Encyclopedia of Biological Chemistry, 3rd Edition
Biochemistry of thiamine and thiamine phosphate compounds Bettendorff Lucien and Wins Pierre
Corresponding Author Name Bettendorff Lucien
GIGA-Neurosciences, University of Liège, Liège, Belgium Quartier Hôpital, Bât. B36
Avenue Hippocrate, 15
4000 Liège 1 (Angleur - Sart Tilman) Belgium
L.Bettendorff@uliege.be Tel.: +3243665967 Co-Author Author Name Wins Pierre
GIGA-Neurosciences, University of Liège, Liège, Belgium Quartier Hôpital, Bât. B36
Avenue Hippocrate, 15
4000 Liège 1 (Angleur - Sart Tilman) Belgium
winspierre9@gmail.com Tel.: +3243665967
Keywords
Alcohol, beriberi, coenzyme, Krebs cycle, mitochondria, 2-oxoglutarate dehydrogenase complex, oxidative metabolism, pentose phosphate shunt, pyruvate dehydrogenase complex, thiamine deficiency disorders, thiamine diphosphate, thiamine triphosphate, transketolase, Wernicke-Korsakoff syndrome.
Abstract
Thiamine (vitamin B1) is an essential molecule for all living organisms. It is the precursor for several phosphorylated derivatives, the most important being the coenzyme thiamine diphosphate (ThDP). Thiamine is transported into cells by specific transporters and pyrophosphorylated to ThDP in the cytosol. ThDP is an essential cofactor for cellular oxidative energy metabolism. Thiamine deficiency disorders lead to severe nervous system lesion and cardiac failure. ThDP is the precursor for two triphosphorylated derivatives, thiamine triphosphate and adenosine thiamine triphosphate, with unknown function but present in all three kingdoms of life.
1. Introduction
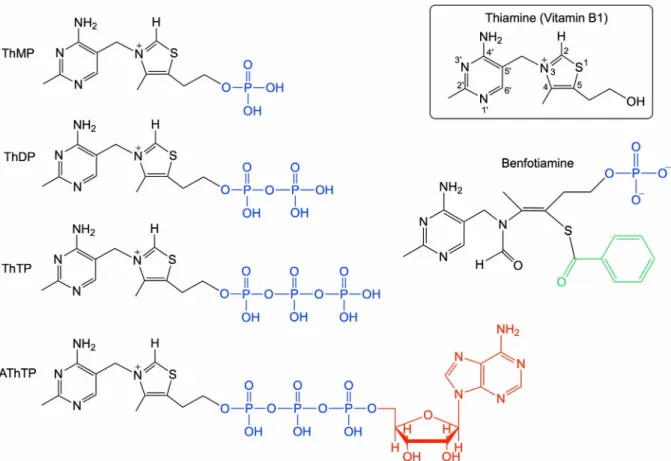
Thiamine (thiamin, vitamin B1), also formerly called aneurin (anti-neuritic factor), is a water-soluble vitamin of the B group. It is an indispensable molecule for intermediate metabolism in all life forms. Or at least it was considered as such until it was shown that the spirochaete Borrelia burgdorferi, responsible for a form of Lyme’s disease, does not require thiamine (Zhang et al., 2016). Such exception should however remain extremely rare and restricted to a few parasitic organisms. This is mainly because its diphosphorylated derivative (thiamine pyrophosphate or diphosphate, ThDP, see Figure 1) is required as a cofactor for central metabolic reactions such as the oxidative decarboxylation of pyruvate or oxoglutarate, key steps in energy metabolism. Thiamine is synthesized in microorganisms and plants but animals, including humans, have to rely on exogenous dietary sources.
As can be expected from the fundamental role of ThDP in metabolism, thiamine deficiency in animals has very harmful consequences, especially on the nervous system and the heart. This is, in part, because these oragns heavily rely on glucose oxidation for their energy needs, and also because thiamine absorption is rather slow in these cells.
In humans, chronic nutritional thiamine deficiency is responsible for beriberi, a disease causing polyneuritis and paralysis. Beriberi was a major health problem in East Asian countries until the beginning of the 20th century, because the major food source there was polished rice, which contains very little thiamine (Carpenter, 2012). Nowadays, the most common disease linked to thiamine deficiency is Wernicke-Korsakoff syndrome, an encephalopathy generally associated with chronic alcoholism. In developed countries, Korsakoff’s psychosis remains the third cause of dementia (Abdou & Hazell, 2014). In many low and middle-income countries, thiamine deficiency can be a largely underdiagnosed and sometimes fatal diease, especially in infants (Johnson et al., 2019). Subclinical thiamine deficiency is probably more common than generally thought, especially in elderly people and patients with diabetes or AIDS (Butterworth, 2003).
2. Chemical properties of thiamine
The structure of thiamine (Figure 1) is quite unusual as it consists of a pyrimidine moiety (2-methyl-4-amino-pyrimidine) and a thiazolium moiety [5-(2-hydroxyethyl)-4-methylthiazolium] connected through a methylene bridge. The aromatic heterocycle thioazole is rarely found in biological molecules. In living cells, the hydroxyethyl group is generally esterified to form phosphorylated derivatives, the most widespread being the cofactor ThDP.
<Figure 1 near here>
Thiamine and its phosphorylated derivatives are very polar and hydrophilic molecules: one gram of thiamine chloride hydrochloride (C12H17ClN4OS.HCl, molecular mass 337.27 g mol-1) dissolves in 1 ml of water. Thiamine is poorly soluble in organic solvents, including ethanol. It is heat-labile and is partially lost upon cooking. Aqueous solutions are most stable between pH 2 and 4. At pH < 4, thiamine is dicationic, as the N-1’ of the pyrimidine ring (but not the –NH2) is protonated. In alkaline solution, thiamine is unstable: in the pH range 8 to 10, hydroxyl attack on C-2 of the thiazolium ring leads to the relatively slow formation of a “pseudobase” intermediate (compound II in Figure 2) and the subsequent fast opening of the thiazolium ring. This yields a thiolate form (III) that can lose a water molecule to form the so-called “yellow” form (IV) (Hopmann, 1982).
<Figure 2 near here>
Note that, even in relatively strong alkaline medium, the deprotonated carbon 2 or ylide carbanion (which plays a key role in catalysis by ThDP-dependent enzymes, see below) is never observed in aqueous solutions of thiamine, because the estimated pKa of the CH in question is too high (in the range from 14 to 20). Nonetheless, a relative lability of the C-2 proton is demonstrated by the observation that it can be rapidly exchanged with deuterium.
3. Thiamine biosynthesis and riboswitches
Thiamine biosynthesis occurs in prokaryotes, fungi and plants through complex pathways. The thiazole and pyrimidine moieties are synthesized separately before assembling to form ThMP, which is either hydrolyzed to thiamine or phosphorylated to ThDP (Begley, Ealick, & McLafferty, 2012).
