du développement de ce travail et de me faire l’honneur de présider mon jury. Son soutien sans limite et ses qualités humaines resteront gravés dans ma mémoire.
Mes sincères remerciements vont également aux membres du jury de thèse : le Professeur Ahmed ELYAHYAOUI de la Faculté de Sciences de Rabat pour avoir mobilisé son temps et ses compétences pour examiner ce travail, le Professeur Abdelaziz BOUKLOUZE de la Faculté de Médecine et de Pharmacie de Rabat d’avoir accepté de juger et d’examiner mon manuscrit de thèse et le professeur Khalid DIGUA de la Faculté des Sciences et Techniques de Mohammedia qui m’a honoré en acceptant d’être rapporteur.
Je ne peux manquer de remercier toute l’équipe du Laboratoire d’Analyses Chimiques et Biocapteurs au sein de laquelle j’ai effectué mes travaux.
Je suis extrêmement reconnaissante à ma famille et mes amis qui m’ont soutenue et encouragé en tout moment.
Je remercie aussi mes enfants Moussa, Taha et Dina, de ne pas leur avoir consacré tout le temps nécessaire.
INTRODUCTION GENERALE………...………..………..1
CHAPITRE I : SYNTHESE BIBLIOGRAPHIQUE……….………4
A. Généralités sur les éléments à analyser : nitrite, nitrate, ammonium et orthophosphate...5
Introduction……….……5
I. Phénomène de l’eutrophisation………..5
II. L’azote inorganique en milieu aquatique……….7
II.1. Cycle de l’azote………...7
II.2. Origine des pollutions azotées……….8
II.3. Conséquences des pollutions azotées………..9
II.3.1. Pollution par les nitrites et nitrates………...9
a. Voies d’exposition………..……..9
b. Toxicité des nitrates………..9
c. Toxicité des nitrites……….10
II.3.2. Pollution par l’ammonium……….…11
III. Le phosphore………12
III.1. Le phosphore en milieu aquatique………...12
III.2. Le phosphore dans les biofilms de cyanobactéries……….…….13
B. Généralités sur les techniques utilisées pour la détermination des ions : Nitrate, nitrite, orthophosphate et ammonium ………..……….15
I. Méthodes d’analyse des nitrates et nitrites………..…………15
I.1. Méthodes spectroscopiques………..………..17
I .2. Méthodes électrochimiques………..……….19
I.3. La chromatographie ionique………..…….……21
II. Méthodes d’analyse des ions ammoniums……….………22
II.1. Méthodes spectrophotométriques………..…………22
II.2. Méthodes enzymatiques………23
II.3. Méthodes électrochimiques………...24
III. Méthodes d’analyse des phosphates………...………..……27
III.1. Méthodes spectrophotométriques……….……27
III.2. Méthodes électrochimiques………..28
Références……….29
CHAPITRE II : TECHNIQUES TE CONDITIONS EXPERIMENTALES………34
Introduction………..….35
I. Techniques électrochimiques………..………..35
I.1. Voltamétrie à tension linéaire et cyclique………..35
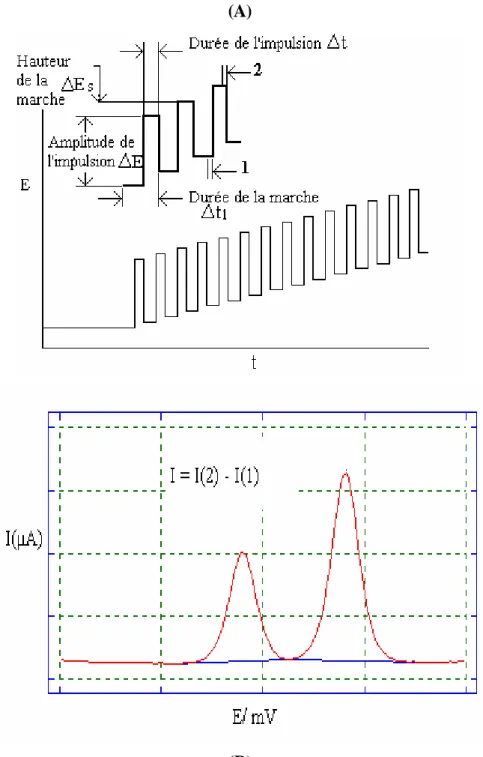
I.2. Voltamétrie impulsionnelle différentielle………..36
I.3. Ampérométrie………....38
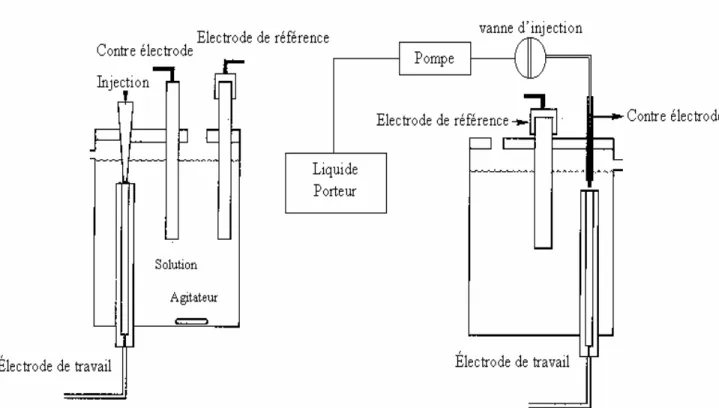
II. Système d’analyse par injection directe en milieu batch « Batch Injection Analysis » (BIA)……….40
II.1. Configuration géométrique de la cellule BIA………40
II.2. Modèle théorique de la réponse du détecteur BIA………44
II.3. Système BIA et applications……….….45
III. Dispositifs et conditions expérimentales……….48
III.1. Cellules électrochimiques………48
III.2. Electrodes de référence………48
III.3. Electrodes de travail……….………49
III.4. Cellule à microélectrode……….………..……50
III.5. Appareils……….….52
Références ………...……….………54
CHAPITRE III : DEVELOPPEMENT D’UNE NOUVELLE METHODE DE DETECTION DES ORTHOPHOSPHATES. …… ……….………..….56
Introduc tion………..….57
I. Matériels et procédures………..…58
I.1. Réactifs et échantillons………..…...58
I.2. Procédures………...…58
I.2.4. Analyse de l’eau de mer………...59
I.2.5. Analyse des échantillons de biofilms de cyanobactéries………..59
I.2.6. Méthode des ajouts dosés……….. …………..60
I.2.7. Calcul du taux de recouvrement……….…..60
II. Résultats et discussion..………61
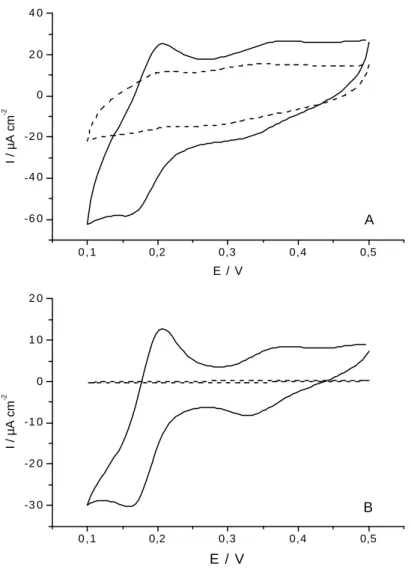
II.1. Analyse en milieu batch……….………61
II.1.1. Choix de l’électrode de travail et du potentiel imposé……...….61
II.1.2. Effet du pH………..63
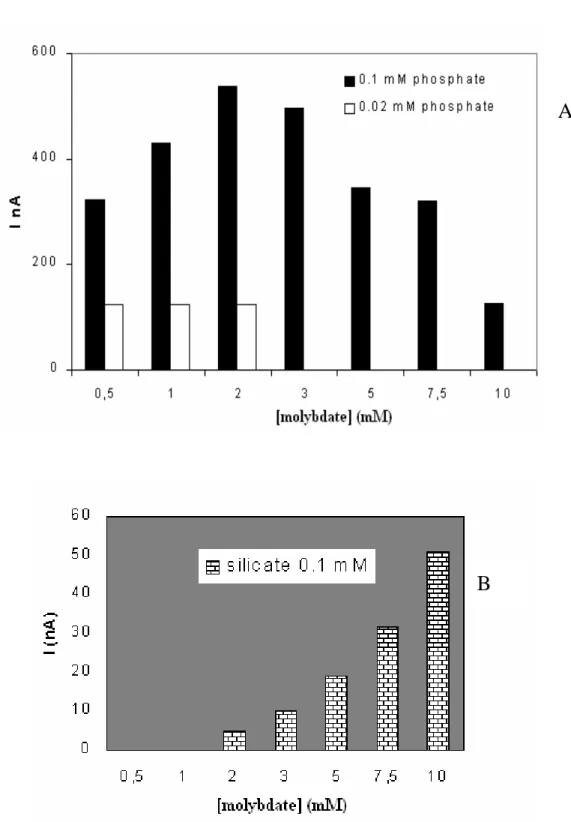
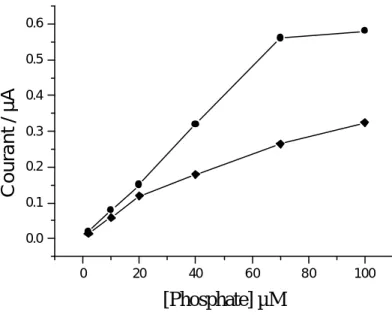
II.1.3. Etude des substances interférentes………..……63
II.1.4. Courbe de calibration………..…66
II.1.5. Effet du solvant organique………..67
II.1.6. Effet du sel………..68
II.2. Système d’analyse par injection directe en milieu batch (BIA)…………69
II.2.1. Détermination des performances analytiques……….……69
II.2.2. Application : Analyse de l’ion PO43- dans l’eau de mer……...71
II. 3. Analyse des phosphates en milieu batch à l’aide de la microélectrode à pâte de carbone………....……….…73
II.3.1. Performances analytiques……….………..…73
II.3.2. Application : Analyse des phosphates dans des biofilms de cyanobactéries…………..……….…………..74
III. Conclusion………...………...………76
Références ………...……….77
CHAPITRE IV : DETECTION ELECTROCHIMIQUE DE L’ION NITRITE BASEE SUR SA REACTION AVEC LE 2,3- DIAMINONAPHTHALENE ………… 78
Introduction………...…79
I. Techniques et méthodes expérimentales ………...80
I.1. Réactifs et échantillons………...80
I.2. Procédure………81
I.2.1. Mesures électrochimiques pour quantification………81
I.2.2. Analyse des standards de nitrite………...81
I.2.3. Analyse des échantillons réels………..82
II.2. Optimisation des paramètres de la réaction chimique………...85
II.3. Analyse des nitrites à l’aide de la DPV……….86
II.4. Mesure des nitrites à l’aide de la méthode BIA- ampérométrie…………87
II.5. Etude des substances interférentes……….89
III. Application : Analyse des échantillons réels…...………...91
IV. Conclusion………...………...92
Références ………...……….93
CHAPITRE V : DETERMINATION ELECTROCHIMIQUE DES NITRATES DANS DES ECHANTILLONS D’EAU ET DE PRODUITS CARNES………94
Introduction………...………95
I. Expérimentation……….96
I.1.Réactifs et échantillons………96
I.2. Réduction des nitrates en milieu batch………...97 I.3. Préparation de la colonne de cadmium………...………97
II. Résultats et discussion………..98
II.1. Analyse spectrophotométrique………..99
II.1.1. Détermination du temps d’incubation nécessaire pour la réduction des nitrates en milieu batch………99
II.1.2. Influence du pH et de la concentration de la solution tampon sur le rendement de la réduction. ………101
II.1.3. Influence du débit sur le rendement de la réduction………..102
II.1.4. Influence de la concentration initiale du nitrite sur le rendement de la réduction………103
II.1.5. Courbe de calibration………..104
II.1.6. Analyse des nitrites et des nitrates dans des échantillons réels……...105
II.2. Détermination des nitrites et nitrates par voie électrochimique………..107
