Journal o.fNeurochernistry
Lί ppί n cott-R aven Pu blis h e r s, Ph iladelph ia 1995
10 International SocietyforNeuroch emistry
Th
ia
min
e Deficie
n
c
y
-
Indu
ce
d P
a
r
tia
l N
ec
r
os
i
s a
nd
M
itoc
h
o
ndri
a
l Un
co
uplin
g i
n N
e
ur
ob
l
asto
m
a Ce
ll
s
Ar
e
R
a
p
i
dly R
e
v
e
r
se
d
b
y Add
it
i
o
n
o
f Th
ia
min
e
L.
B
ettendodf,*
F.
S
luse, t G. Goesse n s, Ρ.W
ins, and Τ.G
risar Laboratories of Neurochemistry and ϊCellularand Tissular Biology, University (Y'Liège,L
iège;and
*
Laboratory ι#'Bioenergetics, University ofLiège, Sart-Tilman, BelgiumAbstract : Culture of neuroblastoma cells in α med ium of low-thiamine concentration (6 ηΜ) and in the presence of the transport inhibitor am prolium leads to the ap pear-ance of overt signs of necrosis; i.e., the chromatin con-denses in dark patches, the oxygen consumption de-creases, mitochondria are uncoupled, and
t
heir cristae are disorganized . Glutamate formed from glutamine is no longer oxidized and accumulates, suggesting that the thiamine diphosphate-dependent α-ketoglutarate deh y-d rogenase activity is impaired. When thiamine (10 μΜ) is added to the cells, the ΟΖ consumption increases, res-piratory control is restored , and normal cell and mito-chondrίal morphology is recovered within 1 h.Succinate, which is oxidized via the thiamine diphosph ate-indepen-dent succinate dehydrogenase, is also able to restore α normal ΟΖ consumption (with respiratory control) in dig-itonin- permeabilized thiamine-deficient cells. Our results therefore suggest that the slowing of the citric acid cycle is the main cause of the biochemical lesion induced by thiamine deficiency as observed inWernίcke's enceph a-lopathy. Key Words:Thiamine deficiency-Necrosis-Neuroblastoma cells- Mitochond ria-Wernicke's en-cephalopathy.
J.
Neurochem. 65, 2178-2184 (1995) .Thiamine diphosphate (TDP), which is the main
thiamine compound in the mammalian brain, is α r e-quired cofactor for pyruvate and u-ketoglutarate dehy-drogenases. Therefore, thiamine deficiencyshould lead to α decreased turnover
o
f the intermediates of the citric acid cycle and, thus, slow down oxidative metab-olism (for review, see Butterworth , 1993) . TheW
er-nicke-Korsakoff syndrome is α common consequence of thiamine deficiency linked to alcohol abuse in hu-mans (Katzman and Terry, 1983) .W
ernicke's enceph-alopathy is characteri zed by ophthalmoplegia, ataxia, global confusional state, and delusions (Victor et α1 ., 1989) . Polyneuropathies are also observed and, at α later stage, there appears α dementia characterized by anterograde amnesia and disorientation ( Korsakoff 's2178
psychosis ;
M
cEntee and Mair, 1990) . Although sev-eral symptoms ofW
ernicke's encephalopathy (oph-thalmoplegia) are al most fully reversible after adminis-tration of thiamine, others (ataxia) are only partially reversible and theK
orsakoff psychosis generally does not respond to thiamine . These thiamine-nonrespon-sive symptoms probably involve irreversibleneuronal lesions . The reasons for theneuronal loss observed in severely thiamine-deficient (TD) brai n are probably multiple and involve impairede
nergy metabolism (Ai-kawa et α1 ., 1984), acidosis (Hakim and Pappius, 1983), and excitotoxic phenomena ( Langlais and Μαίr, 1990 ; Hazell et α1 ., 1993) .Since the early work of Peters (for review, seeP e-ters, 1936), it is known that the early symptoms of thiamine deficie ncy in birds are rapidly reversible on thiamine administration.
M
oreover, the oxygen con-sumption was lower in minced brains prepared from avitaminous compared with controlpigeons and it was partially restored on addition ofthiamine in vitro. Oxi-dative decarboxy lationo
f pyruvate and α-ketoglutarate was decreased in isolated mitochondria prepared fromthe brains of pyrίthiam ine-treated rats compared with control animals (Gubler, 1961 ; B ennett et α1., 1966) .