Thiamine-degrading enzymes (thiaminases) exist in many organisms. Thiaminase I (EC 2.5.1.2), a pyrimidine transferase, is present in microorganisms, but also in some higher multicellular eukaryotes such as fern, shellfish and fish. Consumption of raw parts of these organisms by animals or man can lead to serious thiamine deficiency syndromes, such as cortical necrosis in ruminants and beriberi in humans. Thiaminase II (EC 3.5.99.2), a hydrolase, might not be involved in the breakdown of thiamine as previously thought, but rather in the hydrolysis of aminopyrimidine to hydroxypyrimidine, a building block for the biosynthesis of thiamine by some bacteria (Jenkins, Schyns, Potot, Sun, & Begley, 2007).
Thiamine biosynthesis is regulated by metabolite-sensing mRNAs, called riboswitches (Padhi, Pradhan, Bung, Roy, & Bulusu, 2019). These small RNA sequences are typically located at the 5’-untranslated region of a mRNA molecule, coding for a protein involved in a biosynthetic pathway. Riboswitches have been identified for vitamins, amino acids and nucleotides. The binding of such small molecules leads to a conformational change of the mRNA, affecting protein expression. The ThDP riboswitch (Thi-box) which is found in many bacteria, fungi and plants, is the most widespread riboswitch known today. It acts by various mechanisms. In E. coli, ThDP binding to the Thi-box leads to the formation of a terminator hairpin, halting the transcription of several thiamine biosynthetic enzymes. In fungi and plants the Thi-box is involved in the control of mRNA splicing rather than transcription or translation. The ThDP riboswitch is the only riboswitch so far described in eukaryotes. The existence of riboswitches, in addition to ribozymes, demonstrates that macromolecules other than proteins are able to bind small
molecules with high specificity, supporting the RNA World hypothesis.
4. Determination of thiamine derivatives and their distribution in various living organisms
Early methods for the determination of thiamine in biological or pharmaceutical preparations included paper, thin-layer or liquid chromatography. Nowadays, mainly high-performance liquid chromatography (HPLC), after derivatization of thiamine compounds to the corresponding highly fluorescent thiochrome derivatives, is used because of its sensitivity, selectivity and resolution (Figure 3). All thiochrome derivatives have approximately the same excitation optimum at 360 nm and the same emission maximum at 430 – 435 nm (Kawasaki, 1992).
<Figure 3 near here>
In most eukaryotic tissues, the cofactor ThDP is the most abundant thiamine compound. Free thiamine and ThMP generally amount to 5 - 15 percent of total thiamine, while ThTP, AThTP and AThDP are even less abundant (but exceptions exist, see below) (L. Bettendorff et al., 2007). In animal cells, such as neurons, ThDP is found in highest amounts in mitochondria, where it is mostly bound to pyruvate and oxoglutarate dehydrogenase complexes. Smaller amounts are found in the cytosol (partly free and partly bound to transketolase). In liver and skeletal muscle, however, ThDP is mostly cytosolic. ThDP is also present in peroxisomes, where it is bound to 2-hydroxyacyl-CoA ligase (Fraccascia, Casteels, De Schryver, & Van Veldhoven, 2011). The triphosphorylated derivatives ThTP and AThTP are generally minor compounds in animal tissues, but ThTP has been consistently found in most organisms, from bacteria to mammals (Makarchikov et al., 2003). The subcellular distribution of ThTP in animal cells varies with the tissue studied. In rodent brain, it is essentially localized in mitochondria, while it is mainly cytoplasmic in skeletal muscle. Those differences are probably related to different mechanisms of ThTP synthesis (adenylate kinase or ATP-synthase, see below). In E. coli, ThTP is not found under optimal growth conditions, but it can accumulate significantly (> 20% of total thiamine) during amino acid starvation (Lakaye, Wirtzfeld, Wins, Grisar, & Bettendorff, 2004).
AThTP is found in several tissues (mammalian liver and heart, etc …) but it is not as widespread as ThTP. In E. coli, it is produced in response to carbon starvation. As to AThDP, it was only found in low amounts in rodent liver and in E. coli (Frédérich et al., 2009).
5. Thiamine transport
Thiamine is found in most food, but whole grains, liver and yeast are particularly rich sources. The Reference Daily Intake is 1.4 mg but depends on carbohydrate intake. Large excess oral intake does not seem to have adverse effects. However, allergic reactions (anaphylactic shock) are a rare complication of intravenous thiamine administration.
Thiamine phosphate esters taken up from the food are hydrolyzed to thiamine in the small intestine, transported as thiamine or ThMP across the brush-border membrane to the portal system, and distributed to the various tissues. There is no true thiamine storage reservoir in the body, though, because of its size and high ThDP content, the liver contains 10 - 20% of total body thiamine. Hence, in
case of reduced thiamine intake, liver ThDP can be hydrolyzed to free thiamine, which can be released and transported via the blood flow to other tissues (in particular the brain) (Volvert et al., 2008). Excess thiamine and some of its metabolites (2-methyl-4-amino-5-pyrimidine carboxylic acid, 4-methyl-thiazole-5-acetic acid) are excreted in the urine. Three kinds of cell membrane thiamine transporters have been described. They all belong to the SLC19A solute carrier gene family. SLC19A1 encodes a reduced folate transporter that may also transport ThMP and ThDP, but not thiamine. THTR-1 (product of the SLC19A2 gene) and THTR-2 (product of the SLC19A3 gene) are thiamine/H+ antiporters, rather ubiquitously expressed in mammalian tissues (Manzetti, Zhang, & van der Spoel, 2014).
Thiamine transporters (especially THTR-2) have a high affinity for thiamine but thiamine transport across cell membranes, particularly the blood-brain barrier, is a relatively slow process counter-balanced by a significant efflux (Lockman, Mumper, & Allen, 2003). Furthermore, plasma levels of thiamine in humans are relatively low compared to other mammalian species (Gangolf et al., 2010). Both factors probably contribute to the fact that marginal thiamine deficiency is more common than initially thought. In order to overcome severe deficiencies, several thiamine precursors (i.e. thiamine disulfides, benfotiamine) with a higher bioavailability were developed. These molecules are generally lipophilic (or in the case of benfotiamine yield a lipid-soluble metabolite), easily crossing the membranes, and their administration results in a rapid and important increase in plasma thiamine levels (Volvert et al., 2008).
6. Metabolism of thiamine phosphates
Thiamine phosphate derivatives can be readily interconverted, but the enzymes involved in these reactions remain poorly characterized (Figure 4). Only two of them, the essential thiamine pyrophosphokinase and the 25-kDa thiamine triphosphatase have been characterized at the molecular level.
