HAL Id: hal-02135208
https://hal.archives-ouvertes.fr/hal-02135208
Submitted on 21 May 2019HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires
Genetic screening of male patients with primary
hypogammaglobulinemia can guide diagnosis and clinical
management
Nicolas Vince, Gael Mouillot, Marion Malphettes, Sophie Limou, David
Boutboul, Angélique Guignet, Philippe Pellet, Pierre-Antoine Gourraud,
Patrice Debré, Eric Oksenhendler, et al.
To cite this version:
Nicolas Vince, Gael Mouillot, Marion Malphettes, Sophie Limou, David Boutboul, et al.. Genetic screening of male patients with primary hypogammaglobulinemia can guide diagnosis and clinical man-agement. Human Immunology, Elsevier, 2018, 79 (7), pp.571-577. �10.1016/j.humimm.2018.04.014�. �hal-02135208�
Title page
1
Genetic screening of male patients with primary hypogammaglobulinemia can guide
2
diagnosis and clinical management
3
Nicolas Vince*1,2 ,3, Gaël Mouillot*4, Marion Malphettes1,5, Sophie Limou2,3 ,6,
4
David Boutboul1, Angélique Guignet1, Véronique Bertrand4, Philippe Pellet4,
5
Pierre-Antoine Gourraud2,3, Patrice Debré4, Eric Oksenhendler5, Ioannis
6
Théodorou4, Claire Fieschi1 ,5; and the DEFI Study Group.
7 8
Abbreviated title: 9
Genetic screening of hypogammaglobulinemia
10 11 Corresponding author: 12 Nicolas Vince, PhD 13
CRTI UMR1064 - ITUN 14
CHU Nantes Hôtel Dieu 15 30 bld Jean Monnet 16 44093 Nantes Cedex 01 17 France 18 nicolas.vince@univ-nantes.fr 19 Phone: +33 2 44 76 82 71 20 21
* These authors contributed equally to this work 22
23
1. EA3963, Université Paris 7 Denis Diderot, centre Hayem, Hôpital Saint-Louis, 1 avenue 24
Claude Vellefaux 75010 PARIS France 25
2. Centre de Recherche en Transplantation et Immunologie UMR 1064, INSERM, Université 26
de Nantes, Nantes, France 27
3. Institut de Transplantation Urologie Néphrologie (ITUN), CHU Nantes, Nantes, France 28
4. Laboratoire Central d’Immunologie Cellulaire et Tissulaire, Hôpital Pitié Salpêtrière et 29
INSERM UMR-S945, Bâtiment CERVI, Paris, France 30
5. Département d'Immunologie Clinique, Hôpital Saint-Louis, AP-HP, 1 avenue Claude 31
Vellefaux 75010 PARIS France 32
6. Ecole Centrale de Nantes, Nantes, France 33
34
Word counts: abstract 199 words, text 2876 words. 35
4 tables, 3 figures, 1 supplemental figure 36
53 references 37
Abstract
1
The precise diagnosis of an immunodeficiency is sometimes difficult to assess, especially due 2
to the large spectrum of phenotypic variation reported among patients. Common variable 3
immunodeficiency disorders (CVID) do not have, for a large part, an identified genetic cause. 4
The identification of a causal genetic mutation is important to confirm, or in some cases correct, 5
the diagnosis. We screened >150 male patients with hypogammaglobulinemia for mutations in 6
three genes involved in pediatric X-linked primary immunoglobulin deficiency: CD40LG, 7
SH2D1A and BTK. The SH2D1A screening allowed to reclassify two individuals with an initial
8
CVID presentation as XLP after mutations identification. All these mutations were associated 9
with a lack of protein expression. In addition, 4 patients with a primary diagnosis of CVID and 10
one with a primary IgG subclass deficiency were requalified as XLA after identifying BTK 11
mutations. Interestingly, two out of these 5 patients carried a damaging coding BTK mutation 12
associated with a lower, but detectable, BTK expression in monocytes, suggesting that a 13
dysfunctional protein explains the disease phenotype in these patients. In conclusion, our results 14
advocate to include SH2D1A and BTK in newly developed targeted NGS genetic testing, to 15
contribute to providing the most appropriate medical treatment and genetic counselling. 16
Keywords
17
Immunodeficiency, BTK, SAP, CD40L, CVID 18
Abbreviations
1
CVID: Common variable immunodeficiency disorders 2
EBV: Epstein-Barr virus 3
HC: healthy controls 4
HIGM: X-linked hyperIgM syndrome 5
Ig: immunoglobulin 6
IgAD: IgA deficiency 7
MFI: mean fluorescent intensity 8
NGS: next generation sequencing 9
PE: Phycoerythrin 10
SAP: SLAM-associated protein 11
XLA: X-linked agammaglobulinemia 12
XLP: X-linked lymphoproliferative disease 13
Introduction
1
Common variable immunodeficiency disorders (CVID) are characterized by a defect in 2
immunoglobulin (Ig) production. Most patients present recurrent infections of the respiratory 3
and gastrointestinal tracts and, less frequently, lymphoid proliferation or autoimmune diseases 4
[1]. The prevalence of CVID has been estimated at 1/30,000 in the European population, and 5
this disorder is the second most frequent immunodeficiency of adulthood, after IgA deficiency 6
(IgAD). About 20% of patients have at least one relative affected by IgAD or CVID, suggesting 7
a genetic origin in such cases. Genetic abnormalities in 24 genes involving different immune 8
pathways have been demonstrated in less than 10% of CVID patients to date [2]. Some genetic 9
variants, such as the ones in TNFRSF13B [3], do not seem sufficient to cause CVID phenotype 10
while others, such as the ones in CD19 [2,4], present a full penetrance toward Ig deficiency. 11
The clinical presentation of patients with CVID is wide an made by exclusion of other 12
possible causes [5], which can sometimes lead to a diagnosis reclassification after identification 13
of genetic mutations or occurrence of new biological evidence or symptoms. Few CVID 14
patients were reclassified to XLA (X-linked agammaglobulinemia) when a mutation in the BTK 15
gene was found (OMIM: *300300) [6–16]. XLA is characterized by a failure during B-cell 16
