Expression et fonction des intégrines liant le
collagène chez les lymphocytes Th1
Thèse
Marc Boisvert
Doctorat en physiologie-endocrinologie
Philosophiae Doctor (Ph. D.)
Québec, Canada
© Marc Boisvert, 2013
Résumé
Les lymphocytes Th17 sont une sous-population de lymphocyte T découverte récemment et dont la particularité est de produire de l’IL-17. Ces cellules ont été impliquées dans le développement de maladies auto-immunes. Toutefois, les mécanismes régulant les fonctions des lymphocytes Th17 dans ces maladies ne sont pas très bien connus. Plusieurs études indiquent que les intégrines liant le collagène sont des molécules de costimulation pour les lymphocytes T effecteurs. De plus, ces intégrines ont été impliquées dans le développement de plusieurs maladies auto-immunes. On les retrouve entre autres sur les lymphocytes T arthritiques du liquide synovial. Cependant, l’expression et les fonctions des intégrines liant le collagène chez les lymphocytes Th17 sont inconnues. Nous avons montré que l’intégrine liant le collagène alpha2beta1 (α2β1) est exprimée sur les cellules Th17, mais que l’intégrine α1β1 ne l’est pas. Nous avons également observé que les collagènes de type I et de type II costimulent la production d’IL-17A, d’IL-17F et d’IFN-γ dans les lymphocytes Th17 humains activés par le récepteur des cellules T (TCR) alors que le collagène de type IV, liant l’intégrine α1β1, n’a pas d’effet sur la production de ces cytokines. Nous avons également montré que les lymphocytes T expriment un autre récepteur pour le collagène, soit DDR1. Nos résultats suggèrent que ce récepteur n’est pas impliqué dans l’adhésion, mais plutôt dans la migration des lymphocytes T dans le collagène tridimensionnel. Nous avons trouvé que les lymphocytes Th17 expriment DDR1, mais que celui-ci n’est pas impliqué dans la costimulation par le collagène. Nos résultats ont mis en évidence le rôle des voies de signalisation MAPkinases ERK, JNK et la voie PI3K/AKT pour l’expression et la production d’IL-17 suite à la stimulation par le TCR alors que la voie MAPkinase p38 n’est importante que pour la costimulation de l’IL-17 par l’intégrine α2β1. Nos résultats ont également montré que l’expression du facteur de transcription RORc, qui joue un rôle central dans la différenciation Th17 et l’expression de l’IL-17, est augmentée par la costimulation du collagène. L’expression de RORc nécessite les voies MAPkinase ERK et PI3K/AKT qui sont aussi augmentées par le collagène. Nos résultats indiquent que l’intégrine α2β1 participe à l’activation des lymphocytes Th17 et de ce fait peut réguler à la hausse le développement des maladies auto-immunes dans les tissus riches en collagène.
Abstract
Th17 cells are a new subset of T cells defined by their ability to produce IL-17. These cells have been implicated in the development of autoimmune diseases. However, the mechanisms regulating the functions of Th17 cells in these diseases are still poorly understood. Several studies have shown that collagen-binding integrins are important costimulatory molecules of effectors T cells. Also, these integrins have been implicated in the development of autoimmune diseases. However, the expression and functions of collagen-binding integrins in Th17 cells are currently unknown. We have shown that the collagen-binding integrin alpha2beta1 (α2β1), but not alpha1beta1 (α1β1), is expressed on Th17 cells. We have demonstrated that both collagen type I and II, by binding to α2β1 integrin, are efficient costimulatory molecules as they increase expression and production of IL-17A, IL-17F and IFN-γ by human effector Th17 cells activated through the T-cell receptor (TCR). However, collagen type IV, which binds α1β1 integrin, did not. We have also shown that T cells express another collagen-binding receptor, namely the discoidin-domain receptor 1 (DDR1). Our results suggest that this receptor is not required for cell adhesion but rather for T cell migration through three dimensional collagen matrices. We found that Th17 cells express DDR1 but that it is not implicated in the costimulation induced by collagen. Our results have also shown that the ERK and JNK MAPkinases pathways, as well as the PI3-K/AKT pathway, are required for the expression and production of IL-17 by the TCR while the p38 MAPKinase pathway is only required for the costimulation of IL-17 by α2β1. Lastly, our results have demonstrated that the expression of transcription factor RORC, the Th17 master regulator, is increased by collagen. This expression requires the ERK MAPKinase and PI3-K/AKT pathways which are both increased by collagen. Thus, our results indicate that the collagen-binding integrin α2β1 is a major costimulatory molecule in the activation of Th17 cells and could regulate the development of autoimmune diseases in collagen-rich tissues.
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vii
Liste des tableaux ... xi
Liste des schémas ... xiii
Liste de figures ... xv
Liste des abréviations ... xvii
Remerciements ... xxiii
Avant-propos ... xxv
Chapitre 1 : Introduction ... 1
1.1 Lymphocytes T ... 1
1.2 Récepteur des cellules T ... 3
1.3 Différenciation des lymphocytes T auxiliaires ... 4
1.3.1 Th1 ... 5
1.3.2 Th2 ... 7
1.3.3 Th9 ... 7
1.3.4 Th17 ... 7
1.3.5 Treg ... 11
1.3.6 Plasticité des lymphocytes T auxiliaires ... 12
1.4 Cytokines proinflammatoires ... 16
1.4.1 IL-17 ... 16
1.4.2 IFN-γ ... 19
1.4.3 IL-21 ... 22
1.4.4 IL-22 ... 22
1.5 Lymphocytes T auxiliaires et développement des maladies auto-immunes. ... 22
1.5.1 Arthrite rhumatoïde ... 23
1.5.2 Sclérose en plaques ... 27
1.5.3 Maladies inflammatoires des intestins ... 29
1.6 Signalisation du TCR ... 30
1.6.1 Initiation de la signalisation ... 31
1.6.2 IP3 et la voie du calcium ... 32
1.6.3 DAG et la voie de PKC... 35
1.6.4 Activation des MAP kinases ... 35
1.6.5 Activation de STAT3 ... 38
1.7 Récepteurs de costimulation et de régulation ... 40
1.7.1 Les récepteurs de la famille du CD28 ... 40
1.7.2 Les récepteurs de la famille du TNF ... 43
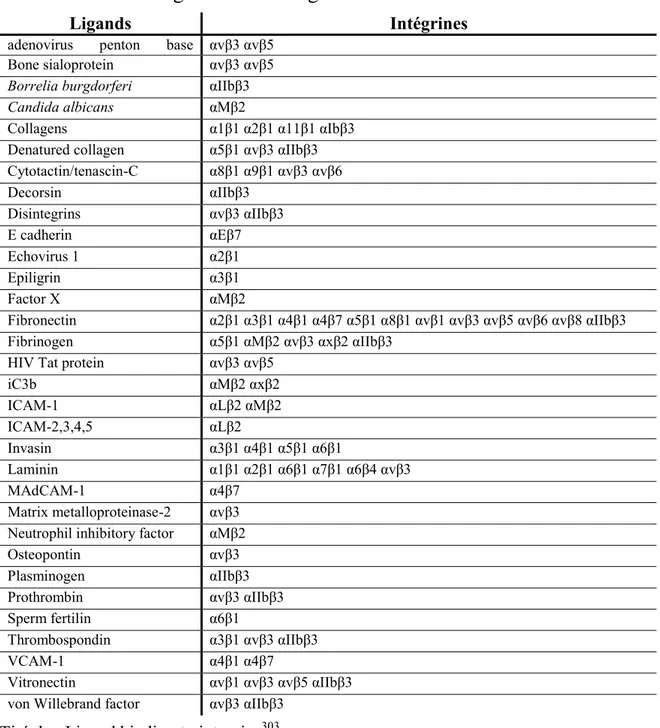
1.8 Intégrines ... 44
1.8.1 Structure et expression des intégrines ... 45
1.8.2 Activation des intégrines ... 49
1.8.3 Signalisation des intégrines ... 51
1.8.4 Fonctions des intégrines ... 53
1.8.4.1 Migration ... 53
1.8.4.2 Prolifération et survie ... 53
1.9 Intégrines et lymphocytes T ... 54
1.9.1 Expression des intégrines chez les lymphocytes T ... 54
1.9.2 Fonctions des intégrines chez les lymphocytes T ... 55
1.9.2.1 Fonctions des intégrines β2 ... 55
1.9.2.2 Fonctions des intégrines β1 ... 56
1.9.3 Intégrines liant le collagène ... 56
1.9.3.1 Expression des intégrines liant le collagène ... 57