II.2.1. Principe de la méthode……….……….107
II.2.2. Procédure expérimentale………...108
II.3. Comparaison entre les résultats obtenus par spectrophotométrie et les résultats obtenus par voie électrochimique………109
CHAPITRE VI : NOUVELLE METHODE DE DETERMINATION DE L’ION AMMONIUM
DANS L’EAU DE DISTRIBUTION……….114
Introduction……….115
I. Réactifs et échantillons………116
II. Résultats et discussion………116
II. 1. Etude sur électrode à pâte de carbone modifiée……….116
II.2. Comportement du système électrochimique étudié………117
II.3. Optimisation des paramètres qui interviennent dans la détermination de l’ion ammonium………...…122
II.3.1. Choix de la solution tampon……….122
II.3.2. Etude de l’influence du pH. …………..………...123
II.3.3. Effet de la force ionique……….………...125
II.3.4. Influence de la concentration du cuivre………125
II.3.5. Choix du potentiel appliqué………..126
II.3.6. Caractéristiques analytiques de la méthode proposée…….…………..127
III. Application : analyse des échantillons réels……….……….128
IV. Conclusion………129
Références ……….……….130
Ag/ AgCl : électrode d’argent / chlorure d’argent
AOAC : en anglais "Association of Official Analytical Chemists"
BIA : "Batch Injection Analysis" c’est l’ injection directe en milieu Batch Cd-Cu: grains de cadmium sur lesquelles le cuivre a été déposé chimiquement CPG : chromatographie en phase gazeuse
CSV : voltamétrie par redissolution cathodique DAN : 2,3-Diaminonaphthalène
DPV : voltamétrie impulsionnelle différentielle ECS : électrode au calomel saturée
EDTA: acide éthylène diamine tétraacétique EPC : électrode à pâte de carbone
FIA : injection dans un flux continue, en anglais "Flow injection analysis" GDH : glutamate déshydrogénase
HMDE : électrode pendante de mercure
HPLC : chromatographie liquide haute performance IC : chromatographie ionique
ISE : électrode sélective aux ions, en anglais "ion selective electrode" LDH : lactate déshydrogénase
NAD : nicotinamide adénine dinucléotide
NADH : forme réduite du nicotinamide adénine dinucléotide
NADP : forme oxydée du nicotinamide adénine dinucléotide phosphate. N° CEE : code international des additifs alimentaires
NTA : 1-H-Naphthotriazole
OMS : organisation mondiale de la santé pKA : constante d’acidité
PTFE : téflon
PVC : Chlorure de polyvinyle R2 : coefficient de détermination RSD : déviation relative standard
SD : déviation standard, c’est l’écart type divisé par la moyenne des mesures S/N : rapport signal/ bruit
UV : ultra-violet
L’orthophosphate (PO43-), le nitrite (NO2-), le nitrate (NO3-) et l’ammonium (NH4+) sont les sels nutritifs les plus utilisés en agriculture moderne et en industrie alimentaire. Présentes en excès dans la nature, ces substances provoquent de sérieux problèmes environnementaux. Le besoin en détecteurs sensibles et sélectifs de ces composés est d’une importance cruciale. Ce besoin qu’entraîne la croissante sévérité des normes dans tous les domaines de chimie et biochimie (environnement, alimentation, pharmacie, sécurité domestique et industrielle…) a incité les scientifiques à développer des techniques chimiques et électrochimiques d’analyse de ces substances de plus en plus performantes.
L’essor de l’électronique et l’extension de l’usage d’électrodes solides ont contribué au développement de méthodes électrochimiques. L’électrochimie offre en effet des perspectives attrayantes quant à la compacité, aux conceptions technologiques simple s, de faible coût et si possible de petite taille permettant une faible consommation d’énergie et donc une utilisation sur site.
Les électrodes à base de carbone occupent une position importante dans l’électroanalyse tant par leur faible coût et leur mise en oeuvre aisée que par leurs performances électrochimiques en analyses organiques et inorganiques [1].
Dans le cadre de ce travail seront présentées de nouvelles méthodes électrochimiques permettant la détermination du phosphate, nitrite, nitrate et ammonium dans les eaux et celle du nitrite et du nitrate dans les eaux et dans les produits carnés.
Notre choix s’est porté sur l’électrode à pâte de carbone parce qu’elle présente des avantages par rapport aux autres électrodes solides classiques tels qu’un renouvellement de surface aisé et exempt d’effet de mémoire et une grande sensibilité vis-à-vis de certaines substances [2].
L’approche expérimentale utilisée dans cette étude repose sur l’utilisation de techniques voltamétriques et ampérométriques avec une instrumentation d’analyse portable. Pour rendre le temps de l’analyse aussi bref que possible, l’Analyse par Injection Directe en milieu Batch (Batch Injection Analysis, BIA) est adoptée. Cette méthode en plus d’être simple et économique de point de vue consommation de réactifs et échantillons, permet une grande cadence de mesure, critère particulièrement important en analyse de routine.
Après un rappel bibliographique permettant d’introduire les différents éléments à analyser ainsi que les méthodes d’analyse développées dans ce sens et citées dans la littérature, nous présenterons au chapitre II les principes et les techniq ues expérimentales utilisés dans la suite de ce travail.
Dans le chapitre III, nous aborderons la détermination des orthophosphates dans l’eau de mer en utilisant la technique BIA couplée à l’ampérométrie et celle du même élément dans des échantillons de biofilms de cyanobactéries à l’aide de la technique ampérométrie sur microélectrode à pâte de carbone. Les conditions optimales pour cette détection et les caractéristiques analytiques de la méthode seront ainsi détaillées.
Le chapitre IV sera consacré à l’élaboration d’une nouvelle technique électrochimique de détection de l’ion nitrite en se basant sur sa réaction avec le 2,3-diaminonaphthalène. L’analyse par injection directe en milieu batch couplée à l’ampérométrie ainsi que la voltamétrie impulsionne lle différentielle sont utilisées pour doser les nitrites dans des échantillons des eaux naturelles et contaminées.
Nous examinerons aussi dans le chapitre suivant la réponse des nitrates, en utilisant la même procédure électrochimique de détection des nitrites, après leur réduction chimique sur colonne de cadmium / cuivre. Les paramètres de l’étape de la réduction seront optimisés pour aboutir à une efficacité maximale. La méthode électrochimique utilisée sera légèrement modifiée pour être couplée avec la réduction sur colonne.