development [17,18], and is usually diagnosed before the age of 18 months, after the loss of 17
maternal antibody protection [19], most of patients present an absence of B-cells and 18
immunoglobulin. Similarly, occasional reports of mutations in SH2D1A were showed in CVID 19
patients [20–24]. Mutations in SH2D1A, encoding SAP (SLAM-associated protein; OMIM: 20
*300490), are found in patients with XLP (X-linked lymphoproliferative disease) [25,26], 21
which is characterized by an extreme susceptibility to Epstein-Barr virus (EBV) infections, 22
resulting in near-fatal infectious mononucleosis, acquired hypogammaglobulinemia, malignant 23
lymphoma or a combination of these conditions [27,28]. 24
In order to identify disease-causing mutations and their potential to support a diagnosis, 1
we explored a large cohort of French hypogammaglobulinemic patients (see figure S1) showing 2
a wide range of primary diagnosis (Table 1). We therefore systematically screened this 3
population for mutations in the SH2D1A and BTK genes, as well as for mutations in CD40LG 4
(OMIM: *300386), a gene responsible for the hyperIgM syndrome (HIGM1) that presents 5
numerous similarities with CVID [29,30]. 6
Materials and methods
1
Patients 2
Investigated patients came from the DEFI cohort, a French cohort of patients with 3
primary hypogammaglobulinemia [1,5]. All patients gave informed consent for DNA 4
sequencing, and the study was approved by the local institutional review board, in accordance 5
with the Helsinki declaration. Initial diagnoses are reported in table 1. 6
Genes 7
The CD40LG gene, located in Xq26.3, encodes for the CD40 ligand. After PCR 8
amplification of the five exons and their flanking intronic regions, the PCR products were 9
directly sequenced in 150 male patients. We identified 3 SNPs (rs11575982, rs3092923 and 10
rs148594123) with a frequency equivalent to the matching 1000 Genomes population 11
(http://www.internationalgenome.org/1000-genomes-browsers/) [31,32]. 12
The SH2D1A gene is located in Xq25 and encodes for SAP. After PCR amplification of 13
the four exons and their flanking intronic regions, we directly sequenced the PCR products in 14
a total of 230 male subjects. We identified 2 SNPs (rs200198093 and rs72610640) with a 15
frequency equivalent to the matching 1000 Genomes population. 16
The BTK gene is located in Xq22.1. After PCR amplification of the 19 exons and their 17
flanking intronic regions, we directly sequenced the PCR products in a total of 181 male 18
patients from the DEFI cohort. 19
Molecular analysis 20
Genomic DNA was extracted from buffy coats. All coding exons and intronic flanking 21
regions of CD40LG, SH2D1A and BTK were amplified (primers available upon request) and 22
sequenced with the Big Dye Terminator kit (Applied Biosystems) on an ABI3730 DNA 23
Total RNA was extracted from PHA T-cell blast and EBV-transformed B-cells with 1
TRIzol (Invitrogen). The mRNA was reverse-transcribed with oligo-dT primers and 2
SuperScriptIII reverse transcriptase (Invitrogen), to obtain cDNA. 3
Cloning of the amplified cDNA 4
The PCR products obtained after RNA extraction and cDNA amplification were 5
inserted into the TOPOÒ TA cloning vector (Invitrogen) and sequenced. 6
Flow cytometry analysis of BTK expression 7
Intracellular BTK staining was performed on thawed PBMC with the Fix and Perm® 8
kit (Caltag), used according to the manufacturer’s instructions. PBMC were first stained with 9
FITC-conjugated anti-CD14 (IgG2a; Immunotech), PerCP-Cyanine5.5-conjugated anti–CD45 10
(IgG1; BD Biosciences) and APC-conjugated anti-CD19 (IgG1; BD Biosciences) for 15 11
minutes at room temperature. The cells were fixed, washed and permeabilized and Fc receptors 12
were blocked by incubation with human serum for 15 minutes at room temperature. PBMC 13
were stained by incubation with Phycoerythrin (PE)-labeled anti-BTK antibody (IgG2a; BD 14
Biosciences) or with an irrelevant Ab (CD10, IgG2a; BD Biosciences) for 15 minutes at room 15
temperature. The stained cells were analyzed with a BD FACS CantoTM flow cytometer (BD
16
Biosciences). The results are expressed by the difference (D) of monocytes (CD45+CD14+) 17
mean fluorescence intensity (MFI) between anti-BTK and irrelevant Ab. Fifteen male healthy 18
controls (HC) presented a median DMFI of 107 [79-173]. 19
Flow cytometry analysis of CD40L expression 20
PHA T-cell blasts were stained by incubation with PerCP-conjugated anti-CD3 (SK7, 21
BD Biosciences) and PE-conjugated anti-CD40L (555700, BD Biosciences) antibodies for 30 22
minutes at 4°C. The stained cells were analyzed in a FACSCalibur flow cytometer (BD 1
Biosciences). 2
SAP Western blotting 3
Lysates of 5.106 PHA T-cell blasts were separated by SDS-PAGE and immunoblotted
4
with a polyclonal rabbit anti-SAP antibody (2778, Cell Signaling Technology) and a polyclonal 5
goat anti-actin antibody (1616, Santa Cruz). 6
Results
1
CD40LG mutation screening
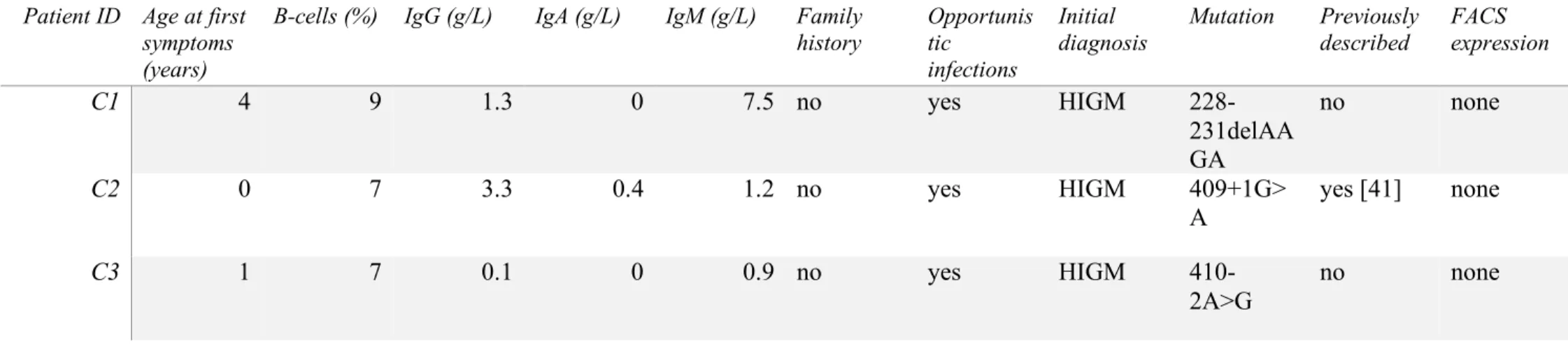
2
Four patients with a primary HIGM diagnosis were included in our cohort for genetic 3
screening. Of those, only 3 presented a mutation in CD40LG: 228_231delAAGA, 409+1G>A 4
and 410-2A>G (Figure 1A). These mutations were associated with an absence of CD40L cell 5
surface detection on activated T-cell (see table 2). The fourth patient with an HIGM diagnosis 6
may carry a mutation in another HIGM-causing gene (e.g. AICDA, CD40) [33,34] and as such 7
should be screened for it. We did not identify any CD40LG mutation in any other patient of the 8
cohort. 9
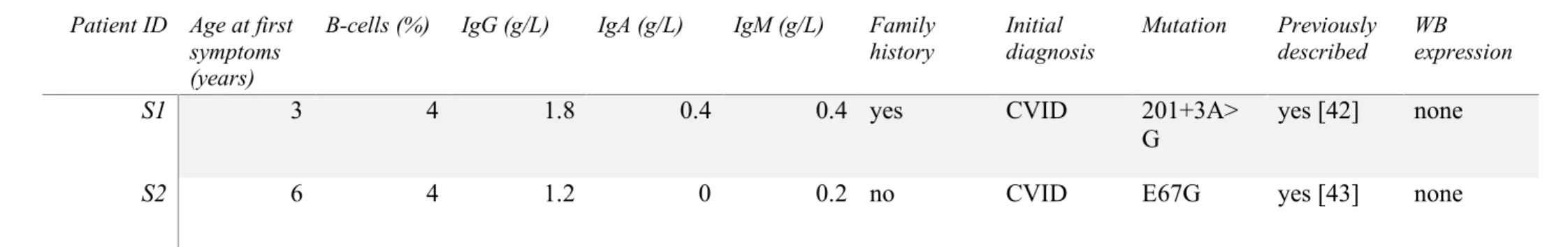