1.9.3.2 Fonctions des intégrines liant le collagène ... 58
1.10 Rôles des intégrines dans le développement des maladies auto-immunes. ... 60
1.10.1 Arthrite rhumatoïde ... 60
1.10.2 Sclérose en plaques ... 61
1.10.3 Maladies inflammatoires des intestins (IBD) ... 62
1.11 Récepteurs à domaine discoïdine ... 63
1.12 Problématique et hypothèse... 64
1.13 Buts ... 65
Chapitre II : L’intégrine alpha2beta1 est la principale intégrine liant le collagène chez les lymphocytes Th17 ... 67
2.1 Résumé ... 67
2.2 Abstract ... 71
2.3 Introduction ... 73
2.4Results ... 75
2.4.1 Th17 cells differentiation preferentially upregulate α2β1 integrin. ... 75
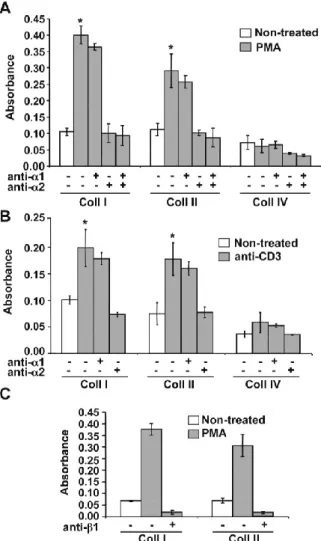
2.4.2 Th17 cell adhesion to collagens is dependent on α2β1 integrin. ... 76
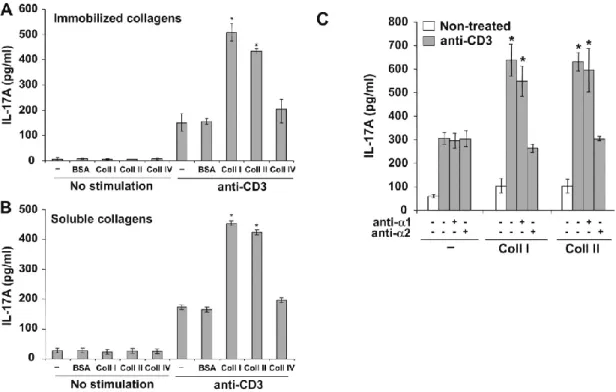
2.4.3 α2β1 enhances the production of Th17 cytokines. ... 77
2.5 Discussion ... 78
2.6 Materials and Methods ... 81
2.6.1 Antibodies and reagents ... 81
2.6.2 Isolation of primary T cells and T helper cell differentiation ... 81
2.6.3 Real-Time RT-PCR analysis ... 82
2.6.4 Cell surface molecule expression ... 82
2.6.5 Staining for integrin receptors and intracellular IL-17 ... 83
2.6.7 T cell costimulation and IL-17 ELISA ... 83
2.7 References ... 85
2.8 Figure and legends ... 89
Chapitre III : L’intégrine alpha2beta1 costimule les lymphocytes Th17 par les voies de signalisation MAPKinase ERK et p38 ainsi que la voie PI3-K/AKT ... 95
3.1 Résumé ... 95
3.2 Abstract ... 99
3.3 Introduction ... 101
3.4 Materials and methods ... 103
3.4.1 Antibodies and reagents ... 103
3.4.2 Isolation of human naïve CD4 T cells and differentiation of Th17 ... 103
3.4.3DDR expression and IL-17 intracellular staining ... 103
3.4.4Real-Time RT-PCR analysis ... 104
3.4.5 Th17 cell activation experiments and IL-17 expression ... 104
3.4.6 Immunoblotting, activation of MAPKs, AKT and STAT3 ... 105
3.5 Results ... 106
3.5.1 The costimulatory effect of collagen is mediated via β1 integrin independently from the discoidin domain receptors. ... 106
3.5.2 α2β1 integrin co-stimulates IL-17 via ERK and p38 MAPK. ... 106
3.5.3 α2β1 integrin weakly co-stimulates the PI3 Kinase /AKT pathway. ... 107
3.5.4 Collagen costimulates RORc in human Th17 cells. ... 107
3.5.5 STAT3 is not involved in IL-17 production. ... 108
3.6 Discussion ... 109
3.7 Figure and legends ... 112
Chapitre IV : L’expression du récepteur à domaine discoïdine 1 chez les lymphocytes T activés est régulée par la voie MAP Kinase ERK ... 123
4.1 Résumé ... 123
4.2 Abstract ... 127
4.3 Introduction ... 129
4.4 Materials and Methods ... 131
4.4.1 Antibodies and reagents ... 131
4.4.2 T cell isolation and activation ... 131
4.4.3 Collagen I binding analysis ... 131
4.4.4 Cell adhesion assay ... 132
4.4.5 Plasmids and Jurkat T Cell transfection ... 132
4.4.6 Immunoblot analysis ... 132
4.4.7 RT-PCR analysis ... 133
4.5 Results ... 134
4.5.2 DDR1 doesn’t enhance T cell adhesion to collagen I. ... 134
4.5.3 CD28 costimulation does not upregulate DDR1 expression. ... 135
4.5.4 TCR-induced DDR1 expression is not regulated by the Ca2+ signaling cascade. ... 135
4.5.5 TCR-induced DDR1 expression is regulated by the Ras/Raf/ERK MAPK signaling pathway. ... 136
4.5.6 TCR- induced DDR1 expression is dependent on PKC pathway. ... 136
4.5.7 Inhibition of ERK MAPK and PKC reduces the ability of activated T cells to bind collagen I. ... 137
4.6 Discussion ... 138
4.7 Figures and legends ... 141
4.8 References ... 149 Chapitre V : Discussion ... 153 5.1 Perspective ... 161 Bibliographie ... 163 Annexe I ... 189 A.1 Abstract ... 191 A.2 Introduction ... 193
A.3 Expression of VLA-2 on T cells ... 194
A.4 VLA-2-mediated T cell adhesion to collagen ... 195
A.5 Role of VLA-2 in T cell survival ... 196
A.5.1 Regulation of activation-induced cell death (AICD) in T cells ... 196
A.5.2 Inhibition of Fas-L expression ... 197
A.5.3 Inhibition of Fas-induced signaling ... 198
A.5.4 Modulation of other forms of apoptosis ... 201
A.6 Role of VLA-2 signaling in T cell costimulation (cytokine production and proliferation) ... 202
A.7 VLA-2 signal transduction ... 204
A.7.1 VLA-2-mediated activation of the MAPK/ERK signaling pathway ... 204
A.7.2 VLA-2 signaling in the context of TCR ... 206
A.8 Role of VLA-2 in inflammatory diseases ... 209
A.9 Concluding remarks ... 210
Liste des tableaux
Tableau 1 : Cytokines impliquées dans la différenciation Th17 ... 10 Tableau 2 : Sous unités des intégrines selon la classification CD ... 46 Tableau 3 : Les intégrines et leurs ligands ... 48
Liste des schémas
Schéma 1 : Différenciation des lymphocytes T auxiliaires ... 6
Schéma 2 : Méthylation des gènes de facteurs de transcription selon le phénotype ... 15
Schéma 3 : Rôle de l’IL-17 dans le développement de l’arthrite rhumatoïde ... 26
Schéma 4 : Signalisation induite lors de l’activation du TCR ... 34
Schéma 5 : Activation de ERK par le TCR ... 37
Schéma 6 : Activation de p38 et JNK par le TCR ... 39
Schéma 7 : Combinaison des différentes sous-unités α et β des intégrines ... 47
Schéma 8 : Liaison des intégrines avec le cytosquelette ... 50
Schéma 9 : Signalisation des intégrines ... 52
Liste de figures
Chapitre II
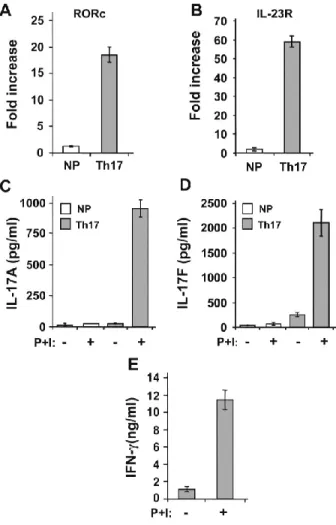
Figure 1 : Th17 differentiation of human naïve CD4+ T cells. ... 89
Figure 2 : Expression of α1, α2 and β1 integrins on human naïve CD4+ T cells cultured under Th17-polarizing conditions. ... 90
Figure 3 : Expression of α1 and α2 integrin chains on human Th 17 cells. ... 91
Figure 4 : Cell adhesion of human Th17 cells to collagen matrices. ... 92
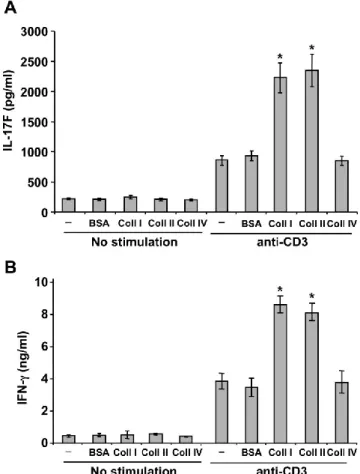
Figure 5 : Costimulation of human Th17 cells by collagens. ... 93
Figure 6 : Collagen costimulation of IL-17F and IFN-γ production. ... 94
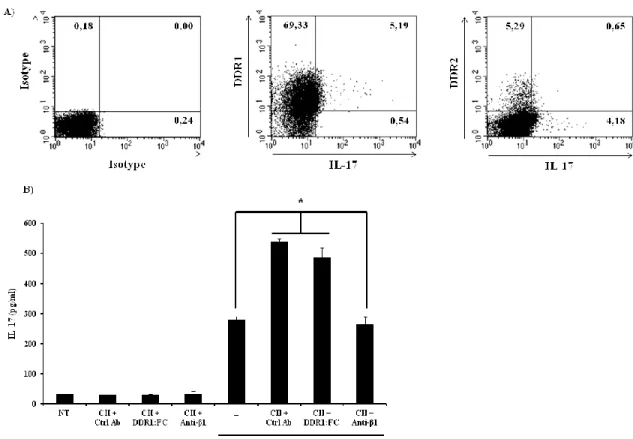
Chapitre III Figure 1 : Collagen co-stimulatory effect does not require DDR1 or DDR2. ... 112
Figure 2 : Collagen costimulates IL-17 expression via ERK and p38 MAPK. ... 113
Figure 3 : Collagen activation of ERK, JNK and p38 MAPKs. ... 114
Figure 4 : Collagen co-stimulation of IL-17 involves the PI3 Kinase/AKT pathway. ... 115
Figure 5 : Collagen co-stimulates the expression of RORc via MAPK/ERK and PI3 kinase/AKT. ... 116
Figure 6 : STAT3 activation is not required for IL-17 co-stimulation by collagen. ... 117
Figure 7 : Model of cooperation between α2β1 integrin and TCR/CD3 signalling in human Th17 cells. ... 118
Chapitre IV Figure 1 : DDR1 expressed by activated T cells binds to collagen I. ... 141
Figure 2 : DDR1 does not enhance T cell adhesion to collagen I. ... 142
Figure 3 : CD28 costimulation does not upregulate DDR1 expression. ... 143