Une méthode d’analyse rapide de l’ion ammonium sera élaborée dans la dernière partie de ce travail. Elle repose sur la réaction de l’ion NH4+ avec les ions Cu2+ en milieu basique pour former le complexe électroactif Cu(NH3)n2+. Les techniques expérimentales utilisées au cours de cette étude sont la voltamétrie impulsionnelle différentielle et la chronoampérométrie.
Références bibliographiques
[1] C. Urbaniczky et K. Lundstrom, J. Electroanal. Chem., 1984, 167, 169.
Chapitre I
A. Généralités sur les éléments à analyser : Nitrite, nitrate, ammonium et
orthophosphate
Introduction
L’utilisation intensive des fertilisants artificiels en agriculture (engrais, minéraux, lisiers) ainsi que l’accroissement des rejets urbains ont augmenté notablement les apports terrigènes d’éléments nutritifs en milieu aquatique. Ces apports ont conduit, entres autres, à un enric hissement en nitrate (issu principalement du lessivage des terres agricoles), en ammonium et en phosphate (très abondants dans les rejets urbains), ce qui a causé un déséquilibre du milieu aquatique, appelé eutrophisation.
Les éléments qui nous intéressent au cours de cette étude sont : l’azote inorganique dissous (nitrite, nitrate et ammonium) et l’orthophosphate. Ces éléments peuvent, en plus de leur contribution au phénomène d’eutrophisation, avoir des effets nocifs pour l’Homme et pour l’environnement. Les effets des composés azotés se résument principalement dans la synthèse des nitrosamines cancérigènes et la formation de la méthémoglobine chez les nourrissons, en présence de fortes teneurs en nitrates et en nitrites dans les eaux de consommation.
Le passage des composés azotés (ammoniac et aérosols de nitrate, protoxyde d’azote,…) du sol vers l’atmosphère conduit à de sérieux problèmes non seulement pour la santé de l’homme mais aussi pour l’environnement (brouillard et pluie acides).
I. Phénomène de l’eutrophisation
L’eutrophisation se manifeste par une formation importante d’algues, ce qui conduit à une augmentation de la charge naturelle de l’écosystème en matière organique à dégrader. La décomposition des algues par les bactéries consommatrices d’oxygène engendre une diminution du taux d’oxygène dans l’eau. Parallèlement, la matière organique morte non décomposée s’accumule dans les sédiments. Un déséquilibre se produit alors entre les eaux de surface oxygénées par aération et photosynthèse et les eaux profondes où le développement des organismes est limité.
En résumé, l’eutrophisation résulte de l’enrichissement d’une eau en sels minéraux entraînant des déséquilibres écologiques tels que la prolifération de la végétation aquatique ou l’appauvrissement du milieu en oxygène. Les différentes étapes du phénomène (figure 1) sont :
1. Apport massif de substances nutritives (nitrates, phosphates)
2. Augmentation de la production primaire: stimulation de la croissance du phytoplancton. A ce stade on note une augmentation des Diatomées marquée par une croissance de larve sur le fond.
3. Enrichissement du cycle biologique: augmentation du zooplancton, des poissons, forte croissance des plantes enracinées
4. Mort progressive des algues en suspension, augmentation de turbidité, sédimentation importante des matières organiques vers les couches profondes et consommation importante de l’oxygène dissous dans l’eau
5. Putréfaction de la vase (diffusion de produits toxiques, H2S, NH3, CH4) due au développement des bactéries anaérobies
6. Désoxygénation des couches inférieures et mort des poissons, prolifération du phytoplancton en surface
7. La turbidité empêche la photosynthèse de s’effectuer: Ceci augmente l’appauvrissement de l’eau en oxygène et la reproduction des poissons peu exigeants en oxygène.
L’eutrophisation entraîne les nuisances suivantes :
- Détérioration de la qualité de l’eau avec déstabilisation des chaînes trophiques. - Diminution de la valeur commerciale de l’aquaculture et de la pêche.
- Toxicité et risques chroniques ou intermittents pour la santé. - Diminution de la valeur esthétique et récréative des eaux affectées.
Elle débute donc par une prolifération anormale d’algues et se termine par l’asphyxie et la destruction de l’ensemble de l’écosystème.
Figure 1 : Différentes étapes du phénomène d’eutrophisation [1].
II. L’azote inorganique en milieu aquatique
II.1. Cycle de l’azote
L’azote représente 78% de l’atmosphère gazeux et 4 à 6% du poids sec d’un animal. L’ensemble des réactions biologiques de croissance requiert la présence de l’élément azote. Le cycle d’azote est parfaitement connu et fait intervenir des réactions de fixation, d’assimilation, d’ammonification, de nitrification et de dénitrification.
La fixation de l’azote correspond à la conversion de l’azote atmosphérique en azote utilisable par les plantes et les animaux. Elle se fait par certaines bactéries qui vivent dans les sols et dans l’eau et qui réussissent à assimiler l’azote diatomique N2. Il s’agit en particulier des cyanobactéries et de certaines bactéries vivant en symbiose avec les plantes. La réaction chimique type est :
2N2 (g) + 3( CH2O) + 3 H2O 4 NH4+ + 3 CO2
Dans les sols où le pH est élevé, l’ammonium se transforme en ammoniac gazeux: NH4+ + OH- NH3 (g) + H2O
La réaction nécessite un apport d’énergie de la photosynthèse.
La nitrification transforme (NH4+, NH3) en nitrites et nitrates. C’est une réaction d’oxydation qui se fait par catalyse enzymatique reliée à des bactéries dans le sol et dans les eaux. La réaction chaîne est :
2 NH4+ + 3O2 2 NO2- + 2 H2O + 4 H+ 2 NO2- + O2 2 NO3
-La dénitrification fait revenir l’azote à l’atmosphère sous forme N2; il s’agit d’une réaction de réduction de NO3- par l’intermédiaire des bactéries transformant la matière organique:
4 NO3- + 5(CH2O) +4H+ 2N2(g) + 5CO2(g) + 7H2O
L’activité humaine perturbe ce cycle par l’introduction de grandes quantités de NH4+, NH3 et NO3-, ce qui se produit suite aux combustions industrielles et domestiques. Par contre, la grande majorité des émissions provient de l’agriculture.
L’azote est considéré comme second polluant des eaux après le carbone et avant le phosphore. On le retrouve sous différentes formes : il est dissous sous forme d’ammoniac, de nitrite et de nitrate et présent dans les molécules organiques comme les acides aminés et les particules en suspension.
II.2. Origine des pollutions azotées :
Les apports en azote viennent s’ajouter aux pollutions naturelles et sont soit diffus soit locaux. Parmi les apports diffus, on peut citer :
- La déposition et l’entraînement par l’eau de pluie de gaz et aérosols contenant l’azote minéral produit par l’industrie (combustion de carburants fossiles, incinérations des ordures…) et par la décharge des composés agricoles.
- Le ruissellement et le lessivage des terres agricoles fertilisées ou les zones de stockage d’excréments animaux, qui apportent essentiellement les formes ammoniaques et nitrate. Les apports ponctuels concernent essentiellement :
- Les industries produisant des émissions d’ammoniaque et les industries utilisant les nitrates dans leurs procédés de fabrication (salaison de viandes, production de fertilisants, de verre,…)
- Les systèmes de traitement des eaux usées et by-pass des stations d’épuration (formes NH4+ et NO3-)
II.3. Conséquences des pollutions azotées
II. 3. 1. Pollution par les nitrites et nitrates
a- Voies d’exposition
L’exposition de la population aux nitrates et aux nitrites se fait principalement par les aliments et occasionnellement par l’eau de consommation (figure 2). Chez l’adulte, la principale source des nitrates et des nitrites provient des légumes et des produits carnés (l’action anti- microbienne des NO3-/NO2- est reconnue depuis longtemps, ils sont utilisés dans la conservation de ces produits). L’apport en nitrate attribuable à l’eau potable devient important lorsque les concentrations en nitrates sont anormalement élevées.
Dans le cas des enfants nourris avec du lait maternisé, l’eau utilisée pour la préparation de ce lait est la seule source des nitrates : elle peut ainsi devenir une source importante d’exposition lorsque l’eau est contaminée.
b- Toxicité des nitrates
Les nitrates ne sont guère toxiques, mais ce n’est qu’à deux conditions que peut se révéler leur toxicité : s’il y a ingestion vraiment massive de ces composés ou s’ils sont transformés en nitrites par la microflore digestive au sein de l’organisme. Une fois ingérés,
les nitrates sont rapidement absorbés au niveau de l’intestin grêle puis distribués dans tout l’organisme. La microflore buccale transforme une partie des nitrates secrétés dans la salive en nitrites. Leur réduction en nitrites peut également survenir au niveau des voies urinaires à la suite d’une infection bactérienne et dans l’estomac.
Figure 2 : voies d’exposition de l’Homme aux nitrates et leur assimilation, élimination ou transformation dans le corps humain en fonction de leurs concentrations.