SH2D1A mutation screening
10
No patient included in our cohort were initially diagnosed with XLP. After identifying 11
a first patient initially diagnosed with CVID but carrying a SH2D1A mutation (patient S1) in 12
the first 150 patients, we expanded our SH2D1A screening to 80 additional individuals and 13
identified a second CVID patient with a SH2D1A mutation (patient S2, table 3 and figure 1B). 14
Patient S1 was mutated in intron 2: 201+3A>G. We identified and sequenced two 15
splicing mRNA variants in activated T-cells by RT-PCR: a first variant where exon 2 were 16
spliced out (cDNA fragment of 323bp) and a second variant where exon 2 and a portion of exon 17
3 were spliced out (cDNA fragment of 268bp). We did not detect SAP protein expression in 18
activated T-cells protein extracts using immunoblot technique (Figure 2). Patient S2 carried a 19
E67G coding mutation in exon 2 that was associated with an absence of SAP protein on the 20
immunoblot performed with activated T-cells protein extract (Figure 2). 21
The clinical presentation of of these 2 patients was as followed. Patient S1 first diagnosis was 22
pediatric CVID at eight years of age after detection of hypogammaglobulinemia during 23
bacterial pneumonia. He also suffered from severe infectious mononucleosis at six years of age. 24
The patient was treated with gammaglobulins, administered intramuscularly for the first 10 1
years, and then intravenously. Patient S1 had 4% circulating B-cells including 87% naive 2
(CD27-IgM+), 3% switched memory (CD27+IgM-) and 7% CD27+IgM+ B-cells (Table 3). 3
He presented a family history suggestive of X-linked disease, indeed, his older brother died 4
with a refractory digestive EBV-driven B large cell lymphoma in the course of an otherwise 5
unremarkable CVID, without previous severe EBV primo-infection. Patient S2 was diagnosed 6
with CVID at the age of 40 based on the detection of profound hypogammaglobulinemia after 7
severe bacterial pneumonia without familial history of X-linked disease. He denied suffering 8
severe EBV primary infection, despite chronic positive plasma circulating EBV viral load. The 9
patient had 4% circulating B-cells including 19% naive (CD27-IgM+), 57% switched memory 10
(CD27+IgM-) and 15% CD27+IgM+ B-cells (Table 3). 11
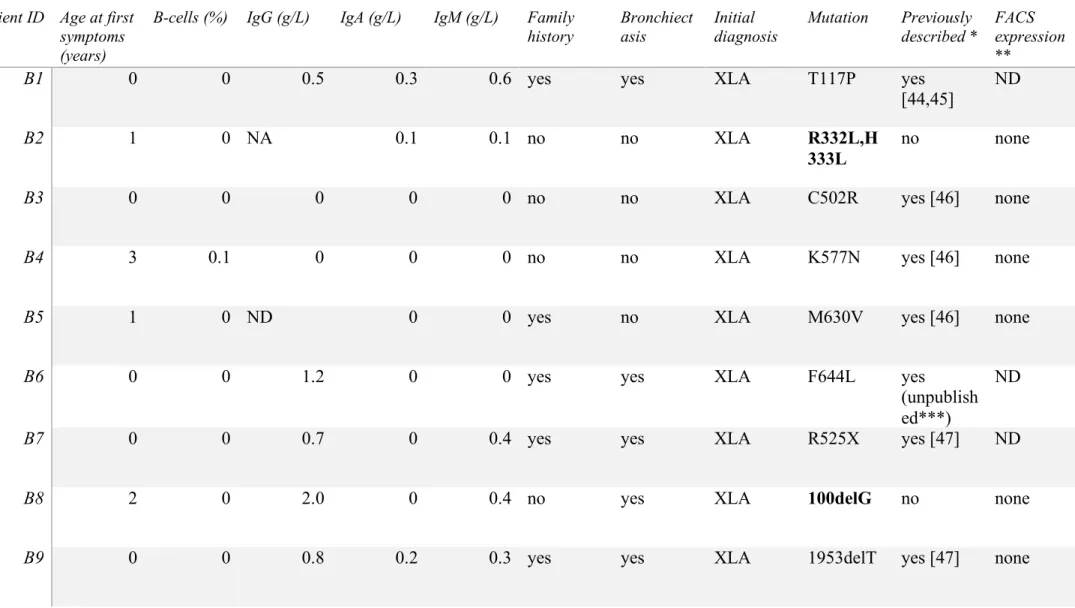
BTK mutation screening
12
In our cohort, 7% of patients presented with an initial diagnosis of XLA (Table 1). As 13
our genetic screening revealed, BTK mutations was found only in patients presenting a very 14
low B-cell count (<1%, see table 4), we included 31 additional hypogammaglobulinemic 15
patients with B-cell counts below 1%. We detected a BTK mutation in 17 out of 20 patients 16
with a primary XLA diagnosis (B1-B17, table 4, Figure 1C). Despite a clinical/immunological 17
phenotype consistent with the diagnosis of XLA, we could not confirm genetically the diagnosis 18
in 3 patients (B18-B20, table 4). These 3 patients may carry a mutation in another gene causing 19
agammaglobulinemia (e.g. BLNK, PI3KR1) [35,36] and as such should be screened for it. 20
Strikingly, we found a BTK mutation in 5 patients (B21-B25) with a primary diagnosis of CVID 21
or IgG subclass deficiency (Table 4, boxes in Figure 1C). Overall, we described 20 different 22
mutations in 22 patients, including 5 novel mutations while the others were previously reported 23
in the BTK database (https://databases.lovd.nl/shared/genes/BTK) [37]. 24
We observed a phenotypic variability in patients carrying BTK mutations despite 1
showing all less than 1% B-cells; indeed, while a majority of patients presented their first 2
symptoms at early age in infancy some were described at later age (up to 18 years old for B22, 3
see table 4). In addition, of all the BTK mutated patients only 3 had an IgG level above 4g/L 4
and all with a primary diagnosis of CVID or IgG subclass deficiency (B21, B22 and B25; see 5
table 4). We hypothesized that this variability could be explained by the impact of each 6
mutation on the BTK protein function and expression. Considering the absence or very low 7
levels of B-cells in XLA patients, we carried out intracellular FACS assay to measure BTK 8
expression in monocytes. We measured the delta between BTK mean fluorescent intensity 9
(MFI) and the irrelevant antibody MFI (i.e. ∆MFI) in 15 healthy controls and observed a median 10
∆MFI of 107 [79-173] (Figure 3A). We then obtained data for patients harboring BTK mutations 11
(17 different mutations out of 21, see table 4 and Figure 3). As expected, BTK protein was 12
undetectable in all 13 patients’ monocytes with a primary diagnosis of XLA, as illustrated with 13
patient B14 on Figure 3A. When considering the 3 patients with a primary diagnosis of XLA 14
but without identified BTK mutation, we detected a lower ∆MFI in 2 patients (B18 and B19, 15
Figure 3B) and a ∆MFI in the normal range for the B20 patient (Figure 3B). 16
Finally, we measured BTK expression in 4 out of 5 patients carrying a BTK mutation 17
but without a primary diagnosis of XLA, as we could not measure BTK expression for patient 18
B24. However, this patient carried the same M630V mutation as for the XLA B5 patient, so we 19
can only extrapolate that he might also present an undetectable BTK expression (Table 4). BTK 20
expression was undetectable in patients B22 and B25 monocytes, who were primarily 21
diagnosed with CVID and IgG subclass deficiency, respectively (Table 4, Figure 3C). Patients 22
B21 and B23 showed a decreased but detectable BTK expression in monocytes (Figure 3C), in 23