Figure 4 : The Ca2+/calcineurin-signaling pathway is not involved in DDR1 expression.. 144
Figure 5 : TCR-induced DDR1 expression is regulated by the Ras/Raf/ERK pathway but not by the p38 or the JNK MAP Kinases. ... 145
Figure 6 : PKC is involved in TCR- and PMA-induced DDR1 expression in T cells. ... 146
Figure 7 : Inhbition of ERK MAPK and PKC reduces the ability of activated T cells to bind collagen I. ... 147
Figure 8 : A model by which TCR/CD3 signaling induces the expression of DDR1 in human T cells. ... 148
Annexe Figure 1 : Model for Coll I/ VLA-2-dependent inhibition of AICD in T cells. ... 200
Liste des abréviations
AICD Mort induite par l'activation (Activation-induced cell death)
AR Arthrite rhumatoïde
ATF-2 Facteur de transcription activateur-2 (Activating Transcription Factor-2)
AP-1 Protéine activatrice 1 (Activator Protein 1)
CD Cluster de Différenciation (Cluster of Differentiation CMH Complexe Majeur d’Histocompatibilité
CPA Cellule Présentatrice d’Antigène
CRAC Canaux dépendant du relargage du calcium (Calcium Release Activated Channels) CTLA-4 Agent cytotoxique des lymphocytes T 4
(Cytotoxic T-Lymphocyte Antigen 4)
DAG Diacyl Glycerol
DDR Récepteur à domaine discoïdine (Discoidin domain receptor) DMARD Drogue anti-rhumatique modifiant la maladie
(Disease Modifying Anti-Rhumatic Drug) EAE Encéphalomyélite Auto-immune Expérimentale
(Experimental Autoimmune Encephalomyelitis) ERK Kinase régulée par des signaux extracellulaires
(Extracellular signal-regulated kinase)
FAK Kinase de l'adhésion focale (Focal adhesion kinase) FasL Ligand de Fas (Fas Ligand)
Foxp3 Forkhead box P3
Grb2 Protéine couplée aux récepteurs de facteurs de croissance-2 (Growth factor receptor-bound protein-2)
IBD Maladie inflammatoire des intestins (Inflammatory Bowel Disease) ICAM-1 Molécule d'adhésion intercellulaire 1
(Intercellular adhesion molecule 1) ICOS Molécule de costimulation inductible
(Inducible costimulator molecule)
IκB Inhibiteur de NF-κB (Inhibitor of NF-κB)
IKK Kinase de IκB (IκB Kinase)
ILK Kinase liée aux intégrines (Integrin-linked kinase)
IFN-γ Interféron-γ
Ig Immunoglobuline
IL Interleukine
IP-3 inositol 1,4,5-triphosphate
ITAM Motif d’activation des immunorécepteurs via une tyrosine (Immunoreceptor Tyrosine-based Activation Motif) iTreg Treg induit (Inducible Treg)
JAK Janus kinase
JNK Kinase de c-Jun en N-terminale (c-Jun N-terminal kinase)
KO Knock Out
LAT Lien d’activation des lymphocytes T (Linker for Activation of T cells) LCMV Virus lymphocytique choriomeningitique
(Lymphocoriomeningitis virus)
LFA-1 Antigène associé à la fonction lymphocytaire (Lymphocyte function-associated antigen)
LPS Lipopolysaccharide
MAPK Protéine kinase à action mitogène (Mitogen activated protein kinases)
MEC Matrice extracellulaire
MEK MAPK/ERK kinase
MMP Métalloprotéinase
nTreg Treg naturel (naturaly occuring Treg) RORc Récepteur orphelin apparenté à RAR c
(RAR-related Orphan Receptor c)
NF-AT Facteur nucléaire des lymphocytes T activés (Nuclear factor of activated T cells)
NF-κB Facteur nucléaire-κB (Nuclear factor-κB)
PD-1 Protéine de mort programmée 1 ( Programmed Death 1)
PHA Phytohémagglutinine
PI3-K Kinase des phosphatidylinositols 3 (Phosphatidylinositol 3-kinase) PIP2 Phosphatidylinositol 4,5 bisphosphate
PIP3 Phosphatidylinositol 3,4,5-trisphosphate
PKB Protéine Kinase B
PKC Protéine Kinase C
PLCγ Phospholipase C γ
PMA Phorbol-12-Myristate 13-Acétate PP2A Protéine phosphatase 2A
PYK2 Tyrosine kinase riche en proline 2 ( Proline-rich Tyrosine Kinase 2) RANK Récepteur activateur de NF-KB
(Receptor-activator of NF-KB)
RANKL RANK ligand
Rap1 Ras-related protein 1
RasGRP Protéine de relargage de guanyle associée à Ras (Ras guanyl-nucleotide-releasing protein) SH2 Homologie à Src-2 (Src homology-2)
SLP-76 Phosphoprotéine contenant un domaine SH-2 des leukocytes de 76 kDa (SH2 domain-containing leukocyte phosphoprotein of 76 kDa)
SNC Système Nerveux Central
Sos Son of Sevenless
STAT Transducteur de Signal et Activateur de la Transcription (Signal Transducer and Activator of Transcription) TCR Récepteur des cellules T (T Cell Receptor)
Th Lymphocyte T auxiliaire (T helper) Tfh Lymphocyte T folliculaire auxiliaire
TGF-β Facteur de croissance transformant β (Transforming Growth Factor β) TNF Facteur de nécrose des tumeurs (Tumor Necrosis Factor)
TNFR Récepteur du TNF (TNF Receptor)
TRAF Facteur associé au récepteur du TNF (TNF receptor-associated factor)
Treg Lymphocyte T régulateur
VCAM-1 Molécule d'adhésion cellulaire vasculaire 1 (Vascular cell adhesion molecule 1)
VLA Antigène très tardif (Very Late Antigen)
Remerciements
En premier lieu, je voudrais remercier mon directeur de recherche, Dr Fawzi Aoudjit pour sa supervision, son enseignement, ses bons conseils et ses encouragements qui m’ont permis de mener à bien mes travaux de recherche. Je lui témoigne toute ma reconnaissance. Merci aux membres de l’équipe, anciens (Nizar Chetoui, Steve Gendron et Marie-Ève Bergeron) et actuels (Dalila Naci et Amine El Azreq), pour les discussions et l’atmosphère agréable du laboratoire. Merci également à tous les membres du CRRI que j’ai côtoyé au cours des années.
Merci aux membres de ma famille d’avoir fait de moi qui je suis et merci à mon amoureuse de rendre ma vie aussi intensément agréable.
Avant-propos
Cette thèse contient la majeure partie de mes travaux de doctorat, paru dans deux articles publiés, un troisième à venir et un chapitre de livre.
Chapitre II : Alpha2beta1 integrin is the major collagen-binding integrin expressed on human Th17 cells.
Eur J Immunol. 2010 Oct;40(10):2710-9.
Dans cet article, j’ai réalisé l’ensemble des manipulations qui ont mené aux figures présentées. J’ai participé à la planification des expérimentations et à l’analyse des résultats ainsi qu’à la rédaction et la correction du manuscrit. Steve Gendron a mis au point le protocole de génération des Th17 et Nizar Chetoui a contribué à l’analyse des résultats de cytométrie de flux (Figure 2 et 3).
Chapitre III : Alpha2beta1 integrin co-stimulates Th17 cells via ERK, p38 MAPKs and PI3 Kinase/AKT signaling pathways.
Non publié (en préparation)
Dans cet article, j’ai réalisé l’ensemble des manipulations qui ont mené aux figures présentées. J’ai participé à la planification des expérimentations et à l’analyse des résultats ainsi qu’à la rédaction et la correction du manuscrit. Jamila Chakir m’a aidé à réaliser les expérimentations de PCR en temps réel.
Chapitre IV : Discoidin domain receptor 1 expression in activated T cells is regulated by the ERK MAP kinase signaling pathway.
J Cell Biochem. 2011 Dec;112(12):3666-74.