Dans les milieux biologiques, la transformation des ions NO3- en ions NO2- ne peut s’effectuer que sous l’action d’une enzyme : la nitrate réductase qui est présente dans tous les organismes susceptibles de métaboliser le nitrate tels que les plantes, les champignons ainsi que quelques espèces de levures et bactéries.
Eau Nitrates Réduction Absorption Elimination par Additifs alimentaires Légumes Au niveau de l’estomac Par des bactéries nitrato-réductrices
En nitrites NO2
-Effets pathogènes Au niveau des intestins
Voie urinaire
Voie entérohépatique
Circuit salivaire
Méthémoglobinémie
c- Toxicité des nitrites :
Les nitrites consommés directement proviennent des produits carnés. Ils sont autorisés comme conservateurs dans ces produits. Ils inhibent le développement du germe responsable de la toxi- infection alimentaire grave, appelée le botulisme [2]. Ils neutralisent la prolifération des microbes dangereux dans la charcuterie, comme les staphylocoques et le bacille botulique qui est un poison mortel.
La méthémoglobinémie (ou syndrome du bébé bleu) est un problème de santé associé à l’ingestion des nitrites et des nitrates. Dans ce cas, l’ion nitrate est converti en ion nitrite dans l’estomac, puis il est absorbé par la circulation sanguine. Les nitrites oxydent l’hémoglobine sanguine en méthémoglobine qui sous cette forme n’est plus apte à jouer son rôle de transporteur d’oxygène ce qui entraîne une hypoxie au niveau des tissus. Chez l’adulte, l’organisme humain est capable de lutter contre cette agression car il dispose d’un système enzymatique apte à effectuer la réaction inverse et à transformer la méthémoglobine en hémoglobine réduite (méthémoglobine réductase). Par contre, ceci n’est pas le cas chez le nourrisson ne possédant pas cet équipement enzymatique ; ce qui augmente les risques des intoxications graves. Ceci altère la capacité des globules rouges de transporter l’oxygène. Les symptômes de cette maladie sont notamment la cyanose (décoloration bleutée de la peau et de la bouche), la difficulté de respirer et la fatigue (des cas mortels ont été rapportés).
Si le pouvoir cancérigène du nitrite est très discuté, la formation des nitrosamines à pouvoir cancérigène est par contre indiscutable et est possible à partir de nitrite et d’amines secondaires et même tertiaires. Cette formation se produit dans le tube digestif du consommateur.
II.2.2. pollution par l’ammonium
L’ammoniac existe simultanément sous deux formes, NH3 (ou ammoniac non ionisé) et NH4+ (ammoniac ionisé ou ammonium). L’ammoniac total est la somme de NH3 et NH4+. L’équilibre entre les deux est régi en grande partie par le pH et la température. La
solubilité de NH3 dans l’eau pure est élevée et de l’ordre de 900g/L à 0°C [3]. L’ammoniac NH3 étant une base faible dont la réaction d’ionisation dans l’eau est :
NH3 + H2O NH4+ + OH- pKA = 9.26
KA désigne la constante d’acidité de NH4+
Dans la gamme des pH de la plupart des eaux naturelles, l’azote ammoniacal existe principalement sous forme de NH4+.
L’ammoniac est un composé naturel dont ont besoin la plupart des organismes pour la synthèse des protéines. Les humains se servent principalement de l’ammoniac comme source d’azote dans les engrais. Les principales sources quantifiables d’ammoniac dans les écosystèmes aquatiques sont les stations municipales de traitement des eaux usées. L’agriculture est aussi une source de contamination des eaux par l’ammonium.
On trouve aussi l’ammoniac dans un certain nombre de denrées alimentaires (fromage, viande vieillie ou entreposée et les légumes entreposées). Les sels d’ammonium sont largement utilisés dans les aliments cuits au four, les bonbons, la gélatine, les graisses, les huiles, les gelées, les fromages, les fruits traités et les boissons.
La présence de l’ammonium dans les eaux peut engendrer divers inconvénients comme la corrosion des conduites, la diminution de l’efficacité du traitement de désinfection au chlore et le développement de microorganismes responsables de saveurs et d’odeurs désagréables. Elle peut donc révéler l’existence d’une décomposition de matières organiques dans le milieu, ce qui constitue un indice de pollution des nappes phréatiques.
Suite à ce type de pollution, les problèmes suivants peuvent intervenir :
- L’ammoniac est oxydé par les bactéries en nitrite et nitrate conduisant à une baisse de la concentration en oxygène dissous et à la mort des poissons.
- L’ammoniac NH3 et l’ammonium NH4+ sont en équilibre chimique ; l’augmentation de la température et /ou du pH provoque une production de plus en plus importante d’ammoniac qui est toxique pour les poissons et pour l’homme. Si l’ammonium est disponible en excès, la forme ammoniac libre NH3 peut s’accumuler dans l’organisme et
causer des effets néfastes. Il est irritant, affecte souvent les yeux, le nez, la gorge et les poumons. S’il est ingéré, il corrode les parois de la bouche, l’œsophage et l’estomac. - La volatilisation de NH3 contribue aussi à un transfert de l’azote contenu dans les effluents d’élevage vers l’atmosphère. Ce qui entraîne des conséquences potentielles variées sur la santé humaine et animale (asthme, bronchites chroniques, diminution des performances zootechniques) et sur les écosystèmes (acidification et eutrophisation).
III. Le phosphore
III. 1. Le phosphore dans le milieu aquatique
La principale source naturelle du phosphate est la pierre. Il est généralement libéré par l’érosion. Durant son parcours vers la mer, la molécule du phosphate pourra être absorbée par des plantes ou des animaux puis retourne au sol ou à l’eau par le biais de la décomposition végétale, animale ou par voie urinaire. Le cycle du phosphate est par la suite perturbé avec l’arrivée de l’agriculture et de l’urbanisation.
Dans les eaux, le phosphore se trouve principalement sous forme d’orthophosphate (HxPO4x-3), de polyphosphates (polymères d’acide phosphorique) et de forme organique du phosphore. La forme orthophosphate est la prépondérante, en raison de l’hydrolyse des deux autres espèces. L’ion phosphate peut se trouver sous trois états de protonisation:
H3PO4 + OH- H2PO4- + H2O pKA1 = 2.1 H2PO4- + OH- HPO42- + H2O pKA2 = 7.2 HPO42- + OH- PO43- + H2O pKA3 = 12.5
KAi désigne les constantes d’acidité des espèces H3-iPO4i-.
Le phosphore est un élément indispensable à la vie. En soi, il n’est pas toxique mais il provoque l’eutrophisation lorsqu’il est en excès dans l’eau. Le phosphate est la forme sous laquelle le phosphore peut être assimilé par les êtres vivants, en particulier les algues. La prolifération de ces algues peut avoir de nombreux effets néfastes, telles par exemple, l’augme ntation de la turbidité de l’eau, la diminution de l’aspect esthétique et la réduction
des activités de loisirs. Certaines algues (algues bleues) peuvent produire des substances qui empoisonnent le zooplancton, les poissons, les oiseaux aquatiques, le bétail et les humains.
Présentes à forte densité, les algues font augmenter les coûts de traitement de l’eau potable et donnent à l’eau une mauva ise odeur et un mauvais goût [4].
D’un autre point de vue, les algues sont depuis toujours utilisées comme aliments, engrais ou médicaments. Aujourd’hui, elles font l’objet de demandes importantes et variées dans différents domaines : industries alimentaires ou textiles, cosmétologie, thalassothérapie, diététique ou agriculture (compost à base d’algues). Les algues sont aussi utilisées comme bioindicateurs du niveau de radioactivité en milieu marin car elles concentrent certains éléments radioactifs [5].
Le phosphate remplit différentes fonctions dans la formation des détergents, en particulier anticalcaire et anti redéposition. Il est présent dans les eaux sous différentes formes : forme dissoute ou particulaire, organique ou minéral.
III.2. Le phosphore dans les biofilms de cyanobactéries
Le contrôle du phosphore est d’une grande importance pour la protection des héritages culturels. Il a été montré que le phosphate est en partie responsable de la biodéterioration des sites archéologiques causé par la prolifération des cyanobactéries. En effet, une grande variété de microorganismes envahissant la pierre peut être observée au dessous et au dessus de la roche, à l’extérieur comme à l’intérieur des monuments. Ces microorganismes développent des systèmes complexes contenant plusieurs espèces appelées biofilms.
En particulier, dans l’Hypogée Romain archéologique à Rome, l’abondance des nutriments dans des substrats lithiques, les apports de quelques composés par le milieu environnant, le taux d’humidité assez élevé ainsi que l’éclairage artificiels constituent des conditions très favorables à la formation de ces microorganismes.
Les cyanobactéries étant des microorganismes photosynthétiques capables de s’adapter et s’acclimater à des émissions de photons de très faibles et variables énergies
sont les plus capables d’attaquer la surface de la pierre. Exposés à la lumière, ces biofilms causent des dommages chimiques, physiques et esthétiques [6].