normal range for B21 (∆MFI=80) and just below for B23 (∆MFI=69). R28H mutation is 24
described in multiple studies as responsible for the XLA phenotype (Table 4), moreover, it is 25
reported as pathogenic in the ClinVar database 1
(https://www.ncbi.nlm.nih.gov/clinvar/variation/11348/). These suggest strongly that R28H is 2
responsible of patient B21 phenotype. G164D coding mutation is predicted by the CADD 3
bioinformatics tool [38] to be damaging for the protein function (score: 29.1). This mutation is 4
not described in populations databases such as the 1000 Genomes project [31]. These data are 5
not sufficient to describe G164D as responsible of the disease, additional family testing would 6
permit to settle this down. Altogether, these results suggest an alternative pathophysiological 7
mechanism to the absence of detectable BTK protein: lower level of BTK may indicate a 8
dysfunction of the BTK signaling pathway that could consequently alter the development of B-9
cells and promote hypogammaglobulinemia. 10
Discussion
1
We sequenced three genes (CD40LG, SH2D1A, BTK) located on the X chromosome in 2
male patients with primary humoral deficiencies from the French national DEFI cohort (Figure 3
S1). Deleterious mutations in these genes are responsible of profound defects in 4
immunoglobulin production, are usually diagnosed in children and associated with specific 5
phenotypes. We intentionally did not restrict patients’ selection to any particular clinical or 6
biological phenotypes as we intended to identify mutations outside of a commonly recognized 7
clinical presentation. We demonstrated that the systematic screening of these genes is useful to 8
confirm and/or reclassify immune defects diagnosis in adults with humoral primary 9
immunodeficiency. 10
CD40LG genetic screening of 150 male patients did not reveal any mutation outside of
11
the classical clinical presentation of HIGM, indicating that the CD40LG mutations are 12
responsible for a very specific clinical and immunological phenotype that cannot be confused 13
with CVID or any other defects. One patient with a HIGM phenotype did not exhibit any 14
mutation in CD40LG; to further characterize this patient’s disease, additional genetic analyses 15
should be performed for autosomal recessive HIGM genes such as AICDA, CD40 or UNG 16
[33,34]. 17
Here, we characterized 2 patients primarily diagnosed as CVID but presenting a 18
SH2D1A mutation with an absence of SAP detection by immunoblot. XLP main feature is an
19
extreme susceptibility to EBV infection, and one of these 2 patients presented a severe 20
infectious mononucleosis at six years of age. However, the second one claimed he did not suffer 21
any severe EBV primary infection during his childhood and had no familial history of X-linked 22
disease. This indicates that, despite the low percentage of diagnosed patients (2 out of 145 if 23
we consider only CVID), the SH2D1A mutation knowledge is important in all male patients 24
diagnosed with CVID, whether or not they have a medical or familial history consistent with 25
severe EBV infection. Indeed, the XLP diagnosis significantly modifies the therapeutic and 1
follow-up options of the patients. 2
The systematic sequencing of BTK in 181 male patients confirmed the suspected XLA 3
diagnosis in 17/20 patients. Moreover, we identified 5 patients with a primary diagnosis of 4
CVID or IgG subclass deficiency carrying a mutation in the BTK gene. This misdiagnosis could 5
have several explanations: persistence of low levels but detectable IgG, IgA or IgM; insufficient 6
genealogic investigation to detect familial X-linked disease; onset of disease at adulthood; 7
absence of serious pulmonary complications. It emphasizes that all male patients with a 8
diagnosis of primary humoral immunodeficiency and with very low levels of circulating B-cells 9
(<1%), regardless of age at diagnosis, residual Ig concentration or familial history, should be 10
screened for BTK defects, either by FACS detection in monocytes or by direct sequencing of 11
the coding region. 12
The 3 patients referred to us with a diagnosis of XLA and in whom we could not identify 13
BTK mutations are of great interest. B18 and B19 patients do display clinical features consistent
14
with XLA, including an absence of circulating B-cells. BTK expression in monocytes, despite 15
being detectable, was lower in these patients compared to controls. B20 also shows an absence 16
of circulating B-cells but with a normal level of BTK expression and a family history of 17
autosomal recessive disease. Notwithstanding no mutations were found in these individuals, 18
including after screening the 5’ and 3’UTR regulatory regions; at this stage, we cannot exclude 19
the possibility that the weak or undetectable BTK expression may be the result of a mutation in 20
another BTK regulatory region not covered by the sequencing method we implemented. 21
Alternatively, other intracellular proteins in the B-cell lineage may be absent or defective in 22
these patients, accounting for the weak translation of BTK and the low level of BTK activity in 23
B-cell precursors. These patients may carry a mutation in genes responsible of autosomal 24
recessive agammaglobulinemia, and especially B20 considering his family history. Beyond 25
these previously identified genes, an agnostic screening using whole-exome or whole-genome 1
next-generation sequencing (NGS) might also reveal novel disease-causing genes. 2
With the rising of NGS technologies, the possibility of genetic screening has increased. 3