N
ormal activity could berapidly restored after addition of TDP to the mitochondrial preparation (Gubler, 1961) . Parker et α1. ( 1984) reported similar observa-tions in mitochondria isolatedf
rom TDrat brain. State 3 respiration as well as respiratory control was de-creased with substrates such as glutaιnate, α-ketogluta-rate, orc
itrate, butnot withsuccinate, which is metabo-lized independently of TDP-dependent enzymes. Par-ReceivedFebruary 6, 1995; revised manuscript received May 15, 1995 ; accepted May 15, 1995.Address correspondence and reprintrequests toDr.L.Bettendorff at University of Liege, Laboratory of Neurnchemί stry, 17place De-cour, Β-4020 Liege,Belgium.
Abbreviations used: CCCP, carbonyl cyanideιη -chlornphenylh y-drazone; TD, thiaιnine-deficient ; TDA, thiamίne-deficient and treated withamprnlium; TDP, thiamine dίphosphate.
THIAMINE DEFICIENCY AND BIOCHEMICAL
L
ESION ker et α1 . ( 1984) consideredt
hat this situation couldlead to abnormal glutamate levels, which might favor the appearance of excitotoxic lesions.
In cultured neuroblastoma cells, α
r
eduction of the thiamine concentration in the extracellular τnedium leads, withind
ays, to an important loss of intracell ularthiamine compounds, but the cells
k
eep αn
early ηοr-ιηαΙ oxygen consumption ( Bettendorff et α1., 1995) . A ddition of amproli um (αt
hiamine transport inhibitor ) to the culturem
edium leads to α furtherd
ecrease in intracell ulart
hiaιnine, decreased respiration, mito-chοndrial swelling and uncoupling, lactate production, and cell death . The aimo
ft
he present study was to investigate the reversibility of those changes after resti-tution of thiamine to the culture medium.M
ATERIALS AND METHODSChemicals
Thiamine, carbonyl cyanide
m
-chlorophenylhydιazone (CCCP), amprolium,r
otenone, digitonin, and oligomycinwere purchased from Sigma. Cell culture
Νeιιrοb1 αstπniα cells were cultured as previously de-scribed ( Beuendorff and Wins, 1994) in
D
ιιlbecco's modi-Γied Eagle's medium (GIBCO, Ghent,B
elgium) containing ΙΟ μΜthiamine and supplemented with 5%ο fetal calf serum (GIBCO) . TD cells were produced by growing them for at least 2 weeks in αD
ulbecco's modified medium devoid of thiamine . Under these conditions, the only thiamine source was the fetal calf serum and its concentration in the medi um was -7 ηΜ. To further increase thiamine deprivation, am-ρrπΙίιιm (20 μΜ) was added to the Culture medium of TD cells 4 days before the experiment ( "TDA cells" ), asde-by
B
ettendorff et α1. (1995) .Thiamine derivatives were determi ned by an HPLC proce-dure exactly as previously described (Bettendorff et α1., 1991 ) . Protein concentrations were determined by the method of Peterson ( 1977) .
Amino acids were extracted
f
romthe cells asd
escribed by Patel and Hunt ( 1985) and determined by HPLC ( Tapuhi et al ., 1981 ) .Oxygen consumption
Oxygen uptake was measured polarographicαlly in α 2-ml cc[] at 37°C as described by Vayssière et al. ( 1986), con-taining - ΙΟ-20 Χ 10` cells in their respective culture me-dia. Digitnnin pernιeabilization was
p
erformed as describedby Vercesi ct α1 . ( 1991 ) .
E
lectron microscopyM
onolayer cell cultures were scraped offthe dishes and centrifuged at 350 g for 3 min. Small fragments of the pellet were fixed at 4°C in glutαraldehyde (2 .5% in cacodylate hυ ffer) and then pοstfixedin 1 %ο osmium tetrnxide solution. The cells were included in Εροη .U
ltrathin sections mounted on copper grids were stained with uranyl acetate and lead citrate before examination under αJeol CX 100 11 electronmicroscope at 60 kV .
RESULTS AND DISCUSSION
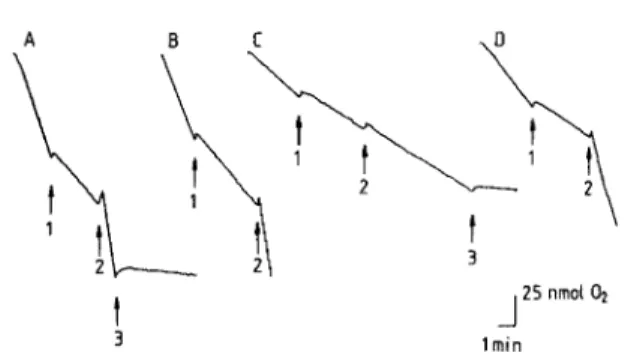
Figure 1 shows the
r
ate of oxygen consumptionb
y neιιroblasto ιna cells under different experimentalcon-2179
FIG. 1. Polarographic recording of oxygen consumption by neu-roblastoma cells in whole culture media containing various amounts of thiamine (Α), TD medium (Β), TDA medium (C), and TDA cells after addition of 10 μΜ thiamine for 60 min (D) . In the case of TDA cells, amprolium (20 μΜ) was added to the culture days before the experiment . The arrows correspond to the addi-tions of various compounds: 1, oligomycin (16 μg/ml) ; 2, CCCP
(5 μΜ) ; 3, KCN (1 mM).