En même temps, la disponibilité de la matière organique, produite à partir de la photosynthèse de cyanobactéries et de la fixation de l’azote N2, favorise la croissance des microorganismes hétérotrophiques (bactéries et champignons au niveau du biofilms), qui sont capables de consommer la matière organique tout en libérant des composés organiques acides. Ces derniers solubilisent les minéraux constituant les roches [7]. Ainsi, les sels nutritifs comme le phosphore peuvent se libérer du substrat, se transformer ou se trouvent stocker dans les cellules de cyanobactéries [8]. Généralement, 0.6% du poids sec de ces cellules est constitué de phosphore qui est principalement sous forme d’orthophosphate [9].
B.
Généralités sur les techniques utilisées pour la détermination des ions : Nitrate, nitrite, orthophosphate et ammoniumI. Méthodes d’analyse des nitrates et nitrites :
Les nitrites et nitrates sont rarement analysés séparément parce qu’on ne peut pas trouver l’un sans trouver l’autre dans un milieu donné. Par conséquent, la majorité de recherches repose sur les interconversions, particulièrement la réduction chimique de l’ion nitrate pour former l’ion nitrite qui est plus réactif. L’essor des techniques d’analyse de ces ions devient de plus en plus important à cause des problèmes environnementaux qui résultent de leur usage excessif.
Selon Moorcroft et ses coll. [10], les méthodes de détection des nitrites / nitrates sont basées sur les réactions représentées sur la figure 3.
Des méthodes électrochimiques associées à l’électrophorèse capillaire permettent la détection simultanée des ions nitrite et nitrate. Chaque ion est mesuré indépendamment de l’autre au cours d’une seule analyse. Les analyses séquentielles reposent sur l’analyse dans une première étape de l’ion nitrite initial, suivie de la réduction de l’échantillon (par exemple en utilisant une colonne Cu/ Cd), ce qui permet la conversion totale du NO3- en NO2-. Une seconde analyse des nitrites totaux permet de déterminer la concentration des ions nitrates égale à la différence entre NO2- total et initial.
Généralement, le NO3- relativement inerte est réduit chimiquement (voie 1, figure 3) en NO2- plus réactif avant d’entamer l’étape de la détection.
Un grand nombre d’agents réducteurs a été étudié dans ce cas tels que : le zinc [11], hydrazine- cuivre [12] et le cadmium cuivré [13]. Les colonnes de cadmium/ cuivre sont les plus efficaces (le rendement de la conversion est proche de 100%).
Takeda et coll. [14] ont développé une nouvelle méthode de réduction à l’aide des radiations UV à des longueurs d’onde comprises entre 200 et 300nm, l’ion nitrate est transformé en ion nitrite et oxygène selon la réaction ci-dessous.
200 – 300nm
NO3- NO2- + 1/2 O2
La chimiluminescence nécessite aussi une étape de conversion des NO3- en NO (voie 3, figure I). Plusieurs agents réducteurs ont été utilisés à ce propos, tels Ti3+ [15, 16], V3+ [15, 17], Mo5+ + Fe2+ et Cr3+ [13].
La plupart des méthodes développées ces dernières années utilisent le système d’analyse par injection en flux continu (FIA) qui permet d’améliorer la sensibilité et la rapidité de l’analyse.
I.1. Méthodes spectroscopiques :
Les méthodes spectroscopiques (UV / Visible [18-26], la chimiluminescence [15, 16, 27] et la fluorimétrie [28- 31]) sont largement utilisées pour la détermination des ions nitrites et nitrates. Pour l’analyse de l’ion NO2-, la méthode de Griess est la plus exploitée. Développée depuis 1879 [32], elle ne cesse d’avoir des applications jusqu’à présent. Cette technique repose sur la réaction de diazotation entre le nitrite acidifié et une amine aromatique. Le produit de la réaction est un complexe hautement coloré dont la densité optique permet de déduire la concentration des ions NO2- (voie 2, figure 3). Le complexe formé présente un maximum d’absorption entre 500 et 600 nm et peut être détecté par un spectrophotomètre visible conventionnel. Les réactifs utilisés sont la sulfanilamide et le N-(1-naphthyl) ethylènediamine.
La limite de détection dans le cas du protocole de Griess varie entre 0.2 et 2µmol/L avec une gamme de linéarité qui s’étend de 1 à 100µmol/L et une longueur du trajet optique qui varie de 1 à 5 cm. Cette technique est simple et sensible mais non fiable dans le cas des milieux complexes : les antioxydants (par exemple l’acide ascorbique) additionnées aux aliments détruisent le nitrite acidifié avant qu’il réagisse avec l’amine aromatique et conduit à une baisse du taux de recouvrement [33]. D’autres problèmes sont aussi rencontrés dans l’analyse des échantillons fortement colorés et turbides. La chromatographie liquide haute pression (HPLC) et le système d’analyse par injection en flux continue (FIA) ont été couplés au détecteur spectrophotométrie reposant sur le protocole de Griess afin de permettre l’analyse des nitrates et nitrites dans des matrices particulièrement complexes tels que les produits alimentaires [34,35].
Les méthodes spectrophotométriques utilisent une variété de réactifs pour former les produits colorés. La réaction entre l’ion nitrite et le 3,6-diaminocridine (proflavin) en milieu acide donne naissance à un composé violet et stable (λmax = 328nm). Cette technique est sensible mais l’ion Fe3+ interfère si sa concentration dépasse 1mg/L [36].
D’autres techniques permettent la mesure des nitrites en mettant à profit sur son effet catalytique vis-à-vis des réactions de complexation. Par exemple, l’oxydation du gallocyanine par le bromate en milieu acide [37] se déroule très lentement mais la présence
des nitrites permet d’accélérer cette réaction. Le complexe ainsi formé est mesuré à 530 nm. L’inconvénient de cette technique est dû à l’interférence d’un grand nombre d’ions lors de la mesure. Dans le même sens, Pettas et coll. [38] ont montré que la réaction d’oxydation du bleu de thymol par le bromate dans un milieu acide est catalysée par l’ion nitrite. L’absorbance est mesurée à 543nm.
La dosage fluorimétrique des ions cérium Ce3+ provenant de l’oxydation des nitrites par Ce4+ représente le protocole indirect le plus simple pour analyser NO2-. Cependant, l’interférence de quelques espèces redox présente un inconvénient pour cette méthode [39]. Lapat et coll. [40] ont étudié deux méthodes fluorimétriques pour analyser l’ion nitrite après complexation par l’acide 2-amino-chloro-1-hydroxybenzéne-6-sulfonique ou le 4-aminofluorescien. Les complexes ainsi obtenus sont hautement fluorescents. De même le diaminonaphthalène réagit en milieu acide avec l’ion nitrite pour former le 2,3-nitronaphthotriazole dont la longueur d’onde d’excitation est égale à 365nm et celle d’émission est d’environs 387nm. La limite de détection dans ce cas est de l’ordre de 0.1 µmol /L [41].
Pour le dosage des ions nitrites par chimiluminescence, la conversion de NO2- en NO gazeux se fait en présence de l’iodure de potassium en milieu acide. Cette méthode permet de déterminer l’ion nitrite dans des échantillons complexes en libérant le monoxyde d’azote. Elle repose sur la réaction entre NO gazeux et l’ozone [42, 43] qui conduit au dioxyde d’azote à l’état excité (NO2*) et l’oxygène moléculaire. La molécule excitée revient spontanément à son état fondamental en émettant une radiation infrarouge de longueur d’onde voisine de 600nm (voie 4, figure 1). La chimiluminescence permet d’analyser les nitrates à condition d’utiliser des réducteurs forts comme Ti3+ [42]. Couplée au système FIA, cette technique a permis d’obtenir des limites de détection très faibles de l’ordre de 10nmol/L [42, 43].
La spectroscopie d’absorption atomique couplée au système d’analyse par injection en flux continu FIA [44] a été aussi utilisée pour le dosage indirecte des ions nitrates et nitrites. Ces ions réagissent avec le chélate du cuivre (I)-neocuproine et forment des paires d’ions qui sont extraits par le methyl- isobutyl-cétone. Le signal du cuivre en phase
organique est proportionnel à la concentration des ions NO3- ou NO2-. Le système permet d’analyser 35 échantillons par heure.
I .2. Méthodes électrochimiques :
Dans le même sens, les méthodes voltamétriques sont élaborées pour la première fois pour réduire électrochimiquement l’ion nitrate à des électrodes de cuivre [45- 47]. D’autres types d’électrodes sont par la suite étudiées et sont en Nickel [48], cadmium [49], platine [50], carbone vitreux [51], en argent [52] et récemment les électrodes de graphite dopée par le bore [53- 54]. Ces méthodes présentent généralement une cinétique de transfert de charge assez lente, montrent souvent une faible sensibilité et une mauvaise reproductibilité.
Par contre, l’ion nitrite est plus réactif et peut être oxydé ou réduit à l’électrode de carbone vitreux [55, 56]. Même si de nombreuses applications sont envisageables à l’aide des électrodes conventionnelles, leur mise en œuvre directe dans des milieux complexes se heurte à des problèmes de surface (salissures…) et à un manque de sélectivité. Afin de résoudre ces problèmes, de nouvelles stratégies destinées à modifier la surface des électrodes furent envisagées pour augmenter leur sensibilité. Il a été aussi montré que ceci peut être réalisé en introduisant un sel de métal approprié dans le milieu. A titre d’exemple, l’ion métallique Cu [47] ou le mélange Cu/Cd [57] est électrolytiquement déposé sur l’électrode et fournit une surface active fraîche au sein de laquelle se produit la réaction de réduction des ions nitrates et nitrites.