Our results advocate to include SH2D1A and BTK genes in newly developed targeted NGS 4
panel genetic testing for all primary immunodeficiency diagnosis including CVID or related 5
hypogammaglobulinemia. This is supported by the fact that such NGS panel are already in 6
place, they include SH2D1A and BTK but also other genes responsible of HIGM, XLP or XLA 7
diseases (e.g. CD40, PIK3R1, etc.). This will further help characterize monogenic forms of 8
CVID, which still count for less than 10% of patients [2], from polygenic forms of CVID. To 9
achieve deciphering the pathophysiology and help improve treatments of these complex forms 10
of CVID, and in addition with NGS, efforts should be combined and integrated from different 11
omics approaches such as epigenomics, proteomics and metabolomics [39,40]. 12
In conclusion, our systematic genetic screening in a large cohort of adults with primary 13
humoral immunodeficiency highlights several key features of CVID and 14
hypogammaglobulinemia genetics: (1) There is a good correlation between CD40LG genotype 15
and phenotype; (2) a SH2D1A mutation can occur in a patient with atypical clinical features; 16
(3) BTK genotype do not always correlates with cellular phenotype (BTK expression in 17
monocytes), immunological phenotype (Ig concentrations at the time of immunodeficiency 18
diagnosis) and clinical phenotype. A knowledge of the genetic status of each patient is therefore 19
crucial for a firm diagnosis, as it leads to the most appropriate medical treatment and genetic 20
counselling. 21
Acknowledgements
1
Authors thank Jean-Claude Brouet and Jean-Christophe Bories for helpful discussions. 2
This work was supported by grants from Programme Hospitalier de Recherche Clinique 2005 3
(E.O., P.D.), GIS maladies rares (E.O.), ANR MRAR-06 (C.F.), Fondation pour la recherche 4
Médicale (CF). N.V. was supported by La Ligue Nationale Contre le Cancer. D.B. was 5
supported by the ARC. 6
Conflicts of interest: none 7
References
1
[1] E. Oksenhendler, L. Gérard, C. Fieschi, M. Malphettes, G. Mouillot, R. Jaussaud, J. 2
Viallard, M. Gardembas, L. Galicier, N. Schleinitz, F. Suarez, P. Soulas-Sprauel, E. Hachulla, 3
A. Jaccard, A. Gardeur, I. Théodorou, C. Rabian, P. Debré, DEFI Study Group, Infections in 4
252 Patients with Common Variable Immunodeficiency, Clin. Infect. Dis. 46 (2008) 1547– 5
1554. doi:10.1086/587669. 6
[2] D.J.A. Bogaert, M. Dullaers, B.N. Lambrecht, K.Y. Vermaelen, E. De Baere, F. 7
Haerynck, Genes associated with common variable immunodeficiency: one diagnosis to rule 8
them all?, J. Med. Genet. 53 (2016) 575–590. doi:10.1136/jmedgenet-2015-103690. 9
[3] U. Salzer, C. Bacchelli, S. Buckridge, Q. Pan-Hammarström, S. Jennings, V. Lougaris, 10
A. Bergbreiter, T. Hagena, J. Birmelin, A. Plebani, A.D.B. Webster, H.-H. Peter, D. Suez, H. 11
Chapel, A. McLean-Tooke, G.P. Spickett, S. Anover-Sombke, H.D. Ochs, S. Urschel, B.H. 12
Belohradsky, S. Ugrinovic, D.S. Kumararatne, T.C. Lawrence, A.M. Holm, J.L. Franco, I. 13
Schulze, P. Schneider, E.M. Gertz, A.A. Schäffer, L. Hammarström, A.J. Thrasher, H.B. 14
Gaspar, B. Grimbacher, Relevance of biallelic versus monoallelic TNFRSF13B mutations in 15
distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody 16
deficiency syndromes, Blood. 113 (2009) 1967–1976. doi:10.1182/blood-2008-02-141937. 17
[4] N. Vince, D. Boutboul, G. Mouillot, N. Just, M. Peralta, J.-L. Casanova, M.E. Conley, 18
J.-C. Bories, E. Oksenhendler, M. Malphettes, C. Fieschi, DEFI Study Group, Defects in the 19
CD19 complex predispose to glomerulonephritis, as well as IgG1 subclass deficiency, J. 20
Allergy Clin. Immunol. 127 (2011) 538-541.e1–5. doi:10.1016/j.jaci.2010.10.019. 21
[5] F.A. Bonilla, I. Barlan, H. Chapel, B.T. Costa-Carvalho, C. Cunningham-Rundles, 22
M.T. de la Morena, F.J. Espinosa-Rosales, L. Hammarström, S. Nonoyama, I. Quinti, J.M. 23
Routes, M.L.K. Tang, K. Warnatz, International Consensus Document (ICON): Common 24
Variable Immunodeficiency Disorders, J. Allergy Clin. Immunol. Pract. 4 (2016) 38–59. 25
doi:10.1016/j.jaip.2015.07.025. 1
[6] H.B. Gaspar, M. Ferrando, I. Caragol, M. Hernandez, J.M. Bertran, X. De Gracia, T. 2
Lester, C. Kinnon, E. Ashton, T. Espanol, Kinase mutant Btk results in atypical X-linked 3
agammaglobulinaemia phenotype, Clin Exp Immunol. 120 (2000) 346–50. 4
[7] S. Hashimoto, T. Miyawaki, T. Futatani, H. Kanegane, K. Usui, T. Nukiwa, S. 5
Namiuchi, M. Matsushita, T. Yamadori, M. Suemura, T. Kishimoto, S. Tsukada, Atypical X-6
linked agammaglobulinemia diagnosed in three adults, Intern Med. 38 (1999) 722–5. 7
[8] A. Jones, L. Bradley, L. Alterman, M. Tarlow, R. Thompson, C. Kinnon, G. Morgan, 8
X linked agammaglobulinaemia with a “leaky” phenotype, Arch Child. 74 (1996) 548–9. 9
[9] H. Kanegane, S. Tsukada, T. Iwata, T. Futatani, K. Nomura, J. Yamamoto, T. 10
Yoshida, K. Agematsu, A. Komiyama, T. Miyawaki, Detection of Bruton’s tyrosine kinase 11
mutations in hypogammaglobulinaemic males registered as common variable 12
immunodeficiency (CVID) in the Japanese Immunodeficiency Registry, Clin Exp Immunol. 13
120 (2000) 512–7. 14
[10] S.J. Kornfeld, R.N. Haire, S.J. Strong, H. Tang, S.S. Sung, S.M. Fu, G.W. Litman, A 15
novel mutation (Cys145-->Stop) in Bruton’s tyrosine kinase is associated with newly 16
diagnosed X-linked agammaglobulinemia in a 51-year-old male, Mol Med. 2 (1996) 619–23. 17
[11] D.M. Stewart, L. Tian, D.L. Nelson, A case of X-linked agammaglobulinemia 18
diagnosed in adulthood, Clin Immunol. 99 (2001) 94–9. 19
[12] M.E. Conley, D.M. Farmer, A.K. Dobbs, V. Howard, Y. Aiba, S.A. Shurtleff, T. 20