<Figure 4 near here>
6.1. Synthesis of ThDP
Thiamine taken up by cells is converted to ThDP by a ubiquitous cytosolic ATP: thiamine diphosphokinase or thiamine pyrophosphokinase (ThDPK or TPK, EC 2.7.6.2). The enzyme has been purified from various prokaryotic and eukaryotic sources, and the molecular characterization of mouse and human TPK has been achieved (Nosaka et al., 1999). The mammalian enzyme consists of 27 kDa subunits associating as dimers in solution. Thiamine is bound at the dimer interface with high affinity (Km for thiamine around 0.2 µM), while the Km for ATP is in the millimolar range. Recent evidence suggests that the equilibrium of the reaction Thiamine + ATP ThDP + AMP is strongly in favor of ThDP production. However, due to regulatory mechanisms, the cytoplasmic ThDP does not accumulate (unpublished results from authors’ laboratory).
6.2. Synthesis of ThTP
The mechanism of ThTP synthesis and its physiological role are very controversial. The first mechanism proposed, a reaction catalyzed by a soluble ThDP-kinase: ATP + ThDP ADP + ThTP, was never confirmed. ThTP can be synthesized by adenylate kinases (EC 2.7.4.3) according to the reaction ADP + ThDP AMP + ThTP. The rate of reaction is 106-107 times slower than the synthesis of ATP according to the reaction 2 ADP AMP + ATP. Thus, synthesis of ThTP by this mechanism is significant only in tissues were adenylate kinase is very abundant, such as skeletal muscle, electric organs or red blood cells. ThTP may be the predominant thiamine compound when 25-kDa ThTPase is absent, such as in electric organs or chicken skeletal muscle. In brain mitochondria and E. coli, ThTP is synthesized according to the reaction ThDP + Pi ThTP, most probably by ATP-synthase, coupled to the respiratory chain through a chemiosmotic mechanism (Gigliobianco et al., 2013).
6.3. Synthesis of AThTP
AThTP can be synthesized by a soluble ThDP adenylyl transferase, according to the reaction ADP (ATP) + ThDP Pi (PPi) + AThTP. Though both ATP and ADP may be the phosphate donor for this reaction, ADP is probably more physiologic as AThTP is synthesized under conditions of energy stress in E. coli. This enzyme has been partially characterized in E. coli (Makarchikov, Brans, & Bettendorff, 2007).
6.4. Hydrolysis of ThDP and ThMP
Several phosphatases may hydrolyze ThDP, but until now no specific thiamine diphosphatase (ThDPase) or thiamine monophosphatase (ThMPase) has been characterized at the molecular level. ThDPases studied so far also hydrolyze nucleoside diphosphates. ThDPase activity has been used as a Golgi marker. ThMPase activity, a classical marker of small-diameter dorsal root ganglia neurons, is probably identical to the transmembrane isoform of prostatic acid phosphatase (Knyihar-Csillik, Bezzegh, Boti, & Csillik, 1986). This enzyme, in fact an ecto-5’-nucleotidase, hydrolyzes adenosine monophosphate to adenosine, which can then activates A1-adenosine receptors, suppressing pain.
6.5. Hydrolysis of ThTP
Membrane-associated and cytosolic thiamine triphosphatases (ThTPase, EC 3.6.1.28) have been described. The membrane-associated ThTPase could not be purified so far. In contrast, a soluble ThTPase, purified from mammalian tissues, has a nearly absolute specificity for ThTP. It is a 25-kDa protein with no homology with other known mammalian proteins (Lucien Bettendorff & Wins, 2013). Its mRNA is widely expressed in human tissues, though at relatively low levels. Orthologs are found in nearly all animals, except birds.
6.6. Thiamine-binding proteins
Thiamine-binding proteins have been described in many animal and plant tissues, but none was characterized at the molecular level. They exist in eggs or plant seeds, probably as thiamine storage proteins.
7. Cofactor role of ThDP
ThDP is an indispensable cofactor for energy metabolism in virtually all cell types. In animals, it is involved in several important enzyme reactions in carbohydrate and amino acid catabolism (Figure 5). Transketolase (TK, EC 2.2.1.1), a cytosolic enzyme, catalyzes a key step in the pentose phosphate pathway. In mitochondria, ThDP is required for oxidative decarboxylation of pyruvate and 2-oxoglutarate. Pyruvate dehydrogenase (PDHc, EC 1.2.4.1) and oxoglutarate dehydrogenase (OGDHc, EC 1.2.4.2) complexes catalyze two essential steps in citric acid metabolism. Oxoacid dehydrogenase (OADHc), only recently identified as different from OGDHc, is involved in lysine-derived 2-oxoadipate catabolism (Jordan, Nemeria, & Gerfen, 2019). ThDP is also required for the catabolism of branched-chain amino acids, being the coenzyme for the mitochondrial branched-branched-chain 2-oxoacid dehydrogenase (BCOADHc, EC 1.2.4.4). In peroxisomes, it is the coenzyme for 2-hydroxyacyl-CoA lyase (HACL1) (Foulon et al., 1999). In yeasts, ThDP is a cofactor for pyruvate decarboxylase (EC 4.1.1.1) catalyzing the non-oxidative anaerobic decarboxylation of pyruvate to acetaldehyde in alcoholic fermentation. ThDP is also the cofactor for several additional enzymes in plants and prokaryotes such as acetolactate synthase (EC 4.1.3.18) or pyruvate oxidase (EC 1.2.3.3) (Costelloe, Ward, & Dalby, 2008).
<Figure 5 near here>
8. Catalytic mechanism involving thiamine diphosphate
ThDP acts as catalyst for the decarboxylation of 2-oxoacids (-cleavage, Eq. 1) and the cleavage (or formation) of -hydroxyketones (Eq. 2):
There is no simple acid-base-catalyzed mechanism for such reactions, hence the need for a special coenzyme (Kluger & Tittmann, 2008).
It is now well established that the first committed step in ThDP-dependent enzyme reactions is the deprotonation of the thiazolium C-2 and attack of the resulting carbanion on a substrate carbonyl. As evidenced by the easy exchange of the C-2 proton with deuterium, this CH group is more acidic than most =CH– groups. Proton removal from C-2 is assisted by the adjacent positive charge (=N+–). In addition, the aminopyridinium moiety of ThDP, presumably involving the imino tautomer, acts as a general base. Finally, a strictly conserved glutamate residue of the apoenzyme forms a hydrogen bond with the weakly basic N-1’ of the aminopyridinium, also facilitating the dissociation of the H atom in C-2 position of the thiazolium (Nemeria, Chakraborty, Balakrishnan, & Jordan, 2009).