Dans cet article, Nizar Chetoui a réalisé les manipulations en lien avec la signalisation (Figure 3 à 6). Amine El Azreq a rédigé le manuscrit et Marie-Ève Bergeron a réalisé l’analyse des interactions entre le collagène et DDR1 par cytométrie de flux (Figure 1 et 7). Pour ma part, j’ai réalisé les tests d’adhésions qui ont mené à la figure 2. J’ai contribué aux manipulations et à l’analyse des interactions entre DDR1 et le collagène (Figure 1 et 7). J’ai également participé à la correction du manuscrit.
Annexe I : VLA-2 signaling on T cells: a new emerging costimulatory pathway in effector/memory T cell subsets?
Effector memory T cells subsets in the extralymphatic tissue-interactions with the extracellular matrix influencing functions. 2009
Cet article de revue fut écrit sous l'invitation du Dr.Ilan Bank de l'Université de Tel-Aviv, pour un livre concernant le rôle des interactions des lymphocytes T effecteurs et mémoires avec la matrice extracellulaire. J'ai contribué à hauteur de 40% à la rédaction du manuscrit. Cette part est égale à celle de Steve Gendron avec qui je partage la place de 1er auteur. Nizar Chetoui a corrigé le manuscrit.
Chapitre 1 :
Introduction
Chez l’humain, le système immunitaire est composé de nombreuses cellules qui interagissent afin de protéger l’organisme contre la myriade de pathogènes présents dans l’environnement. Le système immunitaire peut être divisé en deux volets soit l’immunité innée et l’immunité acquise. L’immunité innée est la première ligne de défense de l’organisme et les cellules qui la composent reconnaissent différentes molécules associées aux pathogènes. Ce volet de l’immunité est rapidement activé en présence de pathogènes. L’activation du système immunitaire inné mènera éventuellement à l’activation de l’immunité acquise. Chaque cellule de l’immunité acquise, contrairement à celles de l’immunité innée, ne reconnaissent qu’un seul antigène. C’est pour cette raison que l’immunité acquise est également connue sous le nom d’immunité spécifique. Les cellules qui la composent sont les lymphocytes B et les lymphocytes T. Ces derniers se divisent en deux catégories soit les lymphocytes T cytotoxiques et les lymphocytes T auxiliaires. Les lymphocytes T cytotoxiques peuvent tuer les cellules infectées de même que les cellules transformées telles que les cellules cancéreuses. Les lymphocytes T auxiliaires quant à eux, vont organiser et diriger la réponse immunitaire acquise en produisant différentes cytokines et chimiokines. En plus de leur rôle dans l’immunité protective, les lymphocytes T sont également impliqués dans la réponse inflammatoire associée aux maladies auto-immunes.
1.1 Lymphocytes T
Les lymphocytes T immatures sont issus de la moelle osseuse, générés par l’hématopoïèse. Ils migrent alors jusqu’au thymus où ils seront sélectionnés pour leur capacité à interagir avec le complexe majeur d’histocompatibilité (sélection positive) sans reconnaître les antigènes du soi (sélection négative). Les lymphocytes T qui ne passent pas ces étapes de sélection sont généralement éliminés par apoptose, mais peuvent également survivre dans un état anergique, c’est-à-dire réfractaire à l’activation1. Ceux qui passent les deux étapes
de sélection deviennent des lymphocytes T matures. Les lymphocytes T matures se divisent en trois groupes que l’on nomme naïfs, effecteurs et mémoires. Les lymphocytes T restent naïfs tant qu’ils n’ont pas rencontré leur antigène, suite à quoi ils deviennent effecteurs. Les lymphocytes T mémoires sont des cellules qui ont rencontré l’antigène et qui persistent après l’élimination du pathogène, participant ainsi à la création de la mémoire immunologique.
Les lymphocytes T naïfs migrent vers les systèmes lymphatiques et circulatoires à la recherche de l'antigène pour lequel ils sont spécifiques. Cet antigène devra être présenté par un complexe majeur d'histocompatibilité (CMH) de type I ou II selon que le lymphocyte T est CD8+ (cytotoxique) ou CD4+ (auxiliaire) respectivement. Les molécules de CMH I sont exprimées par pratiquement toutes les cellules de l'organisme, alors que les molécules de CMH II ne sont exprimées principalement que par les cellules présentatrices d'antigènes (CPA). Les principales CPAs sont les cellules dendritiques et les macrophages. Toutefois, les lymphocytes B, les monocytes et les neutrophiles ont également des fonctions de CPA2–
4. De nombreuses CPAs s’accumulent au niveau des ganglions lymphatiques et c’est
principalement à cet endroit que les lymphocytes T naïfs rencontreront leur antigène. La rencontre entre la CPA présentant l’antigène et le lymphocyte T active ce dernier. Cette activation le fait passer du stade naïf au stade effecteur. Le lymphocyte T effecteur quitte alors le ganglion lymphatique et migre vers le site inflammatoire, guidé par le gradient de chimiokines et de cytokines pro-inflammatoires5.
Le passage du stade naïf à effecteur nécessite deux signaux6,7. Le premier signal est celui qui provient du récepteur des cellules T (TCR) lorsque celui-ci rencontre son antigène présenté par une molécule de CMH dont la classe correspond au type de lymphocyte T. Le deuxième signal qui provient des molécules de costimulation dont les ligands sont exprimés par les CPAs. La costimulation est tout aussi importante que la reconnaissance de l’antigène par le TCR car sans costimulation, les lymphocytes T naïfs subissent le même sort que les lymphocytes T immatures qui ne parviennent pas à passer la sélection thymique. Autrement dit, un lymphocyte T naïf qui rencontre son antigène sans recevoir de signal de costimulation deviendra soit apoptotique soit anergique8, bien que pour les lymphocytes T naïfs, l’anergie est plus fréquente.
La molécule de costimulation principale exprimée par les lymphocytes T est le CD289. Ce dernier possède deux ligands, B7.1 et B7.2, aussi appelés CD80 et CD86 respectivement. On retrouve ces deux molécules sur la plupart des CPAs. Le CD28 n'est cependant pas la seule molécule de costimulation disponible pour le lymphocyte T. De nombreux récepteurs de la famille du TNF ainsi que les molécules d’adhésion peuvent jouer ce rôle8,10,11. La costimulation des lymphocytes T est décrite de façon plus approfondie dans une section ultérieure.
1.2 Récepteur des cellules T
Le TCR est un récepteur membranaire dont le ligand est un antigène associé à une molécule de CMH de type I ou II. La molécule de CMH nécessaire pour présenter l’antigène ne dépend pas du TCR, mais plutôt d’une autre molécule de surface, le CD4 ou le CD8, acquise lors de la maturation thymique12. Ces molécules ne font pas partie du TCR, elles sont très importantes pour l’activation par le TCR13,14. Le CD4 est une glycoprotéine membranaire monomérique contenant quatre domaines extracellulaires immunoglobuliniques. Le CD8 est un dimère αβ bien que des dimères αα puissent exister. Les sous-unités du CD8 sont aussi des glycoprotéines transmembranaires et possèdent un domaine immunoglobulinique. Ces deux molécules possèdent également un domaine intracellulaire dont plusieurs résidus peuvent être phosphorylés. Comme il a été mentionné plus haut, les lymphocytes T matures n’expriment que l’une ou l’autre de ces molécules, ce qui déterminera dans quel contexte le lymphocyte reconnaîtra l’antigène. Le rôle de ces molécules sera décrit plus loin.
Le TCR est un récepteur hétérodimérique composé de sous-unité αβ ou γδ. Le dimère αβ est le plus commun des deux. Les lymphocytes T qui expriment le TCR γδ sont des lymphocytes T qui n’ont pas réussi à exprimer un TCR αβ fonctionnel15. L’expression de γδ est un mécanisme de survie qui permet la génération de lymphocytes T différents, mais qui ont tout de même un rôle important dans la réponse immune16,17. Le TCR fait partie de la super famille des immunoglobulines et, de façon semblable à un anticorps, possède une région variable responsable de la spécificité du récepteur ainsi qu’une région constante qui
permet de l’ancrer à la membrane. Le TCR ne peut cependant activer les lymphocytes T à lui seul, car il ne possède qu’une courte région intracellulaire incapable d’activer les voies de signalisations. Pour ce faire, il s’associe à un complexe moléculaire de surface, le CD3. Le CD3 est composé de trois dimères, soit γε, δε et δδ. Il arrive parfois que le dimère δδ soit remplacé par un dimère δε. Les sous-unités γ, δ et ε font partie de la superfamille des immunoglobulines. Cependant, ces sous-unités ne lient pas l’antigène ni le CMH. Elles font partie des immunoglobulines, car structurellement, elles présentent un repliement que l’on dit immunoglobulinique. Ce repliement permet d’interagir et de se lier au TCR15. Ces trois sous-unités possèdent également un domaine intracellulaire. Les sous-unités δ et ε ne font pas partie de la famille des immunoglobulines. Elles possèdent en fait un très court domaine extracellulaire et un long domaine intracellulaire. Ces deux sous-unités sont les principales responsables de la signalisation induite par le TCR. Les régions intracellulaires des sous-unités du CD3 possèdent des motifs d’activation des immunorécepteurs via une tyrosine (ITAM). Les sous-unités γ, δ et ε en possèdent chacun un alors que les sous-unités δ et ε en possèdent trois. Ces motifs ITAMs sont les sites de phosphorylation qui permettront l’induction de la signalisation intracellulaire18. Leur rôle ainsi que la séquence d’événements qui mène à l’activation seront décrits plus loin. En plus des domaines immunoglobuliniques des sous-unités γ, δ et ε, le complexe TCR/CD3 est maintenu ensemble par une force électrostatique. La région transmembranaire du TCR possède une charge positive alors que la région transmembranaire du CD3 possède une charge négative. Puisque le TCR a absolument besoin du CD3, le terme TCR fait habituellement référence au complexe TCR/CD3. Ce sera le cas ici.