Des protocoles de modification de l’électrode plus délicats ont été élaborés. Ils utilisent toujours des ions métalliques sous forme de complexes [58] pour la réduction des ions nitrates / nitrites : des films de polypyrrole dopés avec l’anion tungstodiphosphate (P2W18O626-) [59], des hétéropolytungstates substitués de fer [60] et des électrodes de film de mercure recouvertes de nafion avec des ions Yb3+ ou UO22+incorporés [61].
Les ions NO2- peuvent aussi être réduits en monoxyde d’azote NO. Le signal ampérométrique est mesuré à une électrode d’or modifié par du PTFE [62].
Dans le cadre des biocapteurs, les enzymes réductases [63] permettent d’améliorer la sélectivité et la sensibilité lors de la réduction des ions nitrates et nitrites. Reshetilov et ses coll. [64] ont développé un biocapteur basé sur l’oxydation des nitrites par la bactérie N. vulgaris strain. L’étape d’oxydation est suivie de mesure ampérométrique du taux d’oxygène formé au cours de cette réaction. Malgré les avantages que présentent les biocapteurs, ils sont rarement utilisés pour le dosage du nitrite / nitrate parce que les réactifs sont très coûteux et le système est très complexe et instable.
Comme mentionné auparavant, dans le cas des méthodes spectrophotométriques, la détermination directe des ions nitrates n’est pas facile. En effet, l’analyse de NO3- nécessite dans la plus part des cas, une étape préliminaire de réduction chimique en utilisant la colonne de cadmium / cuivre ou un autre système réducteur. Ce pendant, cet ion peut aussi être déterminé par l’intermédiaire de ses propriétés chimiques de substitution des composés aromatiques tels que l’acide benzoïque, l’acide salicylique, l’isoquinoline et l’acide thiophène- 2-carboxylique (voie 5 figure 3). Les produits ainsi obtenus (dérivés nitrés) sont dosés en mesurant le courant de réduction à une électrode de carbone vitreux par la voltamètrie linéaire entre +0.0 et +0.5V. Il s’est avéré que l’acide thiophène-2-carboxylique donne la meilleure réponse [65] avec une limite de détection de l’ordre de 0.1µmol/L et quelques problèmes d’interférences, notamment celle de l’oxygène dissous.
Des méthodes indirectes pour l’analyse de nitrite ont été développées en se basant sur la réaction de Griess. Le sel diazonium formé suite à la réaction entre le nitrite acidifié et le phenylènediamine substitué est réduit électrochimiquement [66]. Le signal de réduction est bien défini, intense et situé aux environs de -0.2V où le problème d’interférences des éléments électroactifs ne se pose pas.
Le système iodure / iodate (I- / I3-) peut aussi être exploité dans l’analyse indirecte du nitrite. C’est un système électrochimique réversible à l’électrode d’or et de platine en milieu acide sulfurique [67]. La technique repose sur la réaction chimique entre le nitrite et l’iodure en milieu acide pour former l’ion iodate I3- qui est réduit ampérométriquement entre +0.2 et +0.3V par rapport à l’électrode au calomel saturé (ECS). Des microélectrodes en platine sont aussi utilisées pour mesurer l’ion I3-. Couplé au système FIA, une limite de
détection de l’ordre de 70nmol/L en nitrite a été atteinte. Le nombre d’échantillons analysés par heure est de 60.
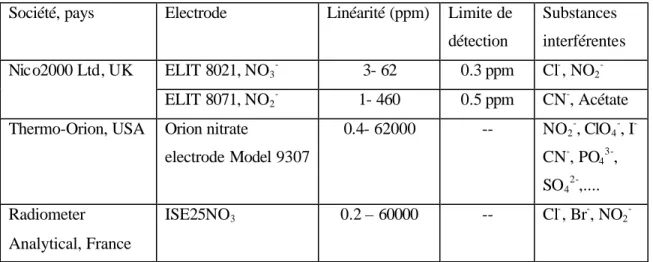
Les méthodes potentiométriques ont été aussi étudiées [68, 69, 70] pour l’analyse des nitrites / nitrates. L’approche la plus connue est l’électrode sélective (ISE : en anglais ion sélective electrode) qui est commercialisée depuis longtemps par diverses sociétés (Tableau 1). La diffusion sélective des espèces chargées de la solution vers la membrane donne une différence de potentiel qui varie en fonction de l’activité des espèces ioniques selon le modèle de Nernst. Par conséquent, la courbe de calibration du potentiel en fonction de la concentration des ions nitrates ou nitrites est obtenue. La durée de vie de l’électrode peut dépasser 15 mois [70].
Tableau 1 : Exemples d’électrodes sélectives commercialisées
Société, pays Electrode Linéarité (ppm) Limite de détection Substances interférentes ELIT 8021, NO3 -3- 62 0.3 ppm Cl-, NO2 -Nico2000 Ltd, UK ELIT 8071, NO2 -1- 460 0.5 ppm CN-, Acétate Thermo-Orion, USA Orion nitrate
electrode Model 9307 0.4- 62000 -- NO2 -, ClO4 -, I -CN-, PO4 3-, SO4 2-,.... Radiometer Analytical, France ISE25NO3 0.2 – 60000 -- Cl -, Br-, NO2
-Pour la plupart des prototypes commercialisés, la limite de détection est insuffisante pour la mesure de faibles concentrations en ion nitrite. Ces détecteurs sont par conséquent utilisés pour des échantillons fortement contaminés. Par contre, l’électrode sélective aux ions nitrates, présente toujours des limites de détection de l’ordre des micromolaires. Son seul désavantage est la présence des éléments interférents.
I.3. La chromatographie ionique
La chromatographie ionique est mise à profit pour la première fois en 1975 lorsqu’elle est couplée à la détection conductimétrique a été réalisé. Cette technique a
connu un succès réel dans la détermination entre autres des ions SO42-, F-, Cl-, PO43-, NO3-, NH4+, Na+. En ce qui concerne les ions NO3- et NO2- qui nous intéressent, la chromatographie ionique a apporté des progrès remarquables dans l’analyse de ces ions. Son principal inconvénient est son coût élevé et son protocole expérimental assez compliqué. En effet, des prétraitements des échantillons sont recommandés. Les détecteurs associés dans ce cas au système chromatographique sont les détecteurs électrochimiques [71, 72].
La détection électrochimique du nitrite est réalisée par l’oxydation de ce dernier à une électrode de carbone vitreux à un potentiel voisin de +0.9V [73]. Les nitrates peuvent être analysés par le même système après leur conversion en nitrite par exposition aux radiations UV. Des limites de détection très faibles ont été obtenues (0.9 nmol/L en nitrite et 4.4 nmol/L en nitrate).
II. Méthodes d’analyse des ions ammoniums :
L’analyse de l’ion ammonium dans les eaux naturelles est d’une grande importance tant que des teneurs élevées en cet élément sont un indice de pollution par les rejets domestiques et l’agriculture. Il est également important d’éliminer NH4+avant l’introduction de l’eau dans le réseau parce qu’il réagit avec le chlore pour produire des chloramines, qui sont des désinfectants moins efficaces et peuvent provoquer des goûts désagréables. Certaines bactéries prolifèrent aussi en transformant l’ammonium en nitrites puis en nitrates.
Le développement de nouvelles techniques d’analyse et de contrôle de l’ammoniac dans l’eau est important car cette espèce est considérée parmi les substances indésirables par l’OMS. On peut déterminer la quantité d’ammoniac / ammonium dans l’eau au moyen de diverses méthodes.
II.1. Méthodes spectrophotométriques
La réaction de Nessler [74] ou la Nesslérisation est une méthode standard d’analyse de NH4+. Le réactif de Nessler synthétisé à partir du chlorure mercurique, d’iodure de potassium et de potasse, ajouté aux échantillons permet de mettre en évidence la présence
de NH4+. Une coloration jaune à brune se développe selon la concentration de cet ion. Cette technique n’est pas très utilisée à cause des problèmes environnementaux engendrés par l’usage du mercure.
La Méthode de Berthelot [75] est par la suite développée. Elle repose sur le bleu l’indophénol qui se forme par réaction des ions ammonium avec l’hypochlorite et le phénol en solution alcaline. L’usage de cette technique est limité à la chimie clinique. Cette réaction est relativement lente, elle est difficilement reproductible et exige des réactifs fraîchement préparés.
La méthode recommandée actuellement [76] est celle de Berthelot améliorée. Elle est basée sur la mesure de l’absorbance à 640nm de l’indophénol fortement coloré et formé suite à la réaction entre l’ammoniac, hypochlorite et le salicylate catalysée par le nitroprusside de sodium.