d
itions. The comτnercialm
edium alway s contained 30 mM gl ucose, 1 ιηΜ pyruvate, and 4 ιηΜ glutamine. 1η each case, we firstm
easuredt
heb
asal oxygen con-sumption . Then, the rate of Ο, concon-sumption was esti-mated i n the presence of added oligomycin, followed by theu
ncoupler CCCP. In the case of cells grown at α high thiamine concentration ( ΙΟ μ,M), a reasonablyh
igh 02 consumption wasm
easured. As expected, oli-gomycin decreased oxygen consumption, whereas in the presence of CCCP it was increased about threefold aboveb
asal level . Oxygen consumption was com-pletely inhibited aftera
ddition of 1 mMK
CN. Α simi-lar pattern was observed with cells grown at α lower thiamine concentration (7 ηΜ ) . Undert
hose condi-tions, total intracellular thiamine content wasd
e-creased from 210 to 13 ρmο1 /mg of protein ( Betten-dorff et α1., 1995) . To further decrease intracellular thiamine concentration, amprolίum (20 μM), α com-petitive inhibitor oft
hiaιnine transport, was added to the TD culturem
edi um ( TDA) for 4d
ays. Pyrithi-amίne is them
ostp
otentk
nown th iamine antίmetabo-lite in vivo;b
ut in this study wep
referred to use am-ρrοΙίυm, though -30-fold highera
mounts of th is com-pound arer
equired to block thiamine transport ( Bettendorff and Wi ns, 1994) . In contrast top
yrίthia-mine, amprolium cannot be phosphoryl αted. Thus, no i nterference with TDP-dependent enzy mes is tob
e ex-pected witha
mprolium.U
nder these conditions, theb
asalr
espi ration was considerablyd
ecreased compared with the untreated cells . Furthermore, oligomycin and CCCP were without effect onr
espiration , suggesting uncoupling of mitochondria.T
hatK
CN (as well as roteno ne; not shown ) still inhibited oxygen consump-tions
uggests that, even under these severeT
D condi-tions, part oft
her
espiration persists and the most con-spicuous effect of thiamined
eficiency is the apparentm
itochondrί alu
ncoupling . When thiamine was added to the TDA cells 60m
inb
efore thep
olarographic deter-.1.Νιυrηι hon- V ιι1. 05, No. 5. 19952180
FIG. 2. Time-dependent recovery of oxygen consumption in TDA cells after addition of 10 μΜ thiamine. Basal oxygen con-sumption ( ι ) ; oxygen concon-sumption in the presence of oligomy-cin (Ο) or CCCP (Ο) . Each point represents the mean ± SD
value for th ree experiments except for the points in the presence of oligomycin, which are the mean values of two experiments.
m
inationo
f oxygen consumption, α nearly completer
ecovery of mitochondrial function was observed; i.e., the basal oxygen consumption increaseda
ndt
he usual effects of oligomycina
nd CCCPr
eturned, suggesting α recoupling of them
itochondria.F
igure 2 shows the time scale of ther
ecovery of mitochondrί alr
espiration. It can be seen that, already 5 mina
ftera
ddition of thiamine, α significant increase in theb
asal and uncoupledr
espiration was observed andt
he effect was complete after 60m
in. Ther
espira-tion in the presence of oligomycinr
emainedun-changed.