1.3 Différenciation des lymphocytes T auxiliaires
L’un des acteurs les plus importants dans l’immunité acquise et l’auto-immunité est le lymphocyte T CD4+ ou Th pour « T helper », car celui-ci peut diriger la réponse immunitaire en fonction de son phénotype.
On connaît plusieurs phénotypes de lymphocytes T auxiliaires différents. En ordre chronologique, les Th1 et Th2 ont été définis par Mosmann et Coffman il y a plus de
vingt-cinq ans19. Par la suite, se sont ajoutés les lymphocytes T régulateurs (Treg) qui étaient connus depuis longtemps sous le nom de lymphocytes T suppresseurs20,21. Plus récemment, les lymphocytes Th1722–24, les lymphocytes T folliculaires (Tfh)25,26 et les Th927,28 ont été ajoutés (Schéma 1). Toutefois, puisque notre travail porte sur les Th17, l'accent sera mis sur ce phénotype.
1.3.1 Th1
Les lymphocytes Th1 sont associés à l’immunité cellulaire. Les Th1 sont activés en réponse aux pathogènes intracellulaires tels que les bactéries et les virus. Ils sont également impliqués dans la réponse inflammatoire associée aux maladies auto-immunes29. Les cytokines qu’ils produisent, tel que l’IFN-γ et le TNF, activent les macrophages et les lymphocytes T cytotoxiques (CD8+) qui détruiront les cellules infectées. Ces mêmes cytokines vont induire un changement de classe d’immunoglobulines (Ig) produites par les lymphocytes B. Ces derniers produiront alors des IgG qui activeront le complément ou opsoniseront les cellules infectées et favoriseront leur phagocytose30. Le rôle de l’IFN-γ dans l’inflammation et l’auto-immunité sera discuté plus loin.
Les cytokines importantes pour la différenciation des lymphocytes Th1 sont l’IL-12 et l’IFN-γ. L’IL-12 est produite principalement par les cellules dendritiques et les macrophages. L’IL-12 active Stat4 qui à son tour active le facteur de transcription t-bet qui est typiquement associé au phénotype Th131. L’activation de t-bet induit l’expression de gènes spécifiques aux lymphocytes Th1 comme l’IFN-γ et les récepteurs de chimiokines CXCR3 et CCR532,33. L’IFN-γ, parmi ses nombreux effets, augmente la production d’IL-12 par les macrophages et les cellules dendritiques tout en augmentant l’activation de t-bet chez les lymphocytes Th1. Un autre rôle de t-bet est d’inhiber la synthèse des cytokines aux autres phénotypes de lymphocytes T auxiliaires, ce qui favorise la différenciation Th1 et contribue à établir la spécificité du phénotype34.
Schéma 1 : Différenciation des lymphocytes T auxiliaires
La présence de différentes cytokines [encadré] permet la différenciation en plusieurs phénotypes de lymphocytes T auxiliaires. Chacun de ces phénotypes possède un facteur de transcriptions qui lui est spécifique (entre parenthèses).
Inspiré de : Plasticity of CD4+ T cell lineage differentiation35 et Development, regulation and functional capacities of Th17 cells36
1.3.2 Th2
Les lymphocytes Th2 sont associés à l’immunité humorale. Ils sont activés en réponse aux pathogènes et aux parasites extracellulaires. Les cytokines produites par les lymphocytes Th2, principalement l’IL-4 et l’IL-5, vont activer la production d’anticorps neutralisant (IgM, IgE et IgG1) par les lymphocytes B30,37. Ces anticorps permettent l’agrégation des pathogènes qui seront ensuite éliminés par les macrophages. Les anticorps peuvent également activer le complément pour éliminer les pathogènes, notamment les parasites trop gros pour être phagocytés par les macrophages.
Les cytokines importantes pour la différenciation des Th2 sont l’IL-4, l’IL-5 et l’IL-10. L’IL-4 active Stat6 qui va activer GATA-3, le facteur de transcription typique des Th238. GATA-3 permet la transcription des gènes associés aux lymphocytes Th2 comme l’IL-4, l’IL5 et les récepteurs de chimiokines CCR3, CCR4 et CCR833,39,40. L’IL-4 produite par les Th2 agit de façon autocrine et favorise la différenciation du phénotype. De plus, GATA-3, de façon semblable à t-bet, inhibe la transcription de cytokines associées aux autres phénotypes41,42.
1.3.3 Th9
Le phénotype Th9 a été décrit récemment, mais le rôle de ces lymphocytes est encore mal défini. Il semblerait toutefois qu’ils aient un rôle à jouer dans la réaction allergique et la protection contre les nématodes27,28.
Les cytokines importantes pour la différenciation des lymphocytes Th9 sont l’IL-4 et le TGF-β. Le TGF-β modifie la signalisation de l’IL-4 et favorise la production d’IL-9. Pour l’instant, seule la production élevée d’IL-9 permet d’évaluer la différenciation Th9, car aucun autre récepteur spécifique n’a été identifié. De plus, aucun facteur de transcription spécifique aux lymphocytes Th9 n’a été identifié28.
1.3.4 Th17
Le phénotype Th17 a été décrit il y a environ 6 ans alors que la cytokine caractérisant ce phénotype, l’IL-17, est connue depuis plus de 15 ans. Bien que le phénotype n’ait été décrit que récemment, il est connu que les lymphocytes Th17 ont un rôle dans l’immunité, par
exemple lors de réponses contre différentes bactéries et champignons43. Cependant, la majorité des publications au sujet des Th17 concernent leur rôle dans le développement de différentes maladies inflammatoires auto-immunes. Ce rôle sera décrit plus en détail dans une section subséquente.
Les lymphocytes Th17 ont été premièrement identifiés chez la souris. Dans cette étude, l’IL-6 et le TGF-β étaient considérées nécessaires et suffisantes à la différenciation des Th1744. Par contre, chez l’humain, la différenciation des Th17 nécessitait la présence de TGF-β, d’IL-6, mais aussi d’Il-23 et d’IL-1. Également, d’autres études ont montré que la présence de TGF-β chez l’humain n’était pas requise et que son rôle serait uniquement d’inhiber la différenciation des Th1 et ainsi favoriser la différenciation des Th1745,46. De plus, certains lymphocytes Th17 humains peuvent exprimer le facteur de transcription t-bet et produire de l’IFN-γ. Ces clones, identifiés Th1/Th17, ont été associés avec les maladies inflammatoires telles que la maladie de Crohn, la colite ulcéreuse et l’arthrite rhumatoïde47,48.
De récentes études ont montré que le TGF-β n’est finalement pas nécessaire, alors que l’IL-23 l’est, pour la différenciation Th17 chez la souris ce qui permet de réconcilier les différences entre les Th17 humains et murins49–51. L’une de ces études a montré que les lymphocytes Th17 différenciés en présence d’IL-1, d’IL-6 et de TGF-β ne sont pas pathogéniques lors de transfert adoptif dans un modèle d’EAE alors que les Th17 différenciés en présence d’IL-1, d’IL-6 et d’IL-23 le sont50. Il semblerait que les lymphocytes Th17 les plus pathogéniques, responsables de l’autoimmunité, sont ceux qui co-expriment l’IL-17 et l’IFN-γ. La différence entre les fonctions des Th17 différenciés en présence de TGF-β ou d’IL-23 a également été étudié par un autre groupe qui a trouvé que la présence de TGF-β favorise des fonctions immunosuppressives52. Ce groupe a montré que seule la présence de TGF-β induit l’expression de CD39 et CD73, des ectonucléotidases qui transforment l’ATP et l’ADP en adénosine et il a été démontré que cette molécule à des propriétés immunosuppressives53. De plus, il semblerait que le TGF-β ait préséance sur l’IL-23 lors de la différenciation des Th17 puisque les cellules différenciées en présence d’IL-1, d’IL-6, d’IL-23 et de TGF-β expriment autant, sinon plus, de CD39 et de CD73 que les cellules différenciées en présence d’IL-1, d’IL-6 et de TGF-β.
D’autres suggèrent que le TGF-β favorise la différenciation des Th17 en inhibant la différenciation des Th1 et des Th246,54,55. Le rôle du TGF-β dans la différenciation Th17 est donc très complexe et n’est toujours pas complètement élucidé. Cependant, une étude récente a montré que certains isoformes de TGF-β peuvent avoir des effets spécifiques sur la différenciation des Th17. En effet, en présence d’IL-6, le TGF-β1 induit un phénotype de Th17 non pathogénique qui requiert la présence d’IL-23 pour le devenir alors que le TGF-β3 induit directement un phénotype pathogénique même en absence d’IL-2356.