As already suggested by Breslow over 50 years ago, the thiazolium dipolar ion (or ylide) formed by this dissociation is the key intermediate in all ThDP-dependent enzyme reactions (Breslow, 1958). Note that the diphosphate group plays no role in catalysis and is only required for tight binding of ThDP to the apoenzymes. The anionic carbon of the thiazolium ring readily reacts with substrates by addition to the carbonyl group, forming a covalent adduct. As a typical example, we shall consider the mechanism of pyruvate decarboxylation that occurs as the first step in the conversion of pyruvate to acetyl-CoA by the PDH complex. This step is catalyzed by the E1 subunits. As shown in Figure 6, the first adduct formed is-lactyl-ThDP. In this complex, the =N+– group of the thiazolium acts as an electron sink to stabilize the formation of the negative charge, which is necessary for decarboxylation. After the release of CO2, the activated aldehyde attached to ThDP is oxidized to form an acetyl group and concomitantly transferred to lipoyl groups attached to the E2 subunits, while the ylide is regenerated (Figure 6). For comparison, the mechanism of non-oxidative pyruvate decarboxylation catalyzed by yeast pyruvate decarboxylase is also represented in figure 6. <Figure 6 near here>
One of best-studied ThDP-dependent enzymes is E. coli pyruvate dehydrogenase. Its mechanism can be considered as the paradigm for the oxidative decarboxylation of 2-oxoacids. This multi-subunit complex catalyzes the conversion of pyruvate to acetyl-CoA, which can then enter the Krebs cycle: Pyruvate + CoA + NAD+ acetyl-CoA + CO2 + NADH + H+
E. coli PDHc is a large complex ( 2390 kDa) composed of multiple copies of three distinct subunits: 24 subunits of pyruvate dehydrogenase (E1, EC 1.2.4.1), 24 subunits of dihydrolipoyl S-acetyltransferase (E2, EC 2.3.1.12) and 12 subunits of dihydrolipoyl dehydrogenase (E3, EC 1.8.1.4). Five different coenzymes (ThDP, lipoic acid, coenzyme A, FAD and NAD+) are involved in the reaction catalyzed by the complex. ThDP is the cofactor for E1, the enzyme catalyzing the decarboxylation of pyruvate and the transfer of the acetyl moiety to the lipoyl cofactor attached to E2. The structures and mechanisms of the OGDHc, OADHc and BCOADHc complexes are quite similar to those of PDHc. ThDP is bound to a conserved amino acid sequence GDG-X26-NN via a Mg2+ cation. This consensus sequence (ThDP-binding motif) is conserved in all ThDP-dependent enzymes (Hawkins, Borges, & Perham, 1989)
9. Non-coenzyme roles of thiamine derivatives
Non-coenzyme roles for thiamine and its phosphate derivatives have been proposed for many years, following the work of Alexander von Muralt in the 30s and 40s. This was based on the observation, made independently by several investigators, that electrical stimulation of isolated nerves led to release of thiamine. Later, several authors suggested non-coenzyme roles for thiamine either related to thiamine itself, hypothetical thiamine metabolites or thiamine triphosphorylated derivatives (L. Bettendorff & Wins, 2009)(Mkrtchyan et al., 2015)(Aleshin, Mkrtchyan, & Bunik, 2019).
9.1. Thiamine triphosphorylated derivatives
The hypothesis of non-coenzyme roles of thiamine drove interest to ThTP present in small amounts in nervous tissues and was sometimes considered as a “neuroactive form” of thiamine. However, interest in this compound rapidly faded as early results were questioned and because reliable detection proved to be difficult. In the early 80s, estimation of ThTP content in brain by HPLC showed that it was less abundant than previously thought, and an earlier hypothesis concerning its possible role in nerve excitation could not be substantiated. However, the discovery of the specific 25-kDa ThTPase with high catalytic efficiency in mammalian tissues (Delvaux et al., 2013), as well as the finding that ThTP can phosphorylate some proteins in brain and electric tissue (Nghiêm, Bettendorff, & Changeux, 2000) still suggests that ThTP may play some physiological role in brain and other animal tissues. In E. coli, it was recently found that ThTP accumulates in large amounts in response to amino acid starvation, suggesting a specific role in the adaptation of bacteria to nutritional downshifts. The discovery of an adenylated thiamine triphosphate derivative (AThTP, or thiaminylated ATP, Figure 1) in 2007 came as a surprise (L. Bettendorff et al., 2007). AThTP accumulates in E. coli during carbon starvation. This compound, like ThTP, might thus be a kind of alarmone, involved in emergency signaling when the cells undergo some forms of metabolic stress. In eukaryotic organisms, the possible biological role(s) of triphosphorylated derivatives (AThTP and ThTP) remains unknown.
9.2. Neuroprotective effects of benfotiamine
Thiamine precursors with higher bioavailabity were first developed to increase blood thiamine levels in patients suffering from thiamine deficiency. However, recent studies have shown that benfotiamine (Figure 1), one of the most widely used precursors, has neuroprotective effects in mouse models of neurodegenerative diseases such as Alzheimer’s disease (Pan et al., 2010) and tauopathies (Tapias et al., 2018). In those models, the transgenic mice used were not deficient in thiamine: the brain content of the coenzyme ThDP was normal and was not increased even after prolonged treatment with high doses of benfotiamine. Thus, the beneficial effects of treatment with this thiamine precursor do not seem to be mediated by effects on ThDP-dependent enzymes (which would result in stimulation of brain energy metabolism).
It should be noted that benfotiamine itself (a polar molecule) never reaches the brain parenchyma and that the neuroprotective effects observed are likely due to either thiamine or other unidentified products of benfotiamine metabolism (Sambon et al., 2019).
10. Human diseases related to thiamine deficiency
Poor diet or problems in absorption can lead to thiamine deficiency. Humans appear to have relatively low plasma and cellular thiamine levels and this may be why humans are particularly sensitive to thiamine deficiency. Severe chronic or acute deficiency leads to two main diseases, beriberi (cardiac and neurological forms) and Wernicke-Korsakoff syndrome. Infantile beriberi has a large specrum of, often nonspecific clinical features with neurological abnormalities but leading to death by heart failure (Abdou & Hazell, 2014). Because of the various, overlapping manifestations of thiamine deficiency through age and individuals, the Gates Foundation-Sackler Institute Thiamine Deficiency Consultation group suggested the use, in particular in infants, of the umbrella term Thiamine Deficiency Disorders (TDD) (Whitfield et al., 2018).
10.1. Beriberi
Beriberi is a polyneuritic syndrome, mainly affecting the lower limbs. Symptoms of “dry” beriberi include weakness and pain in the limbs, weight loss and partial paralysis. In “wet” beriberi, there are also cardiac symptoms, including cardiac insufficiency and enlargement, with tendency to edema. A low transketolase activity of red cells is a reliable diagnostic indicator of severe thiamine deficiency. Beriberi is the classical manifestation of chronic dietary deficiency in vitamin B1. Before the discovery of thiamine, it was a major health problem in East Asian countries, where polished rice (thiamine is mainly present in the husk) was the staple food.
10.2. Wernicke-Korsakoff syndrome
Although severe dietary thiamine deficiency has become rare in Western countries, a special form of deficiency associated with chronic alcoholism remains frequent. This is related to the poor nutritional status of many alcoholics and also to the fact that alcohol interferes to some extent with thiamine absorption and its intracellular conversion to ThDP.