Kurosaki, A minimally hypomorphic mutation in Btk resulting in reduced B cell numbers but 21
no clinical disease, Clin Exp Immunol. 152 (2008) 39–44. 22
[13] K. Morwood, H. Bourne, M. Gold, D. Gillis, E.M. Benson, Phenotypic variability: 23
clinical presentation between the 6th year and the 60th year in a family with X-linked 24
agammaglobulinemia, J Allergy Clin Immunol. 113 (2004) 783–5. 25
[14] J.G. Noordzij, S. de Bruin-Versteeg, N.G. Hartwig, C.M. Weemaes, E.J. Gerritsen, E. 1
Bernatowska, S. van Lierde, R. de Groot, J.J. van Dongen, XLA patients with BTK splice-site 2
mutations produce low levels of wild-type BTK transcripts, J Clin Immunol. 22 (2002) 306– 3
18. 4
[15] D.C. Saffran, O. Parolini, M.E. Fitch-Hilgenberg, D.J. Rawlings, D.E. Afar, O.N. 5
Witte, M.E. Conley, Brief report: a point mutation in the SH2 domain of Bruton’s tyrosine 6
kinase in atypical X-linked agammaglobulinemia, N Engl J Med. 330 (1994) 1488–91. 7
[16] J.R. Sigmon, E. Kasasbeh, G. Krishnaswamy, X-linked agammaglobulinemia 8
diagnosed late in life: case report and review of the literature, Clin Mol Allergy. 6 (2008) 5. 9
[17] S. Tsukada, D.C. Saffran, D.J. Rawlings, O. Parolini, R.C. Allen, I. Klisak, R.S. 10
Sparkes, H. Kubagawa, T. Mohandas, S. Quan, et al., Deficient expression of a B cell 11
cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia, Cell. 72 (1993) 279– 12
90. 13
[18] D. Vetrie, I. Vorechovsky, P. Sideras, J. Holland, A. Davies, F. Flinter, L. 14
Hammarstrom, C. Kinnon, R. Levinsky, M. Bobrow, et al., The gene involved in X-linked 15
agammaglobulinaemia is a member of the src family of protein-tyrosine kinases, Nature. 361 16
(1993) 226–33. 17
[19] J.A. Winkelstein, M.C. Marino, H.M. Lederman, S.M. Jones, K. Sullivan, A.W. 18
Burks, M.E. Conley, C. Cunningham-Rundles, H.D. Ochs, X-linked agammaglobulinemia: 19
report on a United States registry of 201 patients, Med. Baltim. 85 (2006) 193–202. 20
[20] A. Aghamohammadi, H. Kanegane, M. Moein, A. Farhoudi, Z. Pourpak, M. 21
Movahedi, M. Gharagozlou, A.A. Zargar, T. Miyawaki, Identification of an SH2D1A 22
mutation in a hypogammaglobulinemic male patient with a diagnosis of common variable 23
immunodeficiency, Int J Hematol. 78 (2003) 45–7. 24
[21] D. Eastwood, K.C. Gilmour, K. Nistala, C. Meaney, H. Chapel, Z. Sherrell, A.D. 25
Webster, E.G. Davies, A. Jones, H.B. Gaspar, Prevalence of SAP gene defects in male 1
patients diagnosed with common variable immunodeficiency, Clin Exp Immunol. 137 (2004) 2
584–8. 3
[22] M. Morra, O. Silander, S. Calpe, M. Choi, H. Oettgen, L. Myers, A. Etzioni, R. 4
Buckley, C. Terhorst, Alterations of the X-linked lymphoproliferative disease gene SH2D1A 5
in common variable immunodeficiency syndrome, Blood. 98 (2001) 1321–5. 6
[23] K. Nistala, K.C. Gilmour, T. Cranston, E.G. Davies, D. Goldblatt, H.B. Gaspar, A.M. 7
Jones, X-linked lymphoproliferative disease: three atypical cases, Clin Exp Immunol. 126 8
(2001) 126–30. 9
[24] A. Soresina, V. Lougaris, S. Giliani, F. Cardinale, L. Armenio, M. Cattalini, L.D. 10
Notarangelo, A. Plebani, Mutations of the X-linked lymphoproliferative disease gene 11
SH2D1A mimicking common variable immunodeficiency, Eur J Pediatr. 161 (2002) 656–9. 12
[25] A.J. Coffey, R.A. Brooksbank, O. Brandau, T. Oohashi, G.R. Howell, J.M. Bye, A.P. 13
Cahn, J. Durham, P. Heath, P. Wray, R. Pavitt, J. Wilkinson, M. Leversha, E. Huckle, C.J. 14
Shaw-Smith, A. Dunham, S. Rhodes, V. Schuster, G. Porta, L. Yin, P. Serafini, B. Sylla, M. 15
Zollo, B. Franco, A. Bolino, M. Seri, A. Lanyi, J.R. Davis, D. Webster, A. Harris, G. Lenoir, 16
G. de St Basile, A. Jones, B.H. Behloradsky, H. Achatz, J. Murken, R. Fassler, J. Sumegi, G. 17
Romeo, M. Vaudin, M.T. Ross, A. Meindl, D.R. Bentley, Host response to EBV infection in 18
X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding 19
gene, Nat Genet. 20 (1998) 129–35. 20
[26] J. Sayos, C. Wu, M. Morra, N. Wang, X. Zhang, D. Allen, S. van Schaik, L. 21
Notarangelo, R. Geha, M.G. Roncarolo, H. Oettgen, J.E. De Vries, G. Aversa, C. Terhorst, 22
The X-linked lymphoproliferative-disease gene product SAP regulates signals induced 23
through the co-receptor SLAM, Nature. 395 (1998) 462–9. 24
[27] D.T. Purtilo, D. DeFlorio Jr., L.M. Hutt, J. Bhawan, J.P. Yang, R. Otto, W. Edwards, 25
Variable phenotypic expression of an X-linked recessive lymphoproliferative syndrome, N 1
Engl J Med. 297 (1977) 1077–80. 2
[28] T.A. Seemayer, T.G. Gross, R.M. Egeler, S.J. Pirruccello, J.R. Davis, C.M. Kelly, M. 3
Okano, A. Lanyi, J. Sumegi, X-linked lymphoproliferative disease: twenty-five years after the 4
discovery, Pediatr Res. 38 (1995) 471–8. 5
[29] A. Etzioni, H.D. Ochs, The hyper IgM syndrome--an evolving story, Pediatr Res. 56 6
(2004) 519–25. 7
[30] R.C. Allen, R.J. Armitage, M.E. Conley, H. Rosenblatt, N.A. Jenkins, N.G. Copeland, 8
M.A. Bedell, S. Edelhoff, C.M. Disteche, D.K. Simoneaux, et al., CD40 ligand gene defects 9
responsible for X-linked hyper-IgM syndrome, Science. 259 (1993) 990–3. 10
[31] 1000 Genomes Project Consortium, A. Auton, L.D. Brooks, R.M. Durbin, E.P. 11
Garrison, H.M. Kang, J.O. Korbel, J.L. Marchini, S. McCarthy, G.A. McVean, G.R. 12
Abecasis, A global reference for human genetic variation, Nature. 526 (2015) 68–74. 13
doi:10.1038/nature15393. 14
[32] S. Limou, A.M. Taverner, C.A. Winkler, Ferret: a user-friendly Java tool to extract 15
data from the 1000 Genomes Project, Bioinformatics. 32 (2016) 2224–2226. 16
doi:10.1093/bioinformatics/btw147. 17
[33] K. Imai, G. Slupphaug, W.I. Lee, P. Revy, S. Nonoyama, N. Catalan, L. Yel, M. 18