Toutefois, il est actuellement accepté que l’IL-1, l’IL-6, l’IL-23 ainsi que le TGF-β sont requises pour obtenir des lymphocytes Th17 à partir de lymphocytes T CD4+ naïfs.
Toutes ces études montrent la complexité des mécanismes de différenciation des lymphocytes Th17 en comparaison avec celle des Th1 ou des Th2. Il apparait aussi que les différences de différenciation entre l’humain et la souris sont réconciliées, du moins en grande partie. En effet, pour obtenir des Th17 pathogéniques qui produisent aussi bien de l’IL-17 que de l’IFN-γ, les lymphocytes T naïfs nécessitent la présence d’IL-1, d’IL-6 et d’IL-23. La présence de TGF-β pourrait être requise uniquement pour limiter la différenciation des Th1 et favoriser l’émergence des lymphocytes mixtes Th1/Th17.
Les variations observées dans le choix de cytokines peuvent s’expliquer par le rôle de ces cytokines dans la différenciation (Tableau 1). Normalement, comme il le fait chez les iTregs, le TGF-β devrait induire l’expression du facteur de transcription Foxp3. Cependant, l’IL-6, en activant le Transducteur de Signal et Activateur de la Transcription 3 (STAT3), inhibe l’expression de Foxp3 chez les Th17. La combinaison du TGF-β et de l’IL-6 augmente l’expression et l’activation du facteur de transcription RORγt qui est associé aux Th17. De plus, l’IL-6 induit l’expression du récepteur de l’IL-23. L’IL-23, bien qu’elle ne soit pas nécessaire à la différenciation des Th17, active STAT3 et est nécessaire pour obtenir une activation complète de ces lymphocytes. L’IL-1, de son côté, augmente l’expression de RORγt induite par l’IL-6. Finalement, l’IL-21, qui est produite par les Th17, peut remplacer l’IL-6 lors de la différenciation en Th17. Ceci crée donc une boucle d’amplification positive qui peut compenser une perte d’IL-6 dans le milieu.
de RORγt et de STAT3, sans que toutes les cytokines ne soient présentes, ce qui pourrait expliquer les différentes conditions de différenciation des Th17 décrites plus haut.
Tableau 1 : Cytokines impliquées dans la différenciation Th17
Cytokines Rôles
Ref
IL-1 Augmente l’expression de RORγt en conjonction avec l’IL-6 57
IL-6
Inhibe l’activation de Foxp3 par le TGF-β
Augmente l’expression et l’activation de RORγt induite par le TGF-β Active STAT3
Induit l’expression du récepteur de l’IL-23
57,58
IL-21 Peut remplacer l’IL-6 dans la différenciation des Th17 57,59,60
IL-23 Active STAT3 Nécessaire à l’activation complète des Th17 Nécessaire à la survie/prolifération des Th17
57,58
TGF-β
Augmente l’expression du récepteur de l’IL-6
Augmente l’expression de RORγt, mais inhibe son activation Inhibe l’expression de t-bet et GATA-3
46,54,55,5 7
1.3.5 Treg
Les lymphocytes T régulateurs, que l’on nommait auparavant lymphocytes T suppresseurs, ont la capacité de bloquer l’activation des autres lymphocytes T et ainsi favoriser le développement de la tolérance immunitaire. On distingue deux grandes familles de lymphocytes Treg, soit les Tregs naturels (nTreg) et les Tregs induits (iTreg)61.
Les nTregs sont générés dans le thymus. Ce sont des lymphocytes CD4+CD25+ qui expriment fortement le facteur de transcription Foxp3 qui est spécifique aux Tregs62. On croyait à l’origine qu’ils n’étaient impliqués que dans le contrôle de la réponse contre les autoantigènes. On sait maintenant qu’ils sont impliqués dans la réponse ciblant les alloantigènes, les antigènes de tumeur et plusieurs agents infectieux63–67.
Pour activer les nTregs, le TCR n’est pas suffisant à lui seul, la présence d’IL-2 ou d’IL-4 est nécessaire68. Suite à leur activation, les nTregs inhibent la prolifération des lymphocytes auxiliaires et cytotoxiques69,70. Des expériences in vitro ont également montré que les nTregs peuvent inhiber les lymphocytes B71, les cellules NK72, les monocytes et les macrophages73.
Les nTregs inhibent la réaction immunitaire de deux façons. Premièrement, par contact direct avec les cellules. Les nTregs expriment CTLA-4, une molécule de costimulation négative, diminuant ainsi l’activation des cellules avec lesquelles les nTregs entrent en contact74. Les nTregs peuvent également produire de la perforine et du granzyme B et tuer les cellules avec lesquelles ils sont en contact75. Deuxièmement, les nTregs produisent des cytokines qui sont connues pour être anti-inflammatoire telles que l’IL-10, le TGF-β et l’IL-3572,76,77. Ces médiateurs solubles permettent aux nTregs d’atteindre un plus grand nombre de cellules et de créer un microenvironnement où l’activation du système immunitaire est défavorisée.
Il est important de noter que la majorité des études réalisées sur les nTregs ont été effectuées chez la souris, mais que plusieurs groupes ont trouvé des cellules semblables chez l’humain74,78. Un autre groupe suggère toutefois que l’étude des nTregs chez l’humain est plus compliquée, car il existe une importante population de lymphocytes T CD4+ qui exprime un niveau intermédiaire de CD25 et que parmi cette population, seule la fraction
représentant 2% des cellules exprimant le plus haut niveau de CD25 possède des propriétés régulatrices79. Une autre étude a également montré que l’expression de Foxp3 ne garantit pas un phénotype régulateur puisqu’il existe une population significative de lymphocytes T humains exprimant un niveau intermédiaire de CD25 qui exprime également Foxp3, mais ces cellules ne possèdent pas de fonctions régulatrices80.
Les iTregs sont plus près des autres lymphocytes T auxiliaires dans le sens où ils sont générés à partir de lymphocytes T naïfs activés par le TCR. Il existe différents sous-types de lymphocytes iTregs comme les Tr1 et les Th3. Les cytokines nécessaires à la différenciation des iTregs sont l’IL-2 et le TGF-β pour les Th3 ou l’IL-2 et l’IL-10 pour les Tr1. Contrairement aux nTregs, les iTregs n’expriment pas tous le facteur de transcription Foxp3. Par exemple, les Tr1 ne l’expriment pas alors que les Th3 l’expriment, bien que cette expression soit moins forte que chez les nTregs81,82. Les Th3 et les Tr1 utilisent également des moyens différents pour exercer leurs fonctions. Les Th3 utilisent les contacts cellules-cellules ainsi que la production de cytokines, soit le TGF-β82,83. Les Tr1 quant à eux ne répriment pas leurs cibles par contact, mais seulement par la production d’IL-1081.
On constate que les iTregs produisent la même cytokine que celle nécessaire à leur différenciation.
Les Tregs sont aussi associés avec l’auto-immunité où on a observé une diminution de leur expansion ou de leur fonction. Dans le cas de l’arthrite rhumatoïde, le TNF inhibe grandement les fonctions des Tregs en déphosphorylant Foxp384.
1.3.6 Plasticité des lymphocytes T auxiliaires
Suite à la découverte des phénotypes Th1 et Th2, le paradigme dominant était que les lymphocytes T auxiliaires se différenciaient de façon terminale, c’est-à-dire qu’un lymphocyte différencié en Th1 ne pouvait plus changer de phénotype. Ce paradigme découle d’expériences in vitro où les lymphocytes Th1 et Th2, suite à un certain nombre de divisions cellulaires, ne pouvaient plus changer de phénotype même en présence de cytokines associées à l’autre phénotype85. Cependant, depuis la découverte des Th17, mais particulièrement suite aux études qui ont montré que les Tregs, en présence d’IL-6, pouvaient se différencier en Th1786,87, la plasticité des lymphocytes T auxiliaires suscite
beaucoup d’intérêt. Le paradigme voulant que les lymphocytes T auxiliaires se différencient de façon terminale semble de moins en moins probable au fur et à mesure que les différentes études démontrent la capacité d’un certain phénotype à acquérir une ou plusieurs caractéristiques d’un autre phénotype. Par exemple, il a été démontré que les Th17 et les Tregs peuvent produire de l’IFN-γ88,89. De leur côté, les Th17 générés in vitro peuvent devenir Th1 ou Th2 en présence d’IL-12 ou d’IL-4 respectivement90,91. La plasticité n’est pas restreinte aux phénotypes les plus récents. Les Th2 peuvent produire de l’IFN-γ92 et les Th1 peuvent également devenir Th2 et produire de l’IL-4 lorsqu’ils sont placés dans les bonnes conditions91. Comme il a été mentionné plus haut, les Tregs peuvent se différencier en Th17 en présence d’IL-6. Toutefois, la conversion inverse, de Th17 à Tregs, n’a pas encore été observée.