The acute manifestation of the disorder is Wernicke’s encephalopathy, a state of confusion with neurological symptoms such as opthalmoplegia, nystagmus and ataxia. In the absence of prompt therapy with high-dose thiamine, coma and death occur rapidly. In many cases, recovery will not be complete and irreversible sequelae, due to specific brain lesions, will remain. These lesions often result in a state of dementia called Korsakoff’s psychosis. This is an amnesic syndrome, most often including a loss of anterograde memory. It may be accompanied by confusion, apathy, disorientation and fabulations. Surprisingly, the brain lesions are rather specific, mainly affecting thalamic nuclei and mammillary bodies, while (in contrast to Alzheimer’s disease) the cortex is largely spared. Brain damage in Wernicke-Korsakoff syndrome is assumed to be related to impairment of oxidative energy metabolism, neuronal death arising from a combination of excitotoxicity, oxidative stress and inflammatory processes (the latter may primarily affect endothelial cells of the microvessels rather than neurons). However, the reasons for the selective vulnerability of some brain regions remain elusive and are the subject of active investigation.
10.3. Thiamine deficiency in infants
Recently, it has been well documented that in many developing countries newborn infants are particularly at risk of thiamine deficiency, mainly as a results of maternal thiamine deficiency (Whitfield et al., 2018). As the symptoms are very unspecific in the first months of age (irritability, tachycardia, vomiting, crying…) it is often misdiagnosed, resulting in the progression of the disease to congestive heart failure, a major cause of death. In Cambodia for instance, the prevalence of thiamine deficiency in infants (aged 6-59 months) may be as high as 15% - 58%, depending on the cutoff values used. Among the hallmarks of nutritional thiamine deficiency is the rapid reversal of symptoms after thiamine administration. Hence, the importance of early diagnosis of TDDs, requiring reliable biomarkers of thiamine deficiency and cutoff values. Food fortification programs may play an essential role in the prevention of thiamine deficiency in many regions.
10.4. Marginal thiamine deficiency
In addition to the classical disorders beriberi and Wernicke-Korsakoff syndrome, marginal thiamine deficiency is probably quite common, in particular in high-risk groups such as elderly people, lactating women, diabetic and AIDS patients. People with Alzheimer’s disease also have lower blood (Pan, et al., 2016) and cerebral (Mastrogiacomo, Bettendorff, Grisar, & Kish, 1996) ThDP levels and impaired 2-oxoglutarate dehydrogenase activity in the brain (Mastrogiacomo, Bergeron, & Kish, 1993).
11. Defects in thiamine transport and in ThDP-dependent enzymes
Mutations in the genes coding thiamine transporters or ThDP-dependent enzymes may lead to specific diseases in humans.
11.1. Thiamine-responsive megaloblastic anemia
Thiamine-responsive megaloblastic anemia is characterized by megaloblastic anemia, sensorineural hearing loss, and diabetes mellitus. It is caused by mutations in the THTR-1 (product of the SLC19A2 gene) transporter. Relief of the symptoms is generally obtained by high-dose thiamine treatment.
Mutations in the human SLC19A3 gene coding for the high-affinity THTR-2 are responsible for biotin-thiamine responsive basal ganglia disease (Zeng et al., 2005). This a very rare recessive autosomal disorder with onset in the early to late childhood and with brain-specific pathology involving bilateral lesions of the caudate and the putamen. At an early stage, the disease is associated with subacute progressing to acute encephalopathy. The patients respond to biotin, and in some reports to the combination of biotin and thiamine (Alfadhel et al., 2013). Mutations identified in SLC19A3 inhibited the thiamine transport capacity of THTR-2. However the reason why these patients primarily respond to biotin administration remains elusive. It was thought that THTR-2 might transport biotin in addition to thiamine, but this was disproven (Subramanian, Marchant, & Said, 2006).
11.3. Amish lethal microcephaly
The children affected by Amish lethal microcephaly are born with a very small head and an underdeveloped brain, and they do not live beyond 6 month. The disease results from point mutations in the mitochondrial ThDP transporter gene SLC25A19, leading to its inability to transport the cofactor to the mitochondrial matrix (Lindhurst et al., 2006). This causes non-functional PDH and OGDH complexes and impairment of oxidative metabolism.
11.4. Leigh disease (Subacute necrotizing encephalomyelopathy)
Leigh disease is a rare heterogeneous neurodegenerative disorder of early childhood, involving focal, symmetric necrotic brain lesions, leading to death generally before the age of 5. It is clinically and genetically heterogeneous, presenting central and peripheral nervous system abnormalities that result from deficits in respiratory chain mitochondrial complexes (I, II, III, IV, V) or pyruvate dehydrogenase. In a small percentage of patients (< 10%), high-dose thiamine may have beneficial effects (Ortigoza-Escobar et al., 2016)
11.5. Maple syrup urine disease
Maple syrup disease (branched-chain ketoaciduria) is an autosomal recessive disorder caused by mutations in branched-chain 2-oxoacid dehydrogenase (Harris et al., 1990). The toxic effect is due to the accumulation of the branched-chain amino acids (leucine, isoleucine and valine) and their 2-oxoacid degradation products. If not treated by special diets, the patients will develop brain damage after birth, leading to death. High-dose thiamine therapy may be efficient, in particular when the E1 component of branched-chain 2-oxoacid dehydrogenase is affected.
12. Conclusion
It was long considered that the only reason for the existence of thiamine was to form the cofactor ThDP. Although it remains true that the main functions of this vitamin are linked to catalysis by ThDP, the existence of triphosphorylated derivatives such as ThTP and AThTP, whose levels are highly specifically
regulated, strongly suggests that these compounds play a biological role, at least in bacteria. Moreover, free thiamine or other non-phosphorylated derivatives may also play some physiological roles, especially in neuroprotection. Thiamine compounds form a family of molecules that, with respect to their degree of phosphorylation, is reminiscent of nucleotides (though thiamine, lacking a sugar moiety, is not a nucleotide). This suggests that in addition to the classical cofactor role of ThDP, some thiamine derivatives have non-cofactor roles.
Acknowledgement
L. Bettendorff is Research Director of the F.R.S.-FNRS
References
References should be in Harvard style and should include in-text citations. If an update of a previously published article, please replace any further reading with full references.
Abdou, E., & Hazell, A. S. (2014). Thiamine Deficiency: An Update of Pathophysiologic Mechanisms and Future Therapeutic Considerations. Neurochem Res.
Aleshin, V. A., Mkrtchyan, G. V., & Bunik, V. I. (2019). Mechanisms of Non-coenzyme Action of Thiamine: Protein Targets and Medical Significance. Biochemistry. Biokhimiia, 84(8), 829–850. https://doi.org/10.1134/S0006297919080017
Alfadhel, M., Almuntashri, M., Jadah, R. H., Bashiri, F. A., Al Rifai, M. T., Al Shalaan, H., … Al-Twaijri, W. (2013). Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet Journal of Rare Diseases, 8, 83. https://doi.org/10.1186/1750-1172-8-83
Begley, T. P., Ealick, S. E., & McLafferty, F. W. (2012). Thiamin biosynthesis: still yielding fascinating biological chemistry. Biochem Soc Trans, 40(3), 555–560.