D’autres techniques ont également été développées : Ghodratollah Absalam et ses coll. [77] ont mesuré la variation de l’absorbance sous 408 nm, due à la présence de l’ammoniac, du complexe bis (acetylacétoneethylenediamine) tributylphosphin cobalt (III) tétraphenylborate imprégnant une membrane transparente en triacétylcellulose. Le pH du milieu est de 9. La limite de détection est de 5.10-6 mol/L. Cette membrane est utilisée avec succès dans l’analyse de l’eau de robinet.
Conway [78] a procédé à la libération de l’ammoniac par alcalinisation, suivie d’un piégeage en milieu acide et d’une révélation par le réactif de Nessler ou par la réaction de Berthelot, suivi d’une mesure spectrophotométrique. Ces méthodes sont très sensibles. Elles mesurent non seulement NH4+ libre mais également l’ammoniac susceptible d’être dégagé en milieu alcalin.
II.2. Méthodes enzymatiques
En 1963, Kirsten et coll. [79] ont mis au point une méthode enzymatique de dosage de l’ammoniac basée sur la réaction du glutamate déshydrogénase (GDH). Bien que cette méthode ne soit révélée extrêmement spécifique et qu’elle permette une évaluation directe fondée sur la faculté d’absorption molaire du NADH (forme réduite du nicotinamide
adénine dinucléotide), plusieurs problèmes ont surgi. Parmi lesquelles des difficultés à stabiliser les réactifs utilisés.
Da Fonseca-wollheim [80] a amélioré la réaction enzymatique de départ par l’addition de NADPH au mélange réactionnel au lieu de NADH selon l’équation :
α-cétoglutarate + NH4+ + NAD(P)H NAD(P)+ + glutamate + H2O L’utilisation de NADP (forme oxydée du nicotinamide adénine dinucléotide phosphate) au lieu de NAD permet d’améliorer la spécificité de la méthode en éliminant l’interférence potentielle de substrats consommant du NADH comme le pyruvate (en présence de la LDH plasmatique). La modification de la concentration du NAD(P)H est mesurée à 340nm à l’aide d’un spectrophotomètre. Ces techniques sont les plus adéquates pour traiter des échantillons de faible volume.
D’aut res techniques enzymatiques ont utilisé les méthodes électrochimiques comme outil de mesure : J. P. Hart et coll. [81] ont développé un biocapteur ampérométrique à base d’électrodes de carbone sérigraphiées imprégnées de Meldola’s blue (MB). Ces électrodes détectent la réaction d’oxydation du NADH à un potentiel égal à +0.05V (par rapport à l’électrode Ag/AgCl). L’ammoniac est mesuré en recouvrant la surface des électrodes modifiées par du glutamate dehydrogénase, du 2-oxoglutarate et du NADH. Lorsque l’ion NH4+ est présent en solution, le courant anodique diminue suite à la réaction enzymatique de conversion du 2-oxoglutarate en glutamate qui consomme le NADH. La limite de détection est aux environs de 2µmol/L lorsqu’on utilise 4.6 unités d’enzyme.
II. 3. Méthodes électrochimiques
II.3.1. La potentiométrie :
L’ammoniac dissous (NH3aq et NH4+) est converti en NH3aq en ajoutant une base forte aux standards et aux échantillons. La concentration de NH3 est mesurée à l’aide de l’électrode sélective d’ammoniac. Ce détecteur constitué d’électrode de verre équipée d’une membrane perméable à gaz est plus employé [82]. Dans ce système, Le gaz ammoniac produit diffuse à travers une membrane en PTFE (téflon) dans le compartiment
de l’électrode en verre contenant une solution de NH4Cl. Cette diffusion de l’espèce NH3 crée un nouvel équilibre réactionnel et par la suite un changement de pH de la solution située à proximité de la surface de l’électrode.
Pour pallier au problème de fragilité de l’électrode de verre, de nombreuses membranes polymériques contenant des molécules sélectives ont été proposées pour la fabrication de l’électrode sensible à l’ammonium (Meyerhoff [83] et Meyerhoff et coll. [84]). A titre d’exemple, les membranes en chlorure de polyvinyl (PVC) avec des molécules du nonactin incorporées. Ce système présente l’avantage d’être simple, sensible et pas coûteux mais sa durée de vie est très courte (1 semaine).
Les détecteurs potentiométriques développées par la suite sont basés sur des membranes épaisses obtenues par incorporation d’échangeurs d’ions inorganiques à des polymères plastifiés. Ces membranes spécifiques à l’ion NH4+, présentent une grande stabilité thermique et une grande résistance vis-à-vis des acides minéraux et des oxydants forts. Ce pendant, ce type de détecteur n’a pas connu beaucoup d’applications à cause de sa faible sélectivité. Par contre, Saad S. M. Hassan et ses Coll. [85] ont beaucoup gagné en terme de sélectivité en utilisant l’échangeur d’ions zirconium titanium phosphate (ZTP). Ce détecteur a été couplé au système FIA, 30 échantillons peuvent être analysés par heure dans une large gamme de linéarité qui s’étend de 12µmol/L jusqu’à 1 mol/L.
II.3.2. Les techniques voltamétriques
La mise au point de capteurs hautement sélectifs et faciles à manipuler nécessite en général la modification de la surface d’électrodes en utilisant des enzymes immobilisées ou des polymères conducteurs. A titre d’exemple, citons le détecteur ampérométrique développé par B. Strehlitz et coll. [86] qui consiste à mesurer l’ion ammonium à un potentiel égal à +0.3V par rapport à Ag/AgCl en utilisant des électrodes sérigraphies de Platine-carbone modifiées par de la polyaniline. Le même type de capteur a été couplé au système FIA [87].
Il existe peu de techniques voltamétriques pour le dosage de NH4+ : la méthode basée sur sa réaction avec l’hydroquinone en présence de dimethylformamide [87] a
montré que l’équilibre acide base qui existe entre l’hydroquinone et l’ammonium peut être exploité pour détecter NH4+. L’ion ammonium arrache des protons à l’hydroquinone qui s’oxyde vers +1.0V. Ceci permet de déplacer le processus d’oxydation vers un potentiel moins positif (aux environs de +0.5V). Le pic résultant a une intensité proportionnelle à la concentration de NH4+.
Un amalgame d’ammonium peut être obtenu par électrolyse d’une solution de sel d’ammonium à une cathode de mercure [88]. Cet ion est réduit à l’électrode à goutte de mercure à un potentiel de demi vague égal à -1.72V. La solubilité de NH4+ dans le mercure est suffisante (∼10-4mol/L) pour le préconcentrer à la surface de la microélectrode du mercure en utilisant la voltamétrie par redissolution anodique. Gladyshev [89] a montré que cette technique n’est pas efficace pour l’analyse de l’ammoniac et l’ammonium et que cette analyse est possible tout en choisissant un moyen plus simple : mesurer l’ammonium à une électrode à film mince de mercure en utilisant la voltamétrie par redissolution dans une solution de chlorure de potassium. La limite de détection obtenue est de 0.2µmol/L.
Une autre méthode très sensible a été développée par Anne Marie Harbin et Coll. [90]. Elle repose sur la réaction entre l’ammoniac et le formaldéhyde à pH 3.8 donnant naissance au composé méthylenimine. Ce produit est adsorbé à la surface de l’électrode pendante de mercure (HMDE). La voltamétrie impulsionnelle différentielle permet de mesurer le courant de réduction à -0.91V. Après le déroulement de la réaction chimique qui dure 20 minutes, la réponse est enregistrée en utilisant la voltamétrie par redissolution cathodique (CSV). Elle est proportionnelle à la concentration de l’ion ammonium dans l’intervalle 0.01 à 3µmol/L. La réaction de réduction du méthylenimine protonée donnant naissance au méthylanine est la suivante:
III. Méthodes d’analyse des phosphates
III.1. Méthodes spectrophotométriques
Les méthodes d’analyse de l’orthophosphate basées sur sa réaction avec les ions molybdate sont largement mises à profit dans la littérature.
La méthode standard colorimétrique [91] est basée sur la réaction des ions PO43- avec l’acide molybdique pour former le phosphomolybdate, qui est par la suite réduit pour former le complexe fortement coloré : le phosphomolybdate bleu. Cette méthode a connu plusieurs modification notamment en cherchant d’autres agents de réduction du 12-molybdophosphate afin d’améliorer la sélectivité et la stabilité du complexe bleu formé [92, 93]. Le réducteur le plus utilisé est l’acide ascorbique et le tartrate de potassium et d’antimoine comme catalyseur de la réaction [91]. Le seuil minimal de détection est de l’ordre de 10µgP/Lsous une longueur d’onde égale à 880nm, la longueur du trajet optique est de 5cm.
Dans les eaux naturelles, les ions silicates interfèrent avec les ions phosphates. Cette interférence peut être réduite en ajustant les proportions de l’acide et du molybdate en solution [94]. Pour analyser l’orthophosphate dans les eaux naturelles il faut par ailleurs arriver à détecter des teneurs de l’ordre de 1µgP/L. En effet, plusieurs techniques sont développées pour améliorer la sensibilité et la sélectivité de la spectrophotométrie.