Forveille, B. Kavli, H.E. Krokan, H.D. Ochs, A. Fischer, A. Durandy, Human uracil-DNA 19
glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch 20
recombination, Nat Immunol. 4 (2003) 1023–8. 21
[34] P. Revy, T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. 22
Forveille, R. Dufourcq-Labelouse, A. Gennery, I. Tezcan, F. Ersoy, H. Kayserili, A.G. 23
Ugazio, N. Brousse, M. Muramatsu, L.D. Notarangelo, K. Kinoshita, T. Honjo, A. Fischer, A. 24
Durandy, Activation-induced cytidine deaminase (AID) deficiency causes the autosomal 25
recessive form of the Hyper-IgM syndrome (HIGM2), Cell. 102 (2000) 565–75. 1
[35] Y. Minegishi, J. Rohrer, E. Coustan-Smith, H.M. Lederman, R. Pappu, D. Campana, 2
A.C. Chan, M.E. Conley, An essential role for BLNK in human B cell development, Science. 3
286 (1999) 1954–1957. 4
[36] M.E. Conley, A.K. Dobbs, A.M. Quintana, A. Bosompem, Y.-D. Wang, E. Coustan-5
Smith, A.M. Smith, E.E. Perez, P.J. Murray, Agammaglobulinemia and absent B lineage cells 6
in a patient lacking the p85α subunit of PI3K, J. Exp. Med. 209 (2012) 463–470. 7
doi:10.1084/jem.20112533. 8
[37] J. Valiaho, C.I. Smith, M. Vihinen, BTKbase: the mutation database for X-linked 9
agammaglobulinemia, Hum Mutat. 27 (2006) 1209–17. 10
[38] M. Kircher, D.M. Witten, P. Jain, B.J. O’Roak, G.M. Cooper, J. Shendure, A general 11
framework for estimating the relative pathogenicity of human genetic variants, Nat. Genet. 46 12
(2014) 310–315. doi:10.1038/ng.2892. 13
[39] R. Ameratunga, K. Lehnert, S.-T. Woon, D. Gillis, V.L. Bryant, C.A. Slade, R. Steele, 14
Review: Diagnosing Common Variable Immunodeficiency Disorder in the Era of Genome 15
Sequencing, Clin. Rev. Allergy Immunol. (2017). doi:10.1007/s12016-017-8645-0. 16
[40] A.-K. Kienzler, C.E. Hargreaves, S.Y. Patel, The role of genomics in common 17
variable immunodeficiency disorders, Clin. Exp. Immunol. 188 (2017) 326–332. 18
doi:10.1111/cei.12947. 19
[41] J.P. DiSanto, J.Y. Bonnefoy, J.F. Gauchat, A. Fischer, G. de Saint Basile, CD40 20
ligand mutations in x-linked immunodeficiency with hyper-IgM, Nature. 361 (1993) 541–3. 21
[42] Y. Tabata, J. Villanueva, S.M. Lee, K. Zhang, H. Kanegane, T. Miyawaki, J. Sumegi, 22
A.H. Filipovich, Rapid detection of intracellular SH2D1A protein in cytotoxic lymphocytes 23
from patients with X-linked lymphoproliferative disease and their family members, Blood. 24
105 (2005) 3066–71. 25
[43] B. Pasquier, L. Yin, M.C. Fondaneche, F. Relouzat, C. Bloch-Queyrat, N. Lambert, A. 1
Fischer, G. de Saint-Basile, S. Latour, Defective NKT cell development in mice and humans 2
lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product, J Exp 3
Med. 201 (2005) 695–701. 4
[44] M.E. Conley, D. Mathias, J. Treadaway, Y. Minegishi, J. Rohrer, Mutations in btk in 5
patients with presumed X-linked agammaglobulinemia, Am J Hum Genet. 62 (1998) 1034– 6
43. 7
[45] E. Holinski-Feder, M. Weiss, O. Brandau, K.B. Jedele, B. Nore, C.M. Bäckesjö, M. 8
Vihinen, S.R. Hubbard, B.H. Belohradsky, C.I. Smith, A. Meindl, Mutation screening of the 9
BTK gene in 56 families with X-linked agammaglobulinemia (XLA): 47 unique mutations 10
without correlation to clinical course, Pediatrics. 101 (1998) 276–284. 11
[46] J. Väliaho, I. Faisal, C. Ortutay, C.I.E. Smith, M. Vihinen, Characterization of all 12
possible single-nucleotide change caused amino acid substitutions in the kinase domain of 13
Bruton tyrosine kinase, Hum. Mutat. 36 (2015) 638–647. doi:10.1002/humu.22791. 14
[47] I. Vorechovsky, M. Vihinen, G. de Saint Basile, S. Honsova, L. Hammarstrom, S. 15
Muller, L. Nilsson, A. Fischer, C.I. Smith, DNA-based mutation analysis of Bruton’s tyrosine 16
kinase gene in patients with X-linked agammaglobulinaemia, Hum Mol Genet. 4 (1995) 51–8. 17
[48] H.B. Gaspar, L.A. Bradley, F. Katz, R.C. Lovering, C.M. Roifman, G. Morgan, R.J. 18
Levinsky, C. Kinnon, Mutation analysis in Bruton’s tyrosine kinase, the X-linked 19
agammaglobulinaemia gene, including identification of an insertional hotspot, Hum Mol 20
Genet. 4 (1995) 755–7. 21
[49] R.A. Brooimans, A.J. van den Berg, G.T. Rijkers, L.A. Sanders, J.K. van Amstel, 22
M.G. Tilanus, M.J. Grubben, B.J. Zegers, Identification of novel Bruton’s tyrosine kinase 23
mutations in 10 unrelated subjects with X linked agammaglobulinaemia, J. Med. Genet. 34 24
(1997) 484–488. 25
[50] Y. Ohta, R.N. Haire, R.T. Litman, S.M. Fu, R.P. Nelson, J. Kratz, S.J. Kornfeld, M. de 1
la Morena, R.A. Good, G.W. Litman, Genomic organization and structure of Bruton 2
agammaglobulinemia tyrosine kinase: localization of mutations associated with varied clinical 3
presentations and course in X chromosome-linked agammaglobulinemia, Proc. Natl. Acad. 4
Sci. U. S. A. 91 (1994) 9062–9066. 5
[51] B. Duriez, P. Duquesnoy, F. Dastot, P. Bougneres, S. Amselem, M. Goossens, An 6
exon-skipping mutation in the btk gene of a patient with X-linked agammaglobulinemia and 7
isolated growth hormone deficiency, FEBS Lett. 346 (1994) 165–70. 8
[52] M. de Weers, R.G. Mensink, M.E. Kraakman, R.K. Schuurman, R.W. Hendriks, 9
Mutation analysis of the Bruton’s tyrosine kinase gene in X-linked agammaglobulinemia: 10
identification of a mutation which affects the same codon as is altered in immunodeficient xid 11
mice, Hum Mol Genet. 3 (1994) 161–6. 12
[53] M. Velickovic, M.L. Prasad, S.A. Weston, E.M. Benson, Identification of the bruton 13
tyrosine kinase (BTK) gene mutations in 20 Australian families with X-linked 14
agammaglobulinemia (XLA), Hum Mutat. 23 (2004) 398–9. 15
Tables
1
Table 1: Diagnosis provided by the clinical investigators before the genetic screening of CD40LG, SH2D1A and BTK.