Bettendorff, L., & Wins, P. (2009). Thiamin diphosphate in biological chemistry: New aspects of thiamin metabolism, especially triphosphate derivatives acting other than as cofactors. FEBS J., 276, 2917–2925.
Bettendorff, L., Wirtzfeld, B., Makarchikov, A. F., Mazzucchelli, G., Frédérich, M., Gigliobianco, T., … Wins, P. (2007). Discovery of a natural thiamine adenine nucleotide. Nat Chem Biol, 3(4), 211–212. Bettendorff, Lucien, & Wins, P. (2013). Thiamine triphosphatase and the CYTH superfamily of proteins. The FEBS Journal, 280(24), 6443–6455. https://doi.org/10.1111/febs.12498
Breslow, R. (1958). On the mechanism of thiamine action. IV.1 Evidence from studies on model systems. J Am Chem Soc, 80, 3719–3726.
Butterworth, R. F. (2003). Thiamin deficiency and brain disorders. Nutr. Res. Rev., 16(2), 277–284. Carpenter, K. J. (2012). The discovery of thiamin. Annals of Nutrition & Metabolism, 61(3), 219–223. https://doi.org/10.1159/000343109
Costelloe, S. J., Ward, J. M., & Dalby, P. A. (2008). Evolutionary Analysis of the TPP-Dependent Enzyme Family. J Mol Evol, 66(1), 36–49.
Delvaux, D., Kerff, F., Murty, M. R., Lakaye, B., Czerniecki, J., Kohn, G., … Bettendorff, L. (2013). Structural determinants of specificity and catalytic mechanism in mammalian 25-kDa thiamine triphosphatase. Biochim Biophys Acta, 1830(10), 4513–4523.
Foulon, V., Antonenkov, V. D., Croes, K., Waelkens, E., Mannaerts, G. P., Van Veldhoven, P. P., & Casteels, M. (1999). Purification, molecular cloning, and expression of 2-hydroxyphytanoyl- CoA lyase, a peroxisomal thiamine pyrophosphate-dependent enzyme that catalyzes the carbon-carbon bond cleavage during alpha-oxidation of 3- methyl-branched fatty acids. Proc. Natl. Acad. Sci. USA,
96, 10039–10044.
Fraccascia, P., Casteels, M., De Schryver, E., & Van Veldhoven, P. P. (2011). Role of thiamine
pyrophosphate in oligomerisation, functioning and import of peroxisomal 2-hydroxyacyl-CoA lyase.
Biochim Biophys Acta, 1814(10), 1226–1233.
Frédérich, M., Delvaux, D., Gigliobianco, T., Gangolf, M., Dive, G., Mazzucchelli, G., … Bettendorff, L. (2009). Thiaminylated adenine nucleotides. Chemical synthesis, structural characterization and natural occurrence. FEBS J, 276(12), 3256–3268.
Gangolf, M., Czerniecki, J., Radermecker, M., Detry, O., Nisolle, M., Jouan, C., … Bettendorff, L. (2010). Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS One, 5(10), e13616.
Gigliobianco, T., Gangolf, M., Lakaye, B., Pirson, B., von Ballmoos, C., Wins, P., & Bettendorff, L. (2013). An alternative role of FoF1-ATP synthase in Escherichia coli: synthesis of thiamine triphosphate. Sci Rep, 3, 1071.
Harris, R. A., Zhang, B., Goodwin, G. W., Kuntz, M. J., Shimomura, Y., Rougraff, P., … Crabb, D. W. (1990). Regulation of the branched-chain alpha-ketoacid dehydrogenase and elucidation of a molecular basis for maple syrup urine disease. Adv Enzyme Regul, 30, 245–263.
Hawkins, C. F., Borges, A., & Perham, R. N. (1989). A common structural motif in thiamin pyrophosphate-binding enzymes. FEBS Letters, 255(1), 77–82. https://doi.org/10.1016/0014-5793(89)81064-6
Hopmann, R. F. W. (1982). The alkali-induced transformations of thiamin. Ann NY Acad Sci, 378, 32– 50.
Jenkins, A. H., Schyns, G., Potot, S., Sun, G., & Begley, T. P. (2007). A new thiamin salvage pathway.
Nat Chem Biol, 3(8), 492–497.
Johnson, C. R., Fischer, P. R., Thacher, T. D., Topazian, M. D., Bourassa, M. W., & Combs, G. F. (2019). Thiamin deficiency in low- and middle-income countries: Disorders, prevalences, previous
interventions and current recommendations. Nutrition and Health, (in press). https://doi.org/10.1177/0260106019830847
2-Oxoadipate Dehydrogenase Both Generate Superoxide/H2O2 in a Side Reaction and Each Could Contribute to Oxidative Stress in Mitochondria. Neurochemical Research, (in press).
https://doi.org/10.1007/s11064-019-02765-w
Kawasaki, T. (1992). Vitamin B1: Thiamine. In A. P. De Leenheer, W. E. Lambert, & H. J. Nelis (Eds.),
Modern Chromatographic Analysis of Vitamins (2nd ed., pp. 319–354). New York: Marcel Dekker, Inc.
Kluger, R., & Tittmann, K. (2008). Thiamin diphosphate catalysis: enzymic and nonenzymic covalent intermediates. Chem Rev, 108(6), 1797–1833.
Knyihar-Csillik, E., Bezzegh, A., Boti, S., & Csillik, B. (1986). Thiamine monophosphatase: a genuine marker for transganglionic regulation of primary sensory neurons. J Histochem Cytochem, 34(3), 363–371.
Lakaye, B., Wirtzfeld, B., Wins, P., Grisar, T., & Bettendorff, L. (2004). Thiamine triphosphate, a new signal required for optimal growth of Escherichia coli during amino acid starvation. J. Biol. Chem.,
279(17), 17142–17147.
Lindhurst, M. J., Fiermonte, G., Song, S., Struys, E., De Leonardis, F., Schwartzberg, P. L., … Biesecker, L. G. (2006). Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion,
embryonic lethality, CNS malformations, and anemia. Proc. Natl. Acad. Sci. U S A, 103(43), 15927– 15932.
Lockman, P. R., Mumper, R. J., & Allen, D. D. (2003). Evaluation of blood-brain barrier thiamine efflux using the in situ rat brain perfusion method. J Neurochem, 86(3), 627–634.
Makarchikov, A. F., Brans, A., & Bettendorff, L. (2007). Thiamine diphosphate adenylyl transferase from E. coli: functional characterization of the enzyme synthesizing adenosine thiamine
triphosphate. BMC Biochem., 8, 17.
Makarchikov, A. F., Lakaye, B., Gulyai, I. E., Czerniecki, J., Coumans, B., Wins, P., … Bettendorff, L. (2003). Thiamine triphosphate and thiamine triphosphatase activities: from bacteria to mammals.
Cell. Mol. Life Sci., 60(7), 1477–1488.