A titre d’exemples, le phosphomolybdate réagit avec un grand nombre de composés basiques (le vert de malachite, le cristal violet et la rhodamine 6G) en milieu acide. Le complexe extrait est mesuré par spectrophotométrie ou par fluorimétrie. L’association du phosphomolybdovanadate et le vert de malachite a permis de déterminer le phosphate dans l’eau de rivière [95]. Les ions présents dans cette eau n’interfèrent pas. Quant à l’ion silicate, son interférence est plus faible que celle trouvée par les techniques de mesure déjà citées. La limite de détection est de 0.1 µgP/L.
Couplée au système d’analyse par injections en flux continu FIA [96], la méthode spectrophotométrique est très utilisée pour l’analyse des phosphates au laboratoire mais n’est pas applicable pour le contrôle et les mesures sur le terrain.
III.2. Les méthodes électrochimiques
Les méthodes électrochimiques ont été aussi développées : les techniques ampérométriques utilisent le complexe phosphomolybdique comme élément électroactif [97, 98]. Le phosphate a été déterminé à l’aide des méthodes voltamétriques à l’électrode à pâte de carbone [97], à la microélectrode d’or [98], à l’électrode au carbone vitreux [99] et à l’électrode à goutte de mercure [100]. Les signaux obtenus mesurent la réductio n du complexe phosphomolybdique. Les limites de détection sont de quelques micromoles par litre.
Fogg et coll. [101] ont analysé le phosphate sous forme de phosphomolybdovanadate à l’électrode au carbone vitreux. Couplé au système FIA, la procédure permet de détecter 1µmol/L en phosphate avec un coefficient de variation inférieur à 1%. Des meilleures sensibilités ont été aussi atteintes en analyse par injection en flux continu. La limite de détection pour un rapport signal/ bruit égal à 2 est de 0.02µmol/L sachant que le milieu électrolytique contient du méthanol à 30%.
Les biocapteurs électrochimiques sont aussi reportés [102 – 104]. Ces détecteurs utilisent plus qu’une seule enzyme (exemple la xantine oxydase et la nucléoside phosphorylase [104, 105], la phosphorylase A, phosphoglucomutase et la glucose 6-phosphate déshydrogénase [103], la maltose phosphorylase, acide phosphatase et la glucose oxydase [106].
Les ions phosphate ont été aussi déterminés dans l’eau par chromatographie ionique couplé au détecteur conductimétrique [107, 108].
Références
[1]. F. Ramade, Ecologie des ressources naturelles, Masson, 1981. [2]. R. L. Shirly, Bioscience, vol 25, n° 12 1975, 789.
[3]. K. Emerson, R. C. Russo, R. E. Lund and R. V. Thurston. J. Fish. Res. Board Con. 32, 1975, 2379.
[4]. Chambers, P.A., M. Guy, E.S. Roberts, R. Kent, M.N. Charlton, C. Gagnon, G. Grove et N. Foster. 2001. Environnement Canada, Ottawa (Ont.). 271 p.
[5]. http://www.technopole-anticipa.com
[6]. P. Albertano, Giorn Bot. Ital. 127, 1993, 385.
[7]. Th. Warscheid, J. Braams, Int. Biodet. Biodegr. 46, 2000, 343. [8]. P. Albertano, J. Comput. Assist. Microsci. 9, 1997, 81.
[9]. N. G. Carr, B. A. Whitton (Eds.), The biology of cyanobacteria, Botanical Monograph, vol 19, Blackwell Scientific Publications, 1982, p. 105 – 124 (Chapitre 5).
[10]. M. J. Moorcroft, J. Davis, R. G. Compton, Talanta, 54, 2001,785.
[11]. X. L. Su, P. Chen, X. G. Qu, W. Z. Wei, S. Z. Yao, Microchem. J. 59, 1998, 341. [12]. J. Hilton, E. Rigg, Analyst, 108, 1983, 1026.
[13]. J. F. Van Staden, Anal. Chim. Acta, 138, 1982, 403. [14]. K. Takeda, K. Fujiwara, Anal. Chim. Acta. 276, 1993, 25.
[15]. F. Yang, E. Troncy, M. Francoeur, B. Vinet, P. Vinay, G. Czaika, G. Blaise, Clin. Chem., 43, 1997, 657.
[16]. T. Aoki, S. Fukuda, Y. Hosoi, H. Mukai, Anal. Chim. Acta, 349, 1997, 11. [17]. R. S. Braman, S. A. Hendrix, Anal. Chem. 61, 1989, 2715.
[18]. D. Tsikas, F. M. Gutski, S. Rossa, H. Baner, G. Neumann, K. Dockendorff, J. Sandmann, J. C. Frölich, Anal. Biochem. 244, 1997, 208.
[19]. H. W. Jannash, K. S. Johnson, C. M. Sakamoto, Anal. Chem. 66, 1994, 3352. [20]. L. Monser, S. Sadok, G. M. Greenway, I. Shah, R. F. Uglow, Talanta, 57, 2002, 511.
[21]. H. Borcherding, S. Leikefeld, C. Frey, S. Dickmann, P. Steinrucke, Anal. Biochem. 282, 2000, 1.
[22]. I. N. Papadoyannis, V. F. Samanidou, C. C. Nitsos, J. Liq. Chromatogr. R. T. 22, 1999,2023.
[23]. H. Chen, Y. Fang, T. An, K. Zhu, J. Lu; Int. J. Environ. Anal. Chem. 76, 2000, 89. [24]. M. N. Abbas, G. A. Mostafa; Anal. Chim. Acta; 410, 2000, 185.
[25]. M. Miro, A. Cladera, J. M. Estela, V. Cerda; Analyst 125, 2000, 943.
[26]. Z. Legnerová, P. Solich, H. Sklenárová, D. Šatinský, R. Karlicek; Wtar Research, 36, 2002, 2777.
[27]. Z. K. He, B. Fuhrmann, U. Spohn, Fres. J. Anal. Chem.; 367, 2000, 264. [28]. R. T. Masserini, Jr. K. A. Fanning; Marine Chemistry, 68, 2000, 323.
[29]. T. P. Misko, R. G. Schilling, D. Salvemini, W. M. Moore, M. G. Currie; Anal. Biochem. 214, 1993, 11.
[30]. T. Odake, M. Tabuchi, T. Sato, H. Susaki, T. Korenaga; Analytical Sciences, 17, 2001, 535.
[31]. X. - Q. Zhan, D.-H. Li, H. Zheng, J.-G. Xu, Y.-Q. Zhoo, Talanta, 58, 2002, 855. [32]. J. P. Griess, Ber. Dtsch. Chem. Ges. 12, 1879, 426.
[33]. M. Badea, A. Amine, M. Benzine, A. Curulli, D. Moscone, A. Lupu, G. Volpe, G. Palleschi; Microchimica Acta, 147, 2004, 51.
[34]. P. F. Pratt, K. Nithipatikom, W. B. Campbell; Anal. Biochem. 231, 1995, 383. [35]. M. J. Ahmed, C. D. Stalikas, S. M. Tzouwara-Karayanni, M. I. Karayannis; Talanta, 43, 1996, 1009.
[36]. R. S. Guerrero, C. G. Benito, J. M. Calatayud, Talanta, 43, 1996, 239. [37]. A. A. Ensafi, A. Kazemzadeh; Anal. Chim. Acta, 382, 1999, 15.
[38]. I. A. Pettas, S. I. Lafis, M. I. Karayannis; Anal. Chim. Acta, 37,6, 1998, 331. [39]. S. H. Lee, L. R. Field, Anal. Chem. 56, 1984, 2647.
[40]. A. Lapat, L. Székelyhidi, I. Hornyàk, Biomed. Chromatogr. 11, 1997, 102. [41]. P. Damiani et G. Burini, Talanta, vol 33, n° 8, 1986, 649.
[42]. T. Aoki, S. Fukuda, Y. Hosoi, H. Mukai; Anal. Chim. Acta, 349, 1997, 11. [43]. K. Yoshizumi, K. Aoki, Anal. Chem. 57, 1985, 737.
[44]. M. Gallego, M. Silva, M. Valcarcel, Fres. Z. Anal. Chem. 323, 1986, 50. [45]. M. Shibata, K. Yoshida, N. Furuya, J. Electrochem. Soc. 387, 1995, 143. [46]. M. Shibata, K. Yoshida, N. Furuya, J. Electrochem. Soc. 145, 1998, 287. [47]. N. G. Carpenter, D. P. Plecher, Anal. Chim. Acta 317, 1995, 287.
[48]. J. O. Bockris, J. Kim; J. Electrochem. Soc. 143, 1996, 3801. [49]. M. E. Bodini, D. Sawyer; Anal. Chem.,49, 1977, 485. [50]. V. Mori, M. Bertotti; Anal.Lett. 32, 1999, 25.

![Figure 2 : Diagramme schématique de la cellule BIA réalisée par Wang et Taha [14]. (A) électrode de travail ; (B) électrode auxiliaire ; (C) électrode de référence ; (D) embout de la micropipette ; (E) orifice ; (F) barreau magnétique et (G) sortie de la solution](https://thumb-eu.123doks.com/thumbv2/123doknet/2187835.11101/50.894.214.701.560.981/schematique-realisee-electrode-electrode-auxiliaire-reference-micropipette-magnetique.webp)