Thiamine by itself
h
as no knowne
ffect onm
itochon-dria)r
espiration andt
he important compound is the cofactor TDP. Thiami ne is,h
owever, actively trans-ported inton
euroblastoma cells and pyrophosphory-lated in the cytoplasm ( Bettendorff and Wins, 1994) . TDP is then transported into mitochondria ( Barile et al ., 1990) where it binds to the pyruvate and α-ketoglu-tarate dehydrogenases. Αn
early 10-fold increase in intracellular TDP is observed within 1 h aftera
ddition of thiamine to the cells (Fig. 3) . The lagp
eriod mayb
e explained by the fact that thiamine pyrophosphoki-nase is dependent on the intracellular ATP concentra-tion with an excepconcentra-tionallyh
igh Κ~, of 7 mM ( Betten-dorff and Win s, 1994) . In TDA cells, intracellular ATP concentrations are lowered by -50%ο compared with normal cells ( Bettendorff et α1., 1995) . Thus, ther
ate of thiamine phosphorylation is slowb
ut increases as mitochondria are producing ATP.We
h
ave previously reported the existence of abnor-malm
itochondria in TDA cells (Bettendorff et α1 ., 1995) . Witho
ngoing thiamined
eficiency, the mito-chondria) matrix became disorgani zed ande
lectront
ranslucent; no intact cristaer
emai ned visible and some mitochondriab
ecame abnormally large.H
owever, wed
i d not observe any significant increase in the number of mitochondria inT
DA cells comparedw
ith control cells .M
itochondria)r
espiration inT
DA cells was es-sentially uncoupleda
nd, as α preincubation witht
hia-mine leads to αr
ecoupling of respiration, we wanted to / . Νe ιι rιιι h e ιιτ ., Vπ /. 65, No. 5, 1995L.
BE
ΤTE
NDORFF
ETAL.
know whether
t
his was accompaniedb
y morphological modifications . Figure 4α and bs
how, with differentm
agnifications, αt
ypical TDA cell with largelye
lec-tron-translucent mitochondria.T
he nucleus shows evi-dent signs of chromatin condensationt
ypical oft
he early abnormalities of necrosis (Wyllie et al ., 1980) . After 1 h int
he presence of 10 μΜ thiamine these abnormalities were essentially reversed ( Fig. 4c and d) ; them
itochondria became electron dense andc
ristae werer
eformed. Them
itochondriar
esembled those of control cells ( Bettendorff et al., 1995) . Note that the cell cycle in neuroblastoma cel ls lasts -υ 24 ha
nd, thus, normalization in cell morphology, within Ιh,
cannot be explainedb
y the generationo
f new cell s through mitoticd
ivision.The
r
easonsr
espiratory control is lost in TDA cells are not clear. The link between uncoupling and the di sorganizationo
fc
ristae maya
ppear obv ious, as the electrochemical proton gradient tends to dissipate when the inner membranes ared
amaged;h
owever, ther
easons the cristaeb
ecomed
isorganized are unclear. Wem
ay considert
he possibility that the phenomenon isr
elated to the lacko
fs
ubstrates able to donate elec-trons to ther
espiratory chain. If thish
ypothesis is true, the direct addition of α permeant substrate shouldr
e-verseu
ncoupling in αm
annera
nalogous to thiamine addition,m
aybe even faster. As shown inF
ig. 5, the additiono
fs
uccinate to cellsp
ermeabili zed with dig-itonini
ndeed restored coupledr
espiration . Aftera
dd i-tion of digitonin toT
DA cells, the oxygen consumptiong
raduallyd
ecreased, as αr
esult of dilution of ther
e-maining substrates . Addition of succinate increased ther
espiration up to sixfoldi
n some experiments. This ΟΖ consumption was slightlyi
nhibitedb
y oligomycin and increasedb
y CCCP as expected. Theu
ncoupledr
espi-ratory control, i .e., ther
atio of Ο, con sumption int
hep
resence of CCCP to the Ο, consumption in the pres-ence ofo
ligomycin, was 1 .8 -+- 0.5 . This uncoupledr
espiratory control value was not significantlyh
igher when normal (instead of TDA) cells wereu
sed.R
ote-none, an inhibitor of respiratory chain complex Ι, was without effect on ΟΖ consumption after succinate addi-tion, but antimycin, an inhibitoro