Manzetti, S., Zhang, J., & van der Spoel, D. (2014). Thiamin function, metabolism, uptake, and transport. Biochemistry, 53(5), 821–835.
Mastrogiacomo, F., Bergeron, C., & Kish, S. J. (1993). Brain alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. J Neurochem, 61(6), 2007–2014.
Mastrogiacomo, F., Bettendorff, L., Grisar, T., & Kish, S. J. (1996). Brain thiamine, its phosphate esters, and its metabolizing enzymes in Alzheimer’s disease. Ann Neurol, 39(5), 585–591.
Mkrtchyan, G., Aleshin, V., Parkhomenko, Y., Kaehne, T., Luigi Di Salvo, M., Parroni, A., … Bunik, V. (2015). Molecular mechanisms of the non-coenzyme action of thiamin in brain: biochemical, structural and pathway analysis. Scientific Reports, 5, 12583. https://doi.org/10.1038/srep12583 Nemeria, N. S., Chakraborty, S., Balakrishnan, A., & Jordan, F. (2009). Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at
individual steps. Febs J, 276(9), 2432–2446.
Nghiêm, H. O., Bettendorff, L., & Changeux, J. P. (2000). Specific phosphorylation of Torpedo 43K rapsyn by endogenous kinase(s) with thiamine triphosphate as the phosphate donor. FASEB J, 14(3), 543–554.
Nosaka, K., Onozuka, M., Nishino, H., Nishimura, H., Kawasaki, Y., & Ueyama, H. (1999). Molecular cloning and expression of a mouse thiamin pyrophosphokinase cDNA. J Biol Chem, 274(48), 34129– 34133.
Ortigoza-Escobar, J. D., Molero-Luis, M., Arias, A., Martí-Sánchez, L., Rodriguez-Pombo, P., Artuch, R., & Pérez-Dueñas, B. (2016). Treatment of genetic defects of thiamine transport and metabolism.
Expert Review of Neurotherapeutics, 16(7), 755–763.
https://doi.org/10.1080/14737175.2016.1187562
Padhi, S., Pradhan, M., Bung, N., Roy, A., & Bulusu, G. (2019). TPP riboswitch aptamer: Role of Mg2+ ions, ligand unbinding, and allostery. Journal of Molecular Graphics & Modelling, 88, 282–291. https://doi.org/10.1016/j.jmgm.2019.01.015
Pan, X., Fei, G., Lu, J., Jin, L., Pan, S., Chen, Z., … Zhong, C. (2016). Measurement of blood thiamine metabolites for Alzheimer’s disease diagnosis. EBioMedicine, 3, 155–162.
https://doi.org/10.1016/j.ebiom.2015.11.039
Pan, X., Gong, N., Zhao, J., Yu, Z., Gu, F., Chen, J., … Xu, T. L. (2010). Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. Brain, 133, 1342–1351.
Sambon, M., Napp, A., Demelenne, A., Vignisse, J., Wins, P., Fillet, M., & Bettendorff, L. (2019). Thiamine and benfotiamine protect neuroblastoma cells against paraquat and ß-amyloid toxicity by a coenzyme-independent mechanism. Heliyon, (in press).
Subramanian, V. S., Marchant, J. S., & Said, H. M. (2006). Biotin-responsive basal ganglia disease-linked mutations inhibit thiamine transport via the human thiamine transporter-2 (hTHTR2): biotin is not a substrate for hTHTR2. Am J Physiol Cell Physiol, 291, C851–C859.
Tapias, V., Jainuddin, S., Ahuja, M., Stack, C., Elipenahli, C., Vignisse, J., … Beal, M. F. (2018). Benfotiamine Treatment Activates the Nrf2/ARE Pathway and is Neuroprotective in a Transgenic Mouse Model of Tauopathy. Human Molecular Genetics, 27(16), 2874–2892.
https://doi.org/10.1093/hmg/ddy201
Volvert, M. L., Seyen, S., Piette, M., Evrard, B., Gangolf, M., Plumier, J. C., & Bettendorff, L. (2008). Benfotiamine, a synthetic S-acyl thiamine derivative, has different mechanisms of action and a different pharmacological profile than lipid-soluble thiamine disulfide derivatives. BMC Pharmacol.,
8(1), 10.
Whitfield, K. C., Bourassa, M. W., Adamolekun, B., Bergeron, G., Bettendorff, L., Brown, K. H., … Combs, G. F. (2018). Thiamine deficiency disorders: diagnosis, prevalence, and a roadmap for global control programs. Ann. N. Y. Acad. Sci., 1430(1), 3–43. https://doi.org/10.1111/nyas.13919
Zeng, W. Q., Al-Yamani, E., Acierno, Jr., J. S., Slaugenhaupt, S., Gillis, T., MacDonald, M. E., … Gusella, J. F. (2005). Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3.
Am J Hum Genet, 77(1), 16–26.
Zhang, K., Bian, J., Deng, Y., Smith, A., Nunez, R. E., Li, M. B., … Li, C. (2016). Lyme disease spirochaete Borrelia burgdorferi does not require thiamin. Nature Microbiology, 2, 16213.
https://doi.org/10.1038/nmicrobiol.2016.213 Change History
Change History: October 2019. L Bettendorff and P. Wins updated Abstract, paragraph 6.1, and added parapgraphs 9.2 and the reference section. Paragraph 10 was totally rewritten. Figures 1, 5 and 6 were changed and updated.
Further Reading
Bettendorff, L. (2013). Thiamine. In J. Zempleni, J. Suttie, 3rd Gregory J. F., & P. Stover (Eds.), Handbook of Vitamins (5th ed., pp. 268–323). Boca Raton: CRC Press.
Bettendorff, Lucien, & Wins, P. (2013). Thiamine triphosphatase and the CYTH superfamily of proteins. The FEBS Journal, 280(24), 6443–6455. https://doi.org/10.1111/febs.12498
Carpenter, K. J. (2000). Beriberi, white rice, and vitamin B: a disease, a cause, and a cure. Berkeley, CA: University of California Press.
Fitzpatrick, T. B., & Thore, S. (2014). Complex behavior: from cannibalism to suicide in the vitamin B biosynthesis world. Curr Opin Struct Biol, 29C, 34–43.
Fitzpatrick, Teresa B., Basset, G. J. C., Borel, P., Carrari, F., DellaPenna, D., Fraser, P. D., … Fernie, A. R. (2012). Vitamin deficiencies in humans: can plant science help? The Plant Cell, 24(2), 395–414. https://doi.org/10.1105/tpc.111.093120
Gibson, G. E., Hirsch, J. A., Fonzetti, P., Jordan, B. D., Cirio, R. T., & Elder, J. (2016). Vitamin B1 (thiamine) and dementia. Annals of the New York Academy of Sciences, 1367, 21–30. https://doi.org/10.1111/nyas.13031
Kraft, C. E., & Angert, E. R. (2017). Competition for vitamin B1 (thiamin) structures numerous ecological interactions. The Quarterly Review of Biology, 92(2), 151–168.