2 CD40LG n=150 SH2D1A n=230 BTK n=181 HIGM 4 4 4 XLP 0 0 0 XLA 11 14 20 CVID 109 145 120 IgAD 0 3 0 IgG-SCl 9 15 13 Good syndrome 4 9 8 Other 13 40 16 3
HIGM: X-linked hyperIgM syndrome. XLP: X-linked lymphoproliferative disease. XLA: X-linked agammaglobulinemia. CVID: Common 4
variable immunodeficiency disorders. IgAD: IgA deficiency. IgG-SCl: IgG subclass deficiency. 5
Table 2: CD40LG mutations and clinical features of the patients
1
2
3
Patient ID Age at first symptoms (years)
B-cells (%) IgG (g/L) IgA (g/L) IgM (g/L) Family
history Opportunis tic infections Initial diagnosis Mutation Previously described FACS expression C1 4 9 1.3 0 7.5 no yes HIGM 228-231delAA GA no none C2 0 7 3.3 0.4 1.2 no yes HIGM 409+1G> A yes [41] none C3 1 7 0.1 0 0.9 no yes HIGM 410-2A>G no none
Table 3: SH2D1A mutations and clinical features of the patients
1
2
See figure 2 for Western blot. 3
4
Patient ID Age at first symptoms (years)
B-cells (%) IgG (g/L) IgA (g/L) IgM (g/L) Family
history Initial diagnosis Mutation Previously described WB expression
S1 3 4 1.8 0.4 0.4 yes CVID 201+3A>
G
yes [42] none
Table 4: BTK mutations and clinical features of the patients
1
Patient ID Age at first symptoms (years)
B-cells (%) IgG (g/L) IgA (g/L) IgM (g/L) Family
history Bronchiect asis Initial diagnosis Mutation Previously described * FACS expression **
B1 0 0 0.5 0.3 0.6 yes yes XLA T117P yes
[44,45]
ND
B2 1 0 NA 0.1 0.1 no no XLA R332L,H
333L
no none
B3 0 0 0 0 0 no no XLA C502R yes [46] none
B4 3 0.1 0 0 0 no no XLA K577N yes [46] none
B5 1 0 ND 0 0 yes no XLA M630V yes [46] none
B6 0 0 1.2 0 0 yes yes XLA F644L yes
(unpublish ed***)
ND
B7 0 0 0.7 0 0.4 yes yes XLA R525X yes [47] ND
B8 2 0 2.0 0 0.4 no yes XLA 100delG no none
B10 4 0 ND 0.1 0 no yes XLA g.2275_13 114del108 38insCC C
no none
B11 4 0 ND 0 0.1 yes no XLA 174_175in
sT
yes [45] none
B12 1 0 3.7 0.1 0.1 no yes XLA 215_216in
sA yes [48] ND B13 1 0 1.5 0 0 yes no XLA 1349+5G >A yes [49] none B14 0 0 ND 0 0 yes no XLA 1566+1G >T yes [45] none B15 0 0 0.8 0 0 no no XLA 1567-12delTTT G yes [50] none
B16 0 0 0 0 0 yes yes XLA 1750+5G
>A yes [51] none
B17 0 0.1 0 0 0.2 no yes XLA 1750+5G
>A yes [51] none
B18 22 0 0 0 0.1 yes (XL) yes XLA No
mutation
- low
B19 14 0.4 0 0 0.1 yes no XLA No
B20 0 0 1 0 0 yes (AR) yes XLA No mutation - yes B21 5 0.3 4.4 1.6 0.4 no no CVID R28H yes [50,52] yes B22 18 0.1 4.4 1.8 0.1 no no CVID F114V no none
B23 15 0.1 2.1 0 0.3 no yes CVID G164D no low
B24 0 0 0 0 0 yes yes CVID M630V yes [46] ND
B25 0 0.1 4.2 1.2 0.4 no yes IgG
s/class 1350-2A>G yes [53] none
1
*Only the oldest description is cited, see the btkbase for a complete list: https://databases.lovd.nl/shared/genes/BTK.
2
**See figure 3 for FACS expression. 3
***See in btkbase. 4
Patients B1 to B17 had clinical and biological features consistent with XLA and an identified BTK mutation. Patients B16 and B17, as well as, 5
B5 and B24 do not belong to the same family. Patients B18 to B20 had an initial diagnosis of XLA based on phenotypic data, no mutation in 6
BTK were found. Newly described mutations are in bold. CVID or IgG subclass deficiency patients’ mutations are boxed. Patients B21 to B25
7
had a primary diagnosis of humoral deficiency, 4 CVID and 1 IgG subclass deficiency. XL: X-linked; AR: autosomic recessive. 8
Figure Legends
1
Figure 1: (A) Schematic diagram of the CD40LG coding region, with 5 coding exons, 783
2
nucleotides (nt) and 261 amino acids (AA). The 3 patients’ mutations are represented. TM: 3
transmembrane domain. Dotted lines are the trimer interface. (B) Schematic diagram of the 4
coding region of SH2D1A, with 4 exons, 387 nt and 128 AA. SH2: Src homology 2 domain. 5
The 2 patients’ mutations are represented. (C) Schematic diagram of the coding region of BTK, 6
with 18 coding exons, 1980 nt and 659 AA. Newly described mutations are in bold. CVID or 7
IgG subclass deficiency patients’ mutations are boxed. PH: pleckstrin homology. ZF: zinc 8
finger. SH3: Src homology 3 domain. SH2: Src homology 2 domain. TK: tyrosine kinase. 9
Figure 2: Immunoblot of SAP in PHA T-cell blasts protein extraction. Actin is represented as
10
a control of protein expression. HC: healthy control. 11
Figure 3: BTK expression in gated monocytes (CD45+, CD14+). (A) healthy control (HC) and
12
a representative XLA patient (B14, absence of BTK detection). We measured the delta between 13
BTK mean fluorescent intensity (MFI) and the irrelevant antibody MFI (i.e. ∆MFI), the 15 HC 14
showed a median of ∆MFI = 107 [79-173]. (B) Patients with XLA as primary diagnosis but 15
without identified BTK mutations (WT: wild type). B18 and B19 patients showed reduced BTK 16
expression but not B20. (C) Patients with CVID or IgG subclass deficiency as primary diagnosis 17
but with BTK mutations. B21 and B23 patients showed reduced BTK expression; B20 show no 18
BTK expression. Ab: Antibody. 19
1 Figure 1 2 3 4 5 6 7 8
1 Figure 2 2 3 4 5 6 7
1
Figure 3 2
Table S1: Diagnosis provided by the clinical investigators before the genetic screening of CD40LG, SH2D1A and BTK. CD40LG n=150 SH2D1A n=230 BTK n=181 HIGM 4 4 4 XLP 0 0 0 XLA 11 14 20 CVID 109 145 120 IgAD 0 3 0 IgG-SCl 9 15 13 Thymoma 4 9 8 Other 13 40 16
HIGM: X-linked hyperIgM syndrome. XLP: X-linked lymphoproliferative disease. XLA: X-linked agammaglobulinemia. CVID: Common