f complex III, nearly completely inhibited oxygen consumption. Thissug-FIG. 3. Time-dependent recovery of intracellular TDP content in TDA cells after addition of 10 μΜthiamine.Each point represents
F
IG. 4. Electron micrographs of TDA cells (α, b)and TDA cells exposed to 10 IVthia-mine for 1 h (ε, d) . Magnification bars: 1 μm in (α) and (c) ; 0.2 μm in (b) and (d) .THIAMINE
D
EFICIENCY AND BIOCHEMICAL LESIONgests that, under
t
hese conditions, the respiration is indeed sustained by succinate. InT
DA cells, at least some oft
hem
itochondriar
emainf
unctional, andt
he apparent uncoupling ( Fig. 1C) appears tob
e linked to t he lack of oxidί zable substrates. A direct consequence of th is lack ofr
educing substrates is that at least com-plexes III and IV willr
emain completel y oxidized for α long time.T
his may somehow lead to disorganization of cristae,b
ut them
olecular mechanisms involved, if any, remain unknown.M
itochondrial swelling with rupture of cristaeh
as longb
eenk
nown to occur after2181
treatment with ascorbate,
f
errous ions ( Hunter et α1 ., 1963 ), ora
fter treatments thatf
avor lipid peroxidation (Shigenaga et α1 ., Ι 994) . Swelling and inner mem-brane damage were also observed after treatment of mitochondria by thiol-blocking alkylati ng agents ( Lê Qunc andL
ê Qu6c, 1985), glutathione deficiency (.lain et α1., 1991), and deficiency in an enzyme in-volved in cαrdίο1ίρίη synthesis (Ohtsuka et α1 ., 1993) . 1 η all cases described so far,h
owever, there is no evi-dence that the respiratory chain is involved .As thiamine
d
eficiencyh
asb
een reported to affect2182
FIG. 5. Respiration in digitonin-permeabilized TDA cells. The cells were sedimentedandthe culture medium replaced by the test medium containing 125 mM sucrose, 65 mM KCI, 10 mM Tris-HCI (pH 7.2), 1 mM MgC12 , 0.33 mM EGTA, 2.5 mM ΚΗΖΡΟ4 , and 2.5 mM ADP. Trace (α) shows the oxygen con-sumption without addition. In traces (b) and (c), 50μΜdigitonin (DIG) was added. Other additions (trace b) are 16 μ,g/ml oligo-mycin (Ο), 50 ηΜ CCCP, 2MM rotenone (ROT), and 2 μ.g/ml antimycin (Α) .
amino acid metabolism in rat brain (Butterworth and H6roux, 1989 ;
P
age et al., 1989; Butterworth, 1993), wed
etermined the concentrations ofi
ntracellularamino acids in our cells (Table 1) . In control cells, the amino acid concentrations were close to those pre-viously
r
eported in culturedn
eurons and astrocytes (Patel and Hunt, 1985) . As expected, glutamate was them
ost abundant amino acid.W
e wereu
nable to detect any GABA in our cells, an observation that is important for the interpretation of our results as this means that they lack glutamated
ecarboxγlase, an en-zyme specific
f
orGABAergien
eurons (Martin, 1986) .This implies that, in our cells, no succinate can
b
e formedthrough the GABA shuntwhen α-ketoglutarate dehydrogenase is inactive due to lacko
f TDP. Thia-mine
d
eficiency increases them
etabolic flux throughthis
p
athway (Page et α1., 1989) and it can thus be expected that cells with α functionally intact GABA shunt would bem
orer
esistant to thiamine deficiency thancells that do notp
ossess thisp
athway.H
owever, ith
as been shown that glutamate decarboxylase activ -ity and GABA levelsr
eversibly decrease in the brainof
T
Dr
ats (H6roux and Butterworth, 1988),w
hichmay cause disturbances in GABAergic neurotransmis-sion and add to the
r
eversible symptoms caused byt
hiamine deficiency in brain.Aspartate, glycine, and alanine levels were sign ifi-cantly increased in TD compared with control cells. That α1αηίηe is increasedsuggests that
p
yruvate dehy-drogenase is already partially inhibited in these cells; i.e ., pyruvate, instead ofentering the citric acidcycle, is
p
artially transaminated to alanine. Additionof am-prolium leads to α large increase in intracellular
gluta-mine and glutamate concentrations. In
n
eurons, α-k e-tnglutarate andglutamater
apidly and reversibly equ ili-brate inp
arallel withoxaloacetate and aspartate, as α !.Νeυrπιheιπ., Vol. 65, No. 5, /995L.