Lonsdale, D. (2019). Thiamin and protein folding. Medical Hypotheses, 129, 109252. https://doi.org/10.1016/j.mehy.2019.109252
Lonsdale, D., & Marrs, C. (2017). Thiamine Deficiency Disease, Dysautonomia, and High Calorie Malnutrition. Academic Press.
Marcé-Grau, A., Martí-Sánchez, L., Baide-Mairena, H., Ortigoza-Escobar, J. D., & Pérez-Dueñas, B. (2019). Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes,
genetics, and functional studies. Journal of Inherited Metabolic Disease, 42(4), 581–597. https://doi.org/10.1002/jimd.12125
McCandless, D. W. (2010). Thiamine deficiency and associated clinical disorders (1st ed.). Humana Press, a part of Springer Science+Business Media, LLC.
Moretti, R., Caruso, P., Ben, M. D., Gazzin, S., & Tiribelli, C. (2017). Thiamine and Alcohol for Brain Pathology: Super-imposing or Different Causative Factors for Brain Damage? Current Drug Abuse Reviews, 10(1), 44–51.
Victor, M., Adams, R. D., & Collins, G. H. (1989). The Wernicke-Korsakoff Syndrome and related Neurological Disorders due to Alcoholism and Malnutrition. Philadelphia: F.A. Davies.
Walsh, C. T. (2019). Biologically generated carbon dioxide: nature’s versatile chemical strategies for carboxy lyases. Natural Product Reports. https://doi.org/10.1039/c9np00015a
Legends to Figures
FIGURE 1 Structural formulas of thiamine, natural thiamine phosphorylated derivatives and benfotiamine.
FIGURE 2 Conversion of thiamine to thiolate forms in alkaline solution. I, thiamine (cationic thiazolium form); II, unstable intermediate (pseudobase); III, thiolate form; IV, “yellow” weakly basic anion.
FIGURE 3 Conversion of thiamine compounds to the corresponding thiochrome derivatives by oxidation in alkaline medium by potassium ferricyanide or cyanogen bromide (Thc, thiochrome).
FIGURE 4 Interconversion of thiamine derivatives in a typical animal cell. 1, thiamine pyrophosphokinase (TPK); 2, thiamine diphosphatases (non specific); 3, thiamine monophosphatases (non specific); 4, mitochondrial ATP-synthase (ThTP synthesizing); 5, cytosolic adenylate kinase; 6, cytosolic 25-kDa ThTPase; 7, ThDP:ADP adenylyl transferase; 8, AThTP hydrolase (hypothetical).
FIGURE 6 Catalytic mechanism for the first step (oxidative pyruvate decarboxylation) of the reaction catalyzed by the PDH complex. Pyruvate decarboxylation is catalyzed by the E1 subunits of the complex that bind ThDP. The mechanism of non-oxidative pyruvate decarboxylation catalyzed by yeast pyruvate decarboxylase is also shown. The catalytic cycle starts with the formation of a ylide whose negatively charged carbanion will act as a nucleophile attacking the carbonyl carbon of pyruvate (1). Then there is formation of the unstable covalent intermediate -lactyl-ThDP (2) that is rapidly decarboxylated leading to a transfer of electrons towards the positively charged thiazolium quaternary nitrogen, resulting in a resonance-stabilized hydroethyl-ThDP carbanion/enamine intermediate, carrying the activated aldehyde (3). In the nonoxidative pathway, such as catalyzed by pyruvate decarboxylase, the carbanion is neutralized to form tetrahedral hydroxyethyl-ThDP (4) and acetaldehyde is released in the last step (5). In the oxidative pathway (pyruvate dehydrogenase), the activated aldehyde is transferred to lipoamide which is reduced while the activated aldehyde is oxidized (6) before transfer to CoA and formation of the thioester acetyl-CoA (7).
Glossary
Ataxia A neurological condition characterized by severe lack of movement coordination. Ataxia is a non-specific manifestation due to lesions in brain regions involved in movement coordination such as the cerebellum and the vestibular system.
Blood-brain barrier (BBB) The layers of epithelial cells separating the brain parenchyma and the cerebrospinal fluid from the blood. The BBB results from tight junctions between endothelial cells forming the walls of the capillaries. Therefore only molecules heaving specific carriers (glucose, vitamins, salts) or lipid soluble molecules (O2, CO2, alcohol, psychoactive drugs…) may cross the barrier, thus strongly limiting the transport of solutes from the bloodstream to the central nervous system. The BBB is essential for the protection and homeostasis of neurons and glia.
Encephalopathy A global dysfunction of the brain and altered mental state. Encephalopathies can have many different causes such as metabolic stress (due to inhibition of enzymes necessary for energy metabolism, mitochondrial dysfunction, liver failure), infectious agents (prion, bacteria, virus), toxins (drugs, industrial chemicals)
Excitotoxicity A pathological process during which receptors for excitatory amino acid
neurotransmitters, in particular the NMDA-type glutamate receptors, are stimulated above normal levels, leading to increased entry of Ca2+ into neurons. Overstimulation of various Ca2+-dependent processes may then lead to neuronal death. This occurs in ischemia, neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases and Wernicke-Korsakoff syndrome.
Nutritional downshift When bacteria are transferred from a rich medium (optimal for growth) to a medium lacking one or several nutrient categories (phosphates, nitrogen, carbon, amino acids…), they respond by producing signaling molecules (alarmones) redirecting the cell metabolism to cope with the induced starvation stress.
Oxidative stress A pathological process during which reactive oxygen species (such as O2•-, HO•, H2O2) are generated. Production of these species itself is a normal physiological process in aerobic
organisms which possess cellular defense mechanisms against these radical or radical-generating molecules. However, in the presence of either environmental factors (toxins, pollutants) or
pathological processes (ischemia, neurodegenerative diseases, infection) the production of reactive oxygen species, which can attack cellular constituents such as proteins, lipids and nucleic acids, can overwhelm cellular defense mechanisms leading to oxidative stress causing inflammation and possibly mutations. Such mechanisms are thought to play a major role in cancer, neurodegenerative
diseases, cardiovascular diseases, rheumatoid arthritis and aging. Antioxidant molecules (vitamin C, vitamin E, cysteine, ß-carotene, phenolic acids, curcuma, tannins) may help combat oxidative stress.
Polyneuritis or peripheral neuropathy A damage of the nerves of the peripheral nervous system caused by trauma (compression), metabolic illness (vitamin deficiencies, diabetes mellitus), genetic illness (Friedreich's ataxia, Charcot-Marie-Tooth syndrome), toxins (heavy metals) or inflammatory disease (Guillain-Barré syndrome). Polyneuritis generally translates into weakness of the limbs (legs and feet) and sensory modifications.
Ylide (or ylid) A molecule wearing opposite charges on adjacent atoms. The negative charge is always on a carbon and it is therefore a particular kind of carbanion. Ylides often appear as reactive