Malgré le fait que tous les phénotypes puissent posséder un certain degré de plasticité, il semblerait que les Th17 soient ceux qui ont la plus grande capacité à changer de phénotype. Des études sur la méthylation des gènes des facteurs de transcription apporte un élément de réponse important à la question de la plasticité des lymphocytes T35. Ces études ont montré
que la triméthylation de l’histone H3 sur la lysine 4 (H3K4me3) favorise la transcription des gènes alors que la triméthylation de l’histone H3 sur la lysine 27 (H3K27me3) diminue la transcription des gènes. Par exemple, le facteur de transcription associé au phénotype, par exemple t-bet chez les Th1, est méthylé de façon à grandement favoriser son expression alors que les trois autres facteurs de transcription, ici GATA-3, Foxp3 et RORγt, sont méthylés de façon à grandement défavoriser leur expression. Chez les Th17, RORγt est également méthylé afin de favoriser son expression. Cependant, la différence réside au niveau des autres facteurs de transcription. Ceux-ci sont soit non méthylés soit méthylés de façon à rendre leur expression légèrement favorable (Schéma 2). Les Th17 pourraient donc être plus susceptibles à un changement de cytokines dans leur environnement où une dose relativement faible d’une cytokine associée à la différenciation d’un autre phénotype pourrait être suffisante pour activer le facteur de transcription en question et ainsi induire un changement de phénotype chez ces Th17. Par exemple, la présence d’IL-12 dans l’environnement des Th17 induira l’expression de t-bet, en plus de RORγt, chez ces cellules et il en résultera des cellules que l’on nomme Th1/Th17, cellules qui produisent de l’IL-17 et de l’IFN-γ et qui représenteraient les lymphocytes Th17 pathogéniques dont nous avons
discuté précédemment47.
Le rôle et l’importance de la plasticité des lymphocytes T CD4+ ne sont toutefois pas encore très bien compris. Une étude a montré que l’activation de cellules dendritiques par le récepteur dectin-1, un récepteur membranaire impliqué dans la reconnaissance des champignons, permet la conversion des lymphocytes Tregs en Th17 lorsque les cellules dendritiques et les Tregs sont mis en coculture93. Puisque les Th17 sont impliqués dans la réponse immunitaire contre les champignons, la plasticité des lymphocytes T pourrait être un mécanisme de protection permettant de diminuer le nombre de lymphocytes T produits tout en maximisant l’efficacité de la réponse.
Mieux comprendre la plasticité des lymphocytes T sera également important pour le traitement des maladies auto-immunes, car l’une des approches suggérées est l’utilisation de lymphocytes Tregs. Vu leur plasticité, il faudra s’assurer que le contexte dans lequel ils seront utilisés n’est pas propice à la conversion en un phénotype inflammatoire et ainsi aggraver la maladie. De plus, puisqu’aucun cas de plasticité de phénotype Th1, Th2 ou Th17 vers Treg n’a été rapporté, il ne semble pas prometteur d’utiliser la plasticité des lymphocytes T pour diminuer l’inflammation en transformant les lymphocytes T inflammatoires en lymphocytes T régulateurs.
Schéma 2 : Méthylation des gènes de facteurs de transcription selon le phénotype
La triméthylation de l’histone H3 sur la lysine 4 (H3K4me3, en vert) favorise la transcription des gènes alors que la triméthylation de l’histone H3 sur la lysine 27 (H3K27me3, en rouge) diminue la transcription des gènes.
1.4 Cytokines proinflammatoires
Les lymphocytes Th17 produisent différentes cytokines proinflammatoire telles que l’IL-17, l’IL-21, l’IL-22 et l’IFN-γ. Ici, l’emphase sera mise sur l’IL-17 et l’IFN-γ puisque ces cytokines sont particulièrement importantes dans le développement de différentes maladies auto-immunes.
1.4.1 IL-17
L’IL-17 est la cytokine signature des lymphocytes Th17. Il s’agit en fait d’une famille de cytokines dont on connait six membres soit l’IL-17A, B, C, D, E et F94,95. L’IL-17A est la cytokine prototypique de la famille IL-17 et on la nomme souvent simplement IL-17. Les autres membres de la famille ont un degré plus ou moins élevé de similitude avec l’IL-17A. C’est l’IL-17F qui a le plus haut degré de ressemblance et c’est également la cytokine dont les effets se rapprochent le plus de l’IL-17A. Fait intéressant, l’IL-17E est l’isoforme qui possède le moins d’homologie avec l’IL-17A et avait été préalablement découverte sous le nom d’IL-2596.
L’IL-17A et F ne sont pas produites uniquement par les lymphocytes Th17, mais ces derniers restent les producteurs les plus importants, particulièrement pour ce qui est de l’IL-17A. De leur côté, les autres membres de la famille IL-17 sont produites par plusieurs types cellulaires et ne sont pas restreintes aux cellules du système immunitaire95.
L’IL-17 est un dimère d’environ 30-35 kDa dont chaque monomère fait 150 acides aminés pour environ 15 kDa. On retrouve des homodimères d’IL-17A, d’IL-17F ainsi que l’hétérodimère IL-17A/F97. Dans la suite du texte, l’IL-17 fera référence à l’homodimère d’IL-17A, sauf mention contraire.
La production d’IL-17 par les Th17 nécessite, comme pour toutes autres cytokines, l’activation de voies de signalisation intracellulaire. Dans le cas de l’IL-17, ces voies de signalisation sont de plus en plus caractérisées. Il a été démontré que le facteur de transcription spécifique aux Th17, RORγt est important pour la transcription de l’IL-1798. Le promoteur du gène de l’IL-17 possède d’ailleurs plusieurs sites de liaison pour RORγt99. Cependant, l’inhibition de RORγt ne bloque pas complètement l’expression de l’IL-17. Une étude qui a utilisé des souris knock-out pour RORγt ainsi que pour RORα a montré que ce
dernier est également important pour la production d’IL-17, bien qu’à un niveau moindre que RORγt, et que le double knock-out de ces deux gènes inhibe complètement l’expression de l’IL-17100. Une autre étude a montré que RORγ, dont RORγt est un isoforme, est requis pour la différenciation Th17. Bien que RORγ soit exprimé par d’autres types cellulaires (muscles, reins) il n’est exprimé dans aucun autre phénotype de lymphocytes T que Th17101. L’activation du TCR est nécessaire pour l’expression de RORγt, mais les voies de signalisation impliquées sont présentement en cours d’études. L’une de ces voies est la voie de STAT3, un autre facteur de transcription impliqué dans la transcription de l’IL-17. Les activateurs principaux de STAT3 dans les Th17 sont l’IL-6, l’IL-21 et l’IL-23. STAT3 intervient directement sur l’expression de l’IL-17102 en se liant à son promoteur103 et indirectement en augmentant l’expression de RORγt et de RORα104. De plus, il a été montré dans un modèle murin que la délétion de STAT3 inhibe la différenciation des lymphocytes T auxiliaires en lymphocytes Th17105. L’absence de STAT3 inhibe également le développement de l’encéphalomyélite aigue expérimentale (EAE), le modèle murin de la sclérose en plaques106,107. Cependant, il semblerait que
STAT3 ait besoin de RORγt pour induire l’expression de l’IL-17, car la transfection d’une forme active de STAT3 dans des cellules de souris knock-out pour RORγt n’induit qu’un niveau très faible d’IL-17108. L’activation de STAT3 a également été associée avec l’expression de l’IL-17 chez l’humain109.
Une autre étude a montré que les voies du facteur de transcription des lymphocytes T activés (NF-AT) et des MAP kinase ERK et JNK peuvent être importantes pour la production d’IL-17. Dans cette étude, les auteurs ont couplé une séquence retrouvée en amont du gène de l’IL-17 humaine à la luciférase qu’ils ont ensuite transfectée dans des cellules Jurkat. À l’aide d’inhibiteurs chimiques, ils ont montré que les voies de ERK, JNK et particulièrement NF-AT sont nécessaires à la transcription de la luciférase et donc de l’IL-17110. Toutefois, le rôle de la voie de ERK est encore controversé. Une étude différente a trouvé que les voies de NF-κB, JNK et p38, mais pas ERK, étaient nécessaires pour l’expression de l’IL-17 chez la souris111. Cependant, il s’agit là d’une activation par l’ostéopontine et non par le TCR. De plus, deux études ont montré un rôle négatif pour ERK chez la souris. La première a montré que l’inhibition de ERK favorise l’expression d’IL-17112 et la deuxième indique que la voie de ERK inhibe l’expression de l’IL-17 en
inhibant l’activation de STAT3 et de RORγt113. Il semblerait donc qu’il y ait une différence entre les voies de signalisation impliquées dans l’expression de l’IL-17 humaine et l’IL-17 murine, particulièrement en ce qui a trait au rôle de la voie ERK. L’importance de p38 dans la production de l’IL-17 a été montrée à plusieurs reprises. Chez la souris, l’utilisation d’un inhibiteur chimique de p38 a diminué grandement la production d’IL-17 chez les lymphocytes Th17114. Cette étude a montré que p38 active le facteur de traduction elF-4E et qu’ainsi, cette MAP Kinase ne serait pas impliquée dans l’expression du gène de l’IL-17, mais favoriserait la traduction de son ARNm en protéine. Le facteur de transcription Batf activé par p38 est un autre facteur important pour la différenciation des Th17 et la production d’IL-17. Chez les souris knock-out pour Batf, les lymphocytes Th1 et Th2 se développent normalement alors que la différenciation en Th17 est grandement diminuée115. Chez ces souris, l’expression de RORα et RORγt ainsi que la production d’IL-17 sont presque totalement inhibées comparativement aux souris normales. Le facteur de transcription c-Maf, lui aussi activé par p38, a été impliqué dans la production d’IL-17, car il favorise l’expression du récepteur de l’IL-23 nécessaire à la pleine activation des lymphocytes Th17116.