B
ETTENDORFF ET AL.r
esult of very fast transamination (Erecifiska et α1., 1993) . In braincells, glutamated
ehydrogenase activity is probably less important than transamination in thef
ormationo
fα-ketoglutaratefrom glutamate (McCar -thy andT
ipton, 1983) . Indeed, glutamated
ehydroge-nase is
m
ore likely to catalyze ther
everser
eaction, especiallyin thepresence of high NH4 ι concentrations. In intact brain, glucose ispracticallythe only substrate crossing the blood-brainbarrier and,h
ence, glutamate is essentially formed from α-ketoglutarated
erived from glucose. Thism
ight explain thatin theT
D braing
lutamate levels ared
ecreased (Plaitakis et al., 1979;Butterworthand H6roux, 1989) . In ourcells,
h
owever, glutamine is taken up from the culture medium and is thedirect source of glutamate. In TDA cells, whereα-ketoglutarate
d
ehydrogenase is probably strongly in-hibited, α-ketoglutarate will accumulate (especially in the absence of the GABA shunt) and the glutamatef
ormed from glutamine is no longeroxidized via the citric acid circle.There is nop
arallel increasein aspar -tate concentration presumably because no oxaloacetate can bef
ormed.Addition ofthiamine to the cells leads, within 1
h,
to α significantd
ecrease in intracellular glutamate level. This would be in agreement with αr
ecoveryo
fα-ketoglutarate dehydrogenase activity and oxidative
m
etabolism. The respiration in thepresence of thia-mine is
n
early completely blockedb
y rotenone (an inhibitoro
f NADH dehydrogenase) in agreement with this observation.One ofthe
m
ostpuzzling observations in thiamine deficiency, in animalm
odels as well as in human pa-thology, is that the early symptoms are sor
apidly r e-versed on thiamine administration (Butterworth, 1993) . This is the "biochemical lesion"d
escribed byP
eters (1936) . Prolonged thiamined
eficiency,h
ow-ever, leads to irreversibleh
istological lesions with ne u-ronal death. In this ando
ur previous work (Bettendorffet α1., 1995), we show that, in cultured
n
eιιroblastoma cells, severe thiamined
eficiencyleads to α decreaseinthe
r
ate of respiration, tom
itochondrial uncoupling,TABLE 1 . Amino acid contents of ηcυrπh/αsυηηα cells
under various conditions of thiamine deficiency
Amino acids were determined by HPLC according to the method
ofTapuhi et α1. (1981). The last group
r
epresents TDA cells incu-bated in the presence of thiamine ( ΙΟμΜ)for Ι h. In each case, the culture medium contained 0.4 mΜ glycine and 4mM glutamίne.M
ean - SD valuesf
or three experiments. ηιηο1/ιηg of protein Gin Asp Gin Gly Αlα Control 4.2 0.9 5.2 ± 0.4 65 ± 8 43 ± 5 23 ± 1 TD 3.4±2.0 Ι 2±3 56±13 66±2 41±4 TDA 21 ±8 9.4± 1 165±42 83±7 22±3 ΤDλ + thiamine 58 ± 13 10 ± 3 118 ± 15 6 1 ± 1 22 ± 3REFERENCES
THIAMINE DEFICIENCY AND BIOCHEMICA L LESION and to morphological abnorm alities corresponding to
the early sympto ms of nec rosi s (Wyllie et α1., 1980) . ATP concentrations aredec reased, lactate pr odu ction increases, the ce ll sbecomedepolarized, and cell mor-tality increases ( Bettendorff et α1., 1995) . The most remarkable pr operty shown in the pr esent st udy is that those cells that sur vive long enough un de r seve red efi-ciencyrespond rapidly to thia mine treatment; i.e ., nor-ιηαΙ respiration as we ll as norm al cell and mitochon-drial morphology are recovered within 1 h. Th eser e-sults suggest that the biochemical lesion obse rved in thiamine deficiency is theres ult of an inabili ty of the cells to oxidi ze substrates . En ergy failure lea ds to α cellular co llapse, whi ch, if not treated, res ul ts in necro-sis and cell death.
In α recentreport, Zhang et α1 . (1995)demonstrated
the existence of disintegrating mitochon dr ia and chr o-ιηαtίη clumping in degenerating neurons of dience-phalic n uclei in pyrίthiamine-treatedrats. Th eseres ults suggest that, in brain, mec hanis ms similar to those described here
f
or cult ured neur οblasto ma cells maybe operating, except that additional phenomena such as excitotoxicity mig ht contribute to the selective vul-nerability of certain brainregions in pyrithiaminerats (Langlais and Μαί r, 1990 ; Hazell et α1., 1993 ;
L
anglais and Zh ang, 1993) . Sof
ar, however, there is no evi-dence that excitotoxic phenomena are important during the ac ute, reversible ph ase of thiamine deficie ncy (Wernicke's enceph alopathy ) .Ac kn owle dgment:We than k theBelgian Natio nal Fu nds for Scientific Research (FNR S)
f
or α grant toL.Β.
P.W. is Research Associate at the FNRS .Λίkτιωα Α . Η., Watanabe Ι. S., Furuse Τ., Iwasaki Υ., Satoyoshi Ε., Sιιιηί Τ., and Μπrοjίί Τ. ( 1984)Low energy levels in thiamine-deficient encephalopathy. ./ . Νeυrηηαthοί. Exp. Νeυrο1. 43,
276-287.
ΒαrίΙε Μ ., Ρassarella S., and Quagliariello Ε. (1990) Thiamine pyro-phosphate uptake into isolated rat liver ιτιitochondria. Arch. Bioc hem. Βiορ h,vs. 280, 352-357 .
Bc ιιnett C. D., Jones 1. Η., an d Nelson J. (1966) The effects of' thiamine deficiency on the metabolism of thebrain : 1. Oxidation of' various substrates in vitro by the liver and brain of normal an d pyr ίthia mίne-fed rats . J. Ne urochem. 13, 449-459. 13ettendorffL.and Wins Ρ. ( 1994)Mechanis m of thiamine transport
in ηeuroblastoma ce lls . Inhibition of α hig h affinity carrierby sodium channel activators anddependence of thiamineuptake on membrane potential and intracellular ATP. .Ι. Biol. Chem. 264, 14379- 14385 .
Iicι tcndorff L., Peeters Μ ., Jουαπ C., Wins Ρ., and Schnffenlels Ε. ( 1991 ) Determination of thia mi n and its phosphate esters in Cultured neurons and astrocytes using an ion-pair reversed phase high-performance liquid chromatog raphic method. Anal. Βίο-(hιιιι . 198, 52-59.
ΒctteηdnrffL.,Goessens G., Sl ιιse F., Wins Ρ., Bu reau Μ.,Laschet Ι ., and GrisarΤ. ( 1995 ) Thiamine deficiency in cultured neuro-hlastnma cells: effect on mitoc hondria] function andperipheral henzodia zepίne rece ptors . J. Ne uroc hem. 64, 2013-2021 .
ButterworthR. F .(1993) Pathophysiological mechanisms responsi-ble for the reversible (thia mine- res ponsive) and irreversible
2183 (thiami ne non-responsive) neurological sy mptoms of Wer-ηί ckε's encephalopathy . Dr ug AlcoholRev . 12, 315-322.
Butterworth R . Ε. an d Wrοιιχ Μ. (1989) Effect of pyrίthiami ne treatment and subsequent thiamine re habilitation on regional cerebral amino acids andthiamine- dependent enzymes . J.New
rοchem. 52, 1079- 1084 .
Erecifiska Μ., Pleas ure D., Nelson D., Nissl m Ι ., and Yudkoff Μ . (1993) Cerebral aspartateutili zation: near-equilibri um relation-ships in aspartate aminotransferasereaction. .Ι. Neurochem. 60,
1696-1706.
Goblet C. 1. (1961) Studies on the physiological functions of thia-mine, Ι.The effects of thiaminedeficiency andthiami ne antago-nists on the oxidation of α-keto acids by rat tissues. ./. Biol. Chem . 236, 3112-3120.
Haas R. Η . (1988) Thiamin and the brain. Anna. Rev. Nutr. 8, 483-515.
Ηαkίιη Α. Μ. and Ραρρίυ s Η. Μ. (1983) Sequence of metabolic, clinical andhί stnlogical events in experimenta l thiamine defi-ciency . Ann. Neurol. 13, 365-375.
Hazel] Α. S., ButterworthR. F., and Hakim Α. Μ. (1993) Cerebral vulnerability is associated with selective increase in extracellu-Iar gluta mate concentrations in experimental thiamine de fi-ciency .J. Neurochem. 61, 1155-1158 .
Héroux Β. and ButterworthR. F. (1988) Reversible alterations of cerebraly-aminobutyric acid in pyrithia mine-treated rats: impli-cations for the pathogenesis of Wern icke's encephalopathy . J. Neurochem. 51, 1221-1226.
Hunter F. Ε.Jr., Gebicki 1. Μ., Hoffsten Ρ. Ε., Wei nstei n 1 ., and Scott Α. ( 1963) Swelling and ly sis ofrat liver mitochondria induced byferrous ions . J. Biol. Chem. 238, 828-835. Jain Α., Mártensson J., Stole Ε., ΑυΙd Ρ. Α . Μ., and Meister Α.
(1991 ) Gl ιιtathione deficiency leads to mitochond ria] da mage in brain. Ρrηι. Nod. Αιαιί. Sci. USA 88, 1913-1917. Katzman R. andTerry R.D. (1983) The Neurology of Aging. F.Α .
Davis, Philadelphia .
Langlaίs Ρ. 1. andΜαίr R. G. (1990) Protective effects of the gluta-ιnate antagonists MK-801 on pyrίthiamine-induced lesions and amino acid changes in rat brain. J .Neurosci. 10, 1664-1674.