Il existe d’autres molécules qui ont été impliquées dans la signalisation menant à l’expression de l’IL-17. On retrouve par exemple la voie de PI3-K/AKT. Celle-ci est nécessaire à la production d’IL-17 suite à l’activation par TCR/CD28 chez la souris117 et en réponse à l’IL-2, l’IL-7 et l’IL-15 chez l’humain118. Cependant, une autre étude a montré que l’activation d’AKT inhibe la différenciation Th17 et la production d’IL-17 chez la souris119. Le rôle de la voie PI3-K/AKT dans la production d’IL-17 reste donc à confirmer. D’autres molécules sont impliquées dans l’expression de l’IL-17, comme la kinase inductible des lymphocytes T (ITK), le récepteur pour l’aryl hydrocarbone (Ahr) ainsi que les facteurs de transcription de la famille de NF-κB, c-Rel et RelA. Les souris knock-out pour ITK produisent moins d’IL-17120. Une étude utilisant un knock-out d’Ahr a montré que la signalisation induite par ce récepteur inhibait la différenciation en Th17 et favorisait la différenciation en Treg121. Cependant, le rôle et le mécanisme de ces molécules restent encore à confirmer. De leur côté, c-Rel et RelA sont impliqués dans l’expression de RORγ et RORγt. En effet, les lymphocytes T de souris knock-out pour l’un ou l’autre des facteurs de transcription produisent beaucoup moins d’IL-17 et cette perte a été associée à une
diminution de l’expression de RORγ et RORγt101.
L’IL-17 est une cytokine pro-inflammatoire aux effets multiples. Ces effets sont médiés par un récepteur membranaire, l’IL-17R. Ce dernier est un dimère dont cinq membres, allant de A à E, ont été identifiés. L’IL-17RA est le récepteur principal pour 17A alors que l’IL-17RC est le récepteur principal pour l’IL-17F bien que l’IL-17A et l’IL-17F puissent respectivement lier l’IL-17RC et l’IL-17RA. Cependant, le récepteur le plus efficace semble être l’hétérodimère IL-17RA/RC qui peut donc très bien lier l’IL-17A, l’IL-17F et l’IL-17A/F122. La distribution des deux isoformes n’est toutefois pas uniforme. Les cellules du système immunitaire expriment généralement une plus grande proportion d’IL-17RA alors que les autres cellules expriment une plus grande proportion d’IL-17RC123. Pour la suite du texte, le récepteur de l’17 ou 17R fera référence à l’hétérodimère IL-17RA/RC. La liaison de l’IL-17 avec son récepteur induit une signalisation intracellulaire qui inclut l’activation du facteur de transcription NF-κB, la voie des MAP kinases ERK, JNK et p38 ainsi que la voie de PI3-K/AKT124.
L’effet principal de l’IL-17 est d’induire la production d’autres cytokines, ainsi que des chimiokines et autres molécules. Puisque pratiquement toutes les cellules expriment l’IL-17R, les molécules produites en réponse à l’IL-17 sont très variées. Cependant, ces molécules ont en commun le fait qu’elles sont impliquées de près ou de loin dans le développement de l’inflammation et l’activation du système immunitaire. Par exemple, chez les fibroblastes, l’IL-17 induit la production d’IL-6, d’IL-8 et de prostaglandine E2125. L’IL-17 est toutefois mieux connue pour son rôle dans le développement des maladies inflammatoires auto-immunes. Les études à ce sujet sont extrêmement nombreuses. Je me restreindrai au rôle des Th17 et de l’IL-17 dans trois maladies soit l’arthrite rhumatoïde, la sclérose en plaques et la malade de Crohn.
1.4.2 IFN-γ
Les interférons (IFN) forment une famille de cytokine dont on connaît trois membres. L’IFN-α et l’IFN-β, des interférons de type I, sont impliqués dans la réponse antivirale. Les mécanismes par lesquels agissent les interférons de type I sont variés, mais impliquent la diminution de la réplication d’ARN viraux126. L’IFN-γ est le seul interféron de type II. Tout
comme les interférons de type I, l’IFN-γ peut protéger les cellules en cas d’infection virale. Cependant, son rôle principal, qui la distingue des interférons de type I, est l’activation de la réponse immunitaire cellulaire et le développement de l’inflammation127,128.
L’IFN-γ est produit par plusieurs types cellulaires dont les cellules NK, les lymphocytes T cytotoxiques, les lymphocytes Th1 et également les lymphocytes Th17. Chez les lymphocytes T, la stimulation par le TCR induit la production d’IFN-γ. Cette production est augmentée chez les lymphocytes Th1 en présence d’IL-12 et d’IL-18. Les voies de signalisation impliquées dans la production de l’IFN-γ incluent les MAP kinases ERK, JNK et p38 de même que la voie PI3- kinase/AKT129–132.
L’IFN-γ est une cytokine de 166 résidus que l’on retrouve sous forme de dimère de 34kDa. Comme mentionné plus haut, l’IFN-γ a de nombreux effets sur le système immunitaire. Entre autres, elle active les cellules présentatrices d’antigènes chez qui elle augmente l’expression des molécules de CMH I et II et des molécules de costimulation ainsi que des protéines nécessaires pour le traitement des antigènes128. L’IFN-γ augmente également la production de cytokines proinflammatoires, dont l’IL-1 et le TNF, et de chimiokines chez les macrophages. De plus, cette cytokine augmente les fonctions lytiques des macrophages en activant la phagocyte oxydase et l’oxyde nitrique synthase qui produisent les intermédiaires réactifs de l’oxygène et de l’azote133. Dans les lymphocytes B, l'IFN-γ stimule le changement de classe des anticorps, favorisant les formes reconnues par les phagocytes et activant le système du complément128. Ainsi, l’IFN-γ augmente la présentation antigénique ce qui permet d’activer un plus grand nombre de lymphocytes T. Vu son importance dans l’inflammation, un dérèglement ou une perte de contrôle de la production d’IFN-γ peut mener au développement de maladies inflammatoires auto-immunes134. Cependant, depuis la découverte de l’IL-17 et de son implication dans l’inflammation et les maladies auto-immunes, de nombreuses maladies que l’on associait préalablement aux Th1 et à l’IFN-γ se sont vues converties en maladie de type Th17. À cela se sont ajoutées des études qui ont montrés que l’IFN-γ pouvait inhiber le développement de certaines maladies auto-immunes135,136. Il n’en reste pas moins que l’IFN-γ est une puissante cytokine pro-inflammatoire. D’ailleurs, après la vague Th17 qui a déferlée sur le monde de l’immunologie, on commence à réaliser que la combinaison IL-17 / IFN-γ est
probablement responsable du développement des maladies inflammatoires auto-immunes et non pas l’une ou l’autre des cytokines individuellement. L’IFN-γ a été associé aux maladies inflammatoires auto-immunes de différentes façons. Premièrement, il peut réactiver les lymphocytes T autoréactifs qui ont préalablement été anergisés137. Deuxièmement, puisque l’IFN-γ active les CPAs et les macrophages, une dérégulation du contrôle de la production d’IFN-γ qui mènerait à une production constante de la cytokine favorisera la perpétuation de la présentation antigénique et par le fait même de l’activation de la réponse immunitaire acquise lorsque l’antigène ne peut être éliminé, tel un antigène du soi. De plus, les macrophages activés par l’IFN-γ produisent de l’IL-1, l’IL-6 et l’IL-23, ce qui peut contribuer au développement de maladies auto-immunes en favorisant la différenciation Th17133. Troisièmement, bien que la réponse immunitaire soit très importante pour la survie de l’organisme, l’inflammation qui y est associée induit un stress qui peut être néfaste pour l’organe inflammé. C’est pourquoi, en temps normal, la réponse immunitaire est étroitement régulée. Lorsque la production d’IFN-γ devient chronique, il en résulte une production constante, par les macrophages, de molécules réactives dérivées d'oxygène et d'azote ainsi que d’enzymes hydrolytiques qui finiront par détruire le tissu ou l’organe inflammé138,139. Des macrophages activés en trop grand nombre ou pendant trop longtemps, comme dans le cas d'inflammation chronique, peuvent causer des dommages tissulaires majeurs.
Bien que l'IFN-γ soit un médiateur de l'inflammation, des études ont démontré qu'il était nécessaire pour mettre fin à la réponse inflammatoire. L'IFN-γ peut sensibiliser les lymphocytes T à l'apoptose en augmentant l'expression des procaspases et ainsi diminuer le nombre de lymphocytes T et réduire l'inflammation137,140. Les lymphocytes T naïfs ou fraîchement activés ne sont pas sensibles à cet effet de l’IFN-γ car ils n’expriment pas la chaîne β du récepteur qui est responsable de l’effet apoptotique de l’IFN-γ chez les lymphocytes141. Les lymphocytes T n’expriment la chaîne β du récepteur de l’IFN-γ qu’après avoir été activés plusieurs fois, ce qui est le cas lors de maladies auto-immunes dépendantes des lymphocytes T.
Ces effets pro- et anti-inflammatoires contribuent à la controverse qui existe présentement quant au rôle de l'IFN-γ dans les maladies auto-immunes, c’est-à-dire que la présence