Langlaίs Ρ. 1. and Zhang S . Χ. (1993 ) Extracellular gl utamate is increased in thalamus du ring thiamine deficienc y-induced le-sions and is bloc ked by MK-801 . J. Ne υrοcheru. 61,
2175-2182,
Lê-Quóc Κ. and Lê-Quδc D. (1985) Crucial role of sulfhydry l groups in the ιnitochondrial inner membrane structure .J. Biol. Chem. 260, 7422-7428.
ΜαιτίηD. L.(1986) Brain gluta matedecarbnx ylase, in Neurotrans-mitter Enzymes (Boulton Α. Α., Baker G. Β., and Υυ Ρ. Η., eds), pp. 261-388. ΗυιηαηαPress, Clifton,NewJersey. McCarthy Α.D.and TiptonΚ.F.(1983) Gl utamate dehydrogenase,
in Glυtιιυιίυ e, Glutamate, and GA BA in the Nervous Sι ste» ι (HertzL., Kva mme Ε.,McGeer Ε. G., and Sc housboe A., Jeds), pp . 19-32. AlanR. Liss, New York.
McEntee W. .Ι. and Μαίr R. G. ( 1990) The Korsakoff syndrome: α neurnc hemίcal perspective. Trends Νeurn.sci. 13, 340-344. Ohts uka Τ., Nlshijima Μ ., Su zuki Κ., and Akamatsu Υ . (1993)
Mitochon dr ia] dysfunction of a cultured Chinesehamster ovary cell mutantdeficient in cardio lipin . J . Biol . Chem. 268, 22914-22919.
Page Μ. G., Αηkοιηα -Sey V., Cou lson W. F., and Benders D. A. (1989) Brain glutamate andy-aιninnbutyrate (GABA)m
etabo-lism in thiamine-deficient rats . Br.J .Nutr. 62, 245-253. Parker W.D. Jr., Haas R., St um pf D. Α., Parks 1., Eguren L. Α.,
andJackson C. (1984) Brain ιnitochondrial metabolis m in
ex-peri mental thiamine deficiency. Neurology 34, 1477-1481 . Patel Α. 1. and Hunt Α. ( 1985 ) Concentration of free amino acids
in primary cultures of neurones andastrnc γtεs. J. Néurochem. 44, 1816-1821.
Peters R. Α. (1936) Th e biochemical lesion in vitamin Β 1 deficiency. Application of modernbiochemical analysis in its diagnosis . Lancet 1, 1161 -1164.
2184
Peterson G.
L.
(1977) Α simplification of the protein assay method ofL
ow ry et α1 . which is more generall y applicable. Anal. Βίο-chem. 83, 346-356.Plaίta kis Α .,NicklasW. 1 .,andBerl S. (1979) Alterations inuptake and meta bolism of as partate and gl utamate inbrainofthi amine deficient animals. Brain Res. 171, 489-502.
Shigenaga Μ . Κ., Hagen Τ. Μ., and Ames Β. Ν. (1994) Oxidative damage and mitoc hondrial decay in aging. Pro( . Natl. Acad. Scί. USA 91 , 10771 -10778 .
Tapu hi V.,
M
iller Ν., an dKarge r Β.L.
(1981)Practical considera-tions in the chiral separation of Dn s-amino-acidsby re versed-phase li quid chro matographyusi ng metalchelate ad ditives .J.
Chromatogr. 205, 325-337.
Vayss ίereJ.
L., L
archerJ. C., BerthelotF., Benlot C.,GrosF.,
and Croi zat Β. (1986)E
ffects on mitochondrial metabolism of.1. Νeιι r-υ(heηι ., Vol. (i5, No. 5, 1995
L.
BEΤTENDORFF ET AL.CCA, one inducer of ne uroblasto ιna differentiation. Bίochem.
Bίophys . Res. Commun. 140,789-796.
Vercesi Α. Ε., Bernardes C.
F.,
Hoffm ann Μ. Ε., GadelhaF. R .,an d DocampoR. (1991 ) Dίgίtπηίη perιneabilizat ίon doesnot affect mitoc hondrial function and allows thedetermination of the mitoc hondrial mem brane potential of Trypanosoma ιrυzί in situ . J. Biol. Chem. 266, 14431 -14434 .
Victor Μ ., AdamsR. D., and Collins G. Η. (1989) TheWernίck
e-Korsakoff Syndrome.
F.
Α. Davis, Philadelphia.Wγllie Α. Η.,Kerr 1 .F.R.,andCurrie Α.R.(1980) Celldeath: the significance of apoptosis. Int. Rev. Cytol. 68, 251-306. Zh ang S. Χ.,Weilersbac her G. S.,
H
ende rso n 5 .W.,Co rso Τ., OlneyJ